

Abstract
Insulin-like growth factor-binding protein-3 (IGFBP-3) regulates IGF bioactivity and also independently modulates cell growth and survival. By using a yeast two-hybrid screen to identify IGFBP-3-interacting proteins, we cloned humanin (HN) as an IGFBP-3-binding partner. HN is a 24-aa peptide that has been shown to specifically inhibit neuronal cell death induced by familial Alzheimer's disease mutant genes and amyloid-β (Aβ). The physical interaction of HN with IGFBP-3 was determined to be of high affinity and specificity and was confirmed by yeast mating, displaceable pull-down experiments with (His)-6-tagged HN, and ligand blot experiments. Coimmunoprecipitation of IGFBP-3 and HN from mouse testes confirmed the interaction in vivo. In cross-linking experiments, HN bound IGFBP-3 but did not compete with IGF-I–IGFBP-3 binding; competitive ligand dot blot experiments revealed the 18-aa heparin-binding domain of IGFBP-3 as the binding site for HN. Alanine scanning determined that F6A-HN mutant does not bind IGFBP-3. HN but not F6A-HN inhibited IGFBP-3-induced apoptosis in human glioblastoma-A172. In contrast, HN did not suppress IGFBP-3 response in SH-SY5Y neuroblastoma and mouse cortical primary neurons. In primary neurons, IGFBP-3 markedly potentiated HN rescue ability from Aβ1–43 toxicity. In summary, we have identified an interaction between the survival peptide HN and IGFBP-3 that is pleiotrophic in nature and is capable of both synergistic and antagonistic interaction. This interaction may prove to be important in neurological disease processes and could provide important targets for drug development.
Although neuronal apoptosis is a normal event during development, many neurodegenerative diseases are thought to involve abnormal cell death that leads to damage of the nervous system (1). Current therapies to treat, e.g., Alzheimer's disease (AD), rely on intensifying the actions of remaining cells but do not halt disease progression or prevent cell loss (2). Consequently, there has been great interest in the use of neurotrophic factors as therapeutic agents to inhibit cell loss associated with AD and other neurodegenerative diseases (1).
Some of the most promising candidates in drug development for neurodegenerative diseases have been the insulin-like growth factors (IGFs), known for their pleiotrophic roles in promoting cellular survival, proliferation, and differentiation (3). Importantly, the levels of circulating IGF-I are known to decrease during aging, and recently a possible link between low-serum IGF-I and accumulation of amyloid-β (Aβ) in the brain has been reported (4). IGF-I may be a key factor in regulating the clearance of Aβ from the brain through carrier-mediated transport (4). Furthermore, IGF-I has been shown to protect against several neurodegenerative conditions both in vitro and in vivo, including Aβ-induced cell death (5).
IGF-binding proteins (IGFBPs) have been shown to have an essential role in the regulation of cell survival, not only through regulation of free bioactive IGFs, but also via several IGF-independent effects. In particular, IGFBP-3 has been shown to induce apoptosis and inhibit cell growth independently of IGFs (6). IGFBP-3 rapidly translocates to the nucleus, where it specifically and functionally interacts with nuclear proteins, including the nuclear retinoid receptor, RXRα (6, 7). In addition, IGFBP-3 is up-regulated by proapoptotic signal transduction pathways, including tumor necrosis factor α, transforming growth factor β, and the tumor suppressor p53 (8, 9).
The functional roles of IGFBPs in the CNS are still being unraveled. The expression of IGFBPs is known to change in the CNS in response to various insults (10, 11). Recently IGFBP-3 mRNA and protein expression was shown to be significantly up-regulated in AD brain tissue and in response to Aβ exposure in human brain pericytes in vitro (12). This up-regulation of proapoptotic IGFBPs may contribute to neuronal degeneration and apoptosis and subsequently to neuronal loss and clinical symptoms in AD. Importantly, IGF-I-induced neuronal protection against Aβ in vitro is IGFBP-3-sensitive: IGFBP-3 abolishes IGF-I-induced survival (5). In the current study, we demonstrate the interaction between IGFBP-3 and an AD-related survival peptide named humanin (HN).
HN is a recently described AD-related survival peptide of major therapeutic potential (13, 14). This survival-promoting peptide was cloned from a cDNA library extracted from surviving neurons of the occipital lobe of Alzheimer brain and was shown to potently and specifically inhibit neuronal death induced by exogenous Aβ and expression of familial AD mutant genes (14). Recently, HN was shown to bind the proapoptotic Bcl-2 family member, Bax, and convey part of its antiapoptotic effects through inhibition of Bax activation (15).
We describe herein the physical association between IGFBP-3 and HN in vitro and in vivo, identify the binding sites for these molecules, and demonstrate functional effects of HN–IGFBP-3 interaction on cell survival and apoptosis.
Materials
Cell culture reagents were obtained from Life Technologies (Rockville, MD) and American Type Culture Collection. HN peptides [HN, S14G-HN (HNG), C8A-HN (HNA), F6A-HN, K21A-HN, and F6/K21A-HN] were synthesized by Peptides International (Louisville, KY) or Genemed Synthesis Biotechnologies (South San Francisco, CA). Recombinant human IGF-I was provided by Amersham Biosciences. Recombinant human IGFBP-3 and IGFBP-3 peptides were a generous gift from D. Mascarenhas (Protigen, Mountain View, CA). Recombinant human 125IIGFBP-3 and affinity-purified anti-human IGFBP-3 antibody were purchased from DSL (Webster, TX). Rabbit polyclonal anti-HN antibody was generated by Harlan Bioproducts for Science (Madison, WI). Aβ (1–43) peptide was from Peptide Institute (Osaka) (see Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org, for more information).
Methods
Yeast Two-Hybrid Screening and Yeast Mating Assays. Briefly, the yeast strain Saccharomyces cerevisiae HF7c was purchased from Clontech. The fusion gene IGFBP-3/BD was constructed by splicing cDNA encoding the human IGFBP-3 gene into the plasmid pGBT9. A HeLa cDNA library with the activation domain of the GAL4 gene was purchased from Clontech and screened by cotransforming yeast with both plasmids (Clontech Matchmaker protocol handbook). Positive cotransformants were selected, and genes encoding IGFBP-3-binding proteins were isolated by plasmid recovery, amplified, sequenced by using a GAL4 activation domain sequencing primer, and compared with known sequences in GenBank by using the MACVECTOR software program (Oxford Molecular, Williamstown, MA) (see Supporting Text for more information).
Ligand Dot Blot. Western ligand blots were used to assess IGFBP-3 binding as described (6).
Cross-Linking. Cross-linking was performed according to standard procedures. Disuccinimidylsuberate at 0.3 mg/ml was used as a cross-linker. Samples were subjected to SDS/PAGE and autoradiography.
Alanine Scanning. pAla-scanned HN plasmids (14) pHN or pL9R-HN (1 μg of DNA) were transfected in triplicate to F11 cells (14) (7 × 104 cells per well on six-well plates) with Lipofectamine plus. Cell lysates were precipitated with M2 antibody, and 1 or 10 nmol rhIGFBP-3 was eluted through the column according to the standard protocols.
Pull-Down. One nanomolar rhIGFBP3 (Upstate Biotechnology, Lake Placid, NY) was mixed with Ni-NTA agarose beads (Qiagen, Hilden, Germany) immobilizing (His)6x-tagged HN peptides. After the beads were washed, immunoblotting with anti-IGFBP-3 specific antibody was performed according to the standard protocols.
Immunoprecipitation and Immunoblotting. For the coimmunoprecipitation of HN and IGFBP-3 in vitro, A172 cells were treated with 10 μM HN overnight in serum-free conditions. Whole-cell extracts from HN-treated cells were immunoprecipitated according to the standard protocols. For the coimmunoprecipitation of HN and IGFBP-3 in vivo, testes of 3-wk-old mice were homogenized and immunoprecipitated according to the standard protocols.
Cell Culture. A172 glioblastoma and SH-SY5Y neuroblastoma cells (American Type Culture Collection) were maintained and subcultured according to standard procedures. F11 cells and mouse cortical neurons were cultured as described (14).
Caspase Assays. Caspase assay was done by using Apo-ONE homogenous caspase-3/-7 assay (Promega) and performed according to manufacturer's instructions.
Terminal Deoxynucleotidyltransferase-Mediated dUTP Nick End Labeling (TUNEL) Staining. TUNEL staining was performed by using the DeadEnd Colorimetric TUNEL system (Promega) according to the manufacturer's instructions.
WST-8 and Calcein Assays. Primary cortical neurons were treated with or without 25 μM Aβ (1–43) in the presence or absence of 10 pM/10 nM HNG and/or 10 nM (0.35 μg/ml) rhIGBBP3 for 72 h. The WST-8 cell viability assay was performed by using Cell Counting Kit-8, as described (14). The calcein assay was performed with calcein AM (Dojindo, Kumamoto, Japan), as described (14).
Statistical Analyses. Experiments are means of triplicates, and each experiment was performed three to four times. Data are expressed as mean ± SEM. Statistical analyses were performed with SPSS (SPSS, Chicago) by using an unpaired nonparametric Mann–Whitney test. Differences were considered statistically significant when P < 0.05.
Results
Cloning of HN as an IGFBP-3 Partner. A yeast two-hybrid system was used to identify IGFBP-3 partners. As a result, we isolated a 1,061-bp cDNA, in which a 723-bp fragment corresponded to positions 514-1235 of the HN cDNA (GenBank accession no. AY029066.1). The ORF for HN codes a 24-aa peptide, MAPRGFSCLLLLTSEIDLPVKRRA (14). Yeast mating experiments confirmed the interaction between IGFBP-3 and HN, and cotransfected colonies disclosed potent β-galactosidase activity, which was completely absent in single transformants or cotransformants of either hybrid with empty vector. The HN clone was one of five positive clones that reproducibly bound IGFBP-3 in the yeast two-hybrid screen. It was cloned out in four separate experiments (see Supporting Text for details).
Confirmation of IGFBP-3 and HN Interaction. To further verify the interaction between HN and IGFBP-3, we used dot blot and pull-down experiments (Fig. 1). HN peptide (0.3, 3.0, or 7.5 nmol), immobilized to poly(vinylidene difluoride) (PVDF) membrane, showed high affinity and specificity in binding to 125I-labeled rhIGFBP-3 (Fig. 1 A). IGF-I (0.15, 1.5, and 3 nmol) and insulin (0.3, 3.0, and 7.5 nmol) were positive and negative controls, respectively. Two additional mutant HN peptides were also tested: HNG (HN-S14G) and HNA (HN-C8A) (both at 3.0 and 7.5 nmol) showed equal binding to IGFBP-3 when compared with native HN peptide (3.0 and 7.5 nmol) (Fig. 1B). The dot blot experiments confirmed our original finding from the yeast two-hybrid screening, demonstrating association of HN and IGFBP-3, and further revealed that, although S14G and C8A mutations in the HN peptide are known to affect the rescue ability against AD-related insults (14), the binding of these mutants to IGFBP-3 is not affected. As an additional independent confirmation, we used Ni-NTA agarose pull-down of IGFBP-3 with C terminally (His)6x-tagged HN-peptide (Fig. 1C). In this experiment, (His)6x-tagged HN-peptide (1 nmol) pulled down rhIGFBP-3 (1 nmol) in a displaceable manner (Fig. 1C). Untagged HN (100× molar excess) displaced (His)6x-tagged HN from IGFBP-3.
Fig. 1.

IGFBP-3 binds HN. (A) A ligand dot blot experiment showing IGFBP-3 binding to HN. HN, IGF-I, and insulin at 0.15–7.5 nmol were immobilized on a PVDF membrane and probed with125I-labeled IGFBP-3. (B) A ligand dot blot showing equal binding of immobilized HNG and HNA (at 3 and 7.5 nmol) to 125I-IGFBP-3. DMSO and insulin (7.5 nmol) are negative controls, and IGF-I (3 nmol) is a positive control. (C) Ni-NTA agarose column with immobilized (His)6x-HN elutes rhIGFBP-3. Western blot with anti-hIGFBP-3 is shown. Lanes: 1, (His)6x-HN (1 nmol) pull down of IGFBP-3 (1 nmol); 2, displacement of IGFBP-3 from (His)6x-HN with 100 molar excess of unlabeled HN; 3, rhIGFBP-3 positive control. (D) HN coimmunoprecipitates with IGFBP-3 (Left). As negative controls, eluates from beads containing protein A/G and normal goat IgG were used. (Right) Testes of 3-wk-old mice were homogenized in immunoprecipitation buffer and immunoprecipitated with either polyclonal goat anti-mouse-IGFBP-3 or normal goat IgG. Mouse HN runs at ≈4.5 kDa. Eluate from beads containing protein A/G plus IgG was used as negative control. Synthetic HN (60 ng of pure peptide) was used as positive control in both experiments. Results are representative of three independent experiments.
To further verify HN–IGFBP-3 interaction, we treated A172 glioblastoma cells with exogenous HN (10 μM) overnight and immunoprecipitated whole-cell lysates with anti-human IGFBP-3 and normal IgG. IGFBP-3 is abundantly expressed in these cells as examined by Western blotting (data not shown) and as also reported earlier (16). The precipitates from A172 cells were subjected to immunoblot analysis with anti-HN antibody. As shown in Fig. 1D, the ≈3-kDa HN coimmunoprecipitates with IGFBP-3 but not with the control IgG.
HN has been shown to be expressed in the testes of mice (15, 17), and to confirm IGFBP-3–HN interaction in vivo, we immunoprecipitated mouse testis lysates with mouse-specific anti-IGFBP-3 antibody and control IgG. As shown in Fig. 1D, a band corresponding to the reported size of 4.5 kDa of rodent HN (18) was detected in α-IGFBP-3-precipitated samples but not in IgG-precipitated samples. Synthetic HN peptide was used as positive control (≈3 kDa). The testis lysates were confirmed to express HN and IGFBP-3 by Western blot analysis (data not shown).
Structural IGFBP-3-Binding Properties of HN. Amino acids Pro-3, Ser-7, Cys-8, Leu-12, Thr-13, Ser-14, and Pro-19 were found to be essential for the rescue function of HN (14, 19). However, the preliminary analysis of HNG and HNA in our dot blot experiment (Fig. 1B) suggested that the essential amino acids of HN with respect to IGFBP-3 binding might differ from those found to be important with respect to AD insults. To further analyze the nature of HN-binding properties, we performed an alanine scanning of the FLAG-tagged HN peptide (Fig. 2 and Fig. 6, which is published as supporting information on the PNAS web site). Two mutant HN peptides, Phe-6 to alanine (F6A-HN) and Lys-21 to alanine (K21A-HN), did not pull down rhIGFBP-3 in a M2 agarose column (Fig. 2 Left; Fig. 6 Upper), whereas all other HN mutants, as well as the native FLAG-tagged HN, did (see Fig. 6). However, when IGFBP-3 concentration was increased from 1 to 10 nmol (Fig. 2 Left, compare lanes 3–6 with 8–11), K21A-HN pulled down IGFBP-3, whereas F6A-HN still did not, suggesting that F6A-HN has a substantially lower affinity to IGFBP-3 than K21A-HN. A dot blot confirmation (Fig. 2 Right) showed that, whereas HN and K21A-HN (2.5 and 5.0 nmol) showed significant binding to125IIGFBP-3, F6A-HN did not. Furthermore, HN with a double mutation (F6/K21A-HN) did not bind 125IIGFBP-3. Thus, F6 is essential for IGFBP-3 binding. Importantly, F6 is conserved in the HN sequence when compared with the recently cloned rat homologue of HN, rattin (18), suggesting that this residue is critical in HN function.
Fig. 2.

F6A-HN shows reduced IGFBP-3 binding. Alanine scanning of FLAG-tagged pHN, pF6A-HN, and p21A-HN revealed that F6A-HN has decreased affinity to IGFBP-3 when compared with K21A-HN. In contrast to 1 nmol IGFBP-3, at 10 nmol IGFBP-3, M2 antibody pulls down IGFBP-3 from pK21A-HN transfected cells, whereas in pF6A-HN transfected cells, IGFBP-3 does not coimmunoprecipitate. Vector alone was used as a negative control, and IGFBP-3 protein (1 nmol) was used as a positive control. Lysates were subjected to Western blotting with M2 antibody to ensure equal loading (data not shown) (Left). A ligand dot blot experiment confirmed that F6 is essential for IGFBP-3 binding (Right). F6A, K21A, and F6/K21A-HN at 2.5 or 5 nmol were immobilized on a PVDF membrane and probed with125I-labeled IGFBP-3. Both F6A and F6/K21A mutants showed significantly reduced binding. HN at 2.5 and 5 nmol was used as a positive control.
To further analyze the binding affinity of HN and F6A to IGFBP-3, we performed a competitive cross-linking analysis with125I-HN (Fig. 6 Lower).125I-HN runs at ≈3 kDa (not shown), but when it is cross-linked, it forms dimers/oligomers as also suggested recently by Terashita et al. (20). We also performed a ligand dot blot experiment that confirmed the self-binding ability of HN to125I-HN (data not shown). As expected, when IGFBP-3 (1 μg) was cross-linked to125I-HN, the band shifted to the expected size of a HN-IGFBP-3 complex at ≈35–36 kDa. We competed125I-HN with HN that was added at decreasing concentrations (2, 1, and 0.1 μg). HN was able to displace125I-HN completely at 2 and 1 μg (i.e., ≈25× and 10× molar excess), whereas F6A-HN significantly competed only at 2 μg (i.e., 25× molar excess). The band intensities were analyzed with STORM PhosphorImager and IMAGEQUANT software (Molecular Dynamics). F6A-HN shows an order of magnitude reduced displacement ability when compared with HN (Fig. 6 Lower).
The C-Terminal Domain of IGFBP-3 Is Involved in HN Binding. To study the involvement of different IGFBP-3 domains in HN binding, we used a competitive dot blot technique, where HN (2.5 nmol) or HNG (7.5 nmol) were dotted equally onto four PVDF membranes and incubated in separate containers with 125IIGFBP-3 (1 × 105 cpm) either in the presence of (i) a 1 μM competing unlabeled IGFBP-3 protein; (ii) unlabeled 20-aa N-terminal IGF-I-binding domain of IGFBP-3 (amino acids 59–78, TELVREPGCGCCLTCALREG, in human IGFBP-3, GenBank accession no. CAA46087); or (iii) an 18-aa C-terminal domain, previously described as heparin-binding domain (HBD) of IGFBP-3 (amino acids 242–259, KKGFYKKKQCRPSKGRKR) (21) (Fig. 3). The N-terminal peptide blocked IGFBP-3 binding to IGF-I (data not shown), but it did not block 125I-IGFBP-3 binding to HN or HNG. In contrast, the HBD-domain peptide did block HN/HNG binding to a similar extent as full-length IGFBP-3 (Fig. 3). This strongly suggests that the primary binding site of HN to the IGFBP-3 protein is within 18 aa of the C-terminal HBD domain. The N terminus of IGFBP-3 is considered the primary IGF-I-binding site, although the distal C terminus of IGFBP-3 is thought to also contribute to IGF-I binding. In separate experiments, we showed that the N-terminal peptide of IGFBP-3 strongly interferes with 125I-IGFBP-3–IGF-I binding. In contrast, the HBD peptide had no effect on 125I-IGFBP-3–IGF-I binding (data not shown).
Fig. 3.

IGFBP-3 and HN binding is blocked by the HBD of IGFBP-3. HN/HNG–IGFBP-3 binding was assessed in a competitive ligand dot blot experiment, where HN (2.5 nmol) or HNG (7.5 nmol) immobilized onto PVDF membranes were probed with125I-rhIGFBP-3 either without competition or in the presence of 1 μM competing unlabeled125I-IGFBP-3, N-terminal 20-aa peptide of IGFBP-3 (IGFBP-3-N), or C-terminal 18-aa HBD peptide of IGFBP-3 (IGFBP-3-HBD). 125I signals were detected by using storm phosphorimager and imagequant software (Molecular Dynamics). HBD-IGFBP-3 competed to a similar extent as full-length rhIGFBP-3, whereas the IGFBP-3-N-terminal peptide did not block125IIGFBP-3–HN binding. The results are shown as a representative from three independent experiments for both HN and HNG. *, P < 0.05.
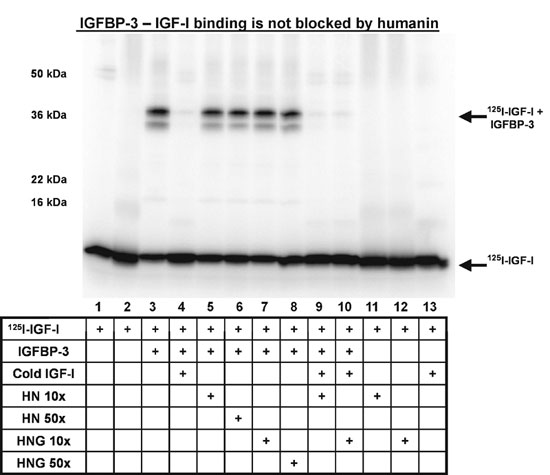
HN Does Not Block IGFBP-3–IGF-I Binding in Vitro. To test whether HN interferes with the IGFBP-3 binding to IGF-I, we performed a cross-linking experiment in the presence of125I-labeled IGF-I (Fig. 7, which is published as supporting information on the PNAS web site). Labeled IGF-I could be found at ≈8 kDa and, when cross-linked to 1 μg of IGFBP-3, it mobility-shifted to ≈38 kDa, corresponding to the size of a rhIGFBP-3–rhIGF-I complex (Fig. 7). Addition of cold IGF-I (1.5 μg) completely displaced the labeled rhIGF-I from rhIGFBP-3 and increased the relative amount of free labeled IGF-I, as determined by densitometric analysis of the bands. A minor decrease in IGFBP-3 bound125I-IGF-I was observed when HN or HNG (1 or 5 μg, i.e., 10× or 50× molar excess to IGFBP-3) was present (Fig. 7). However, the formation of an IGFBP-3–GF-I complex was still substantial. These results indicate that IGF and HN binding occur at different domains of the IGFBP-3 molecule (N-terminal for IGF-I and HBD for HN). Also of note is that no ternary complex of IGF-I, IGFBP-3, and HN was detected.
IGFBP-3 Induces Apoptosis in Neuronal and Glial Cells. IGFBP-3 is known to induce apoptosis and cell growth inhibition in several cell types. In this study, we used a caspase-3/-7-specific fluorometric assay (ApoONE, Promega) to measure the degree of apoptosis induction by IGFBP-3. Overall, the increase in caspase-3/-7 activity varies between 50% and 80% in A172 and SH-SY5Y cells. In A172 glioblastoma cells, the IGFBP-3-induced caspase activation reaches its maximum at 1–6 h, after which it decreases (Fig. 4A). In contrast, SH-SY5Y neuroblastoma cells have a slower response to IGFBP-3 addition, reaching the maximum at 8–12 h and decreasing slowly thereafter (Fig. 4A). Furthermore, IGFBP-3-induced caspase activation in SHSY5Y is blocked with simultaneous addition of IGF-I (100 ng/ml) or phorbol 12-tetradecanoate 13-acetate (TPA) (50 ng/ml) (Fig. 4C). The addition of TPA also blocked “basal” caspase activation, decreasing it below serum-free control level (Fig. 4C). As expected, the addition of IGF-I in serum free conditions also decreases the basal caspase activation (see below and Fig. 4C).
Fig. 4.

HN antagonizes IGFBP-3-induced apoptosis. (A) IGFBP-3 induces caspase-3/-7 activation in the A172 glioblastoma and SH-SY5Y neuroblastoma cells. Cells were treated with exogenous (1 μg/ml) rhIGFBP-3 protein in serum-free (SF) media for indicated times (1, 4, 8, and 24 h). Caspase activity for control was measured at 24 h. (B) IGFBP-3-induced caspase activity in A172 cells is blocked by IGF-I, TPA, and HN. A172 cells were treated in SF media with 1 μg/ml IGFBP-3 alone for 6 h or supplemented with HN (100 nM), TPA (25 ng/ml), and IGF-I (100 ng/ml). (C) SH-SY5Y neuroblastoma cells responded to IGFBP-3 by increase in caspase activity at a later time point than A172, and this activation was not blocked by addition of exogenous HN, HNG, or HNA. The values represent fold change compared with the SF (A172 6 h and SH-SY5Y 16 h) control sample. *, P < 0.05 was considered signifi-cant. The results are from three to four independent experiments. (D) HN but not F6A-HN antagonizes IGFBP-3-induced cell death in A172 cells. TUNEL staining of A172 cells treated for 6 h with IGFBP-3 (4 μg/ml) in SF medium, with or without HN or F6A-HN (500 nM). Cells in SF alone or in 10% serum were used as controls.
HN Specifically Blocks IGFBP-3-Induced Cell Death. To test whether the association of HN to IGFBP-3 has functional effects, we measured IGFBP-3-induced apoptosis in the presence of HN peptide and its derivatives. Our results show that, in addition to specifically binding to IGFBP-3, HN can potently inhibit IGFBP-3-induced apoptosis in vitro. We performed a series of experiments inducing apoptosis by exogenously added IGFBP-3 (1–4 μg/ml) in A172 glioblastoma and SH-SY5Y neuroblastoma cells. As described above, the A172 cells respond to IGFBP-3 more rapidly than SH-SY5Y cells (see above and Fig. 4A), indicating differences in the mechanisms of IGFBP-3 actions between these cell lines. Further experiments revealed that the quick response of A172 glioblastoma cells was HN dependent, because it was potently inhibited by low (100 nM) concentrations of HN peptide or its derivatives, HNG and HNA (Fig. 4B). However, the slow response of SH-SY5Y cells was HN independent, and caspase activity was not affected by addition of HN, HNG, or HNA (100 nM) (Fig. 4C), whereas IGF-I (100 ng/ml) and TPA (25 ng/ml) still had a protective effect against IGFBP-3-induced apoptosis. That SH-SY5Y responded differentially to IGFBP-3, and HN did not affect this response, indicates that the effects of IGFBP-3 differ between cell lines, and that the actions of proapoptotic IGFBP-3 and antiapoptotic HN are interconnected.
To test whether the physical association between HN and IGFBP-3 mediates any of these functional effects, we performed TUNEL staining of A172 cells that were treated with HN (500 nM) and IGFBP-3 (4 μg/ml) for 6 h in serum-free conditions. As expected, addition of IGFBP-3 to cells in serum-free media increased cell death, shown as increased TUNEL staining when compared with cells in serum-free media alone (Fig. 4D). There was no significant increase in staining by serum-free medium alone when compared with samples maintained in 10% serum media. Also no significant differences were observed in cells treated with HN or F6A-HN alone. However, the presence of HN markedly prevented IGFBP-3-induced cell death, with HN-treated cells having similar staining to serum-free control cells. In contrast, addition of F6A-HN, the non-IGFBP-3-binding mutant of HN, was not able to block IGFBP-3-induced cell death in A172 cells, indicating that the IGFBP-3-binding property of HN is an important part of its cell survival protective actions.
Cooperative Interaction Between HN and IGFBP-3. HN, and its more potent derivative, HNG, have been previously shown to specifically protect neuronal cells from Aβ-induced toxicity (14). However, the effects of exogenous IGFBP-3 on primary neuronal cells have not been examined.
First, by using WST-8 and calcein assays, we studied the effects of IGFBP-3 on primary neurons at doses from 1 to 100 nM. Addition of IGFBP-3 at 1–10 nM did not have any significant effects on neuronal viability examined up to 72 h (data not shown). However, addition of 100 nM IGFBP-3 had a modest (20%) decrease in neuronal viability at 72 h, when quantified with WST-8 and calcein assays (data not shown). Addition of HN, HNG, or HNA did not rescue the neurons from IGFBP-3, similar to results obtained in SH-SY5Y cells (see above and Fig. 4C).
Second, to examine the effect of coaddition of IGFBP-3 and HN to Aβ-induced toxicity, we treated primary cortical neurons with Aβ1–43 (25 μM) and HNG either with or without IGFBP-3 (10 nM) (Fig. 5). HNG was used at increasing concentrations ranging from 10 pM to 10 nM. As expected, HNG significantly protected neurons from Aβ1–43-induced death at a concentration of 1 nM and completely suppressed cell death at doses higher than 10 nM. No protection was observed with HNG at doses of 10–100 pM. Surprisingly, coaddition of IGFBP-3 dramatically potentiated HN action: whereas HNG alone at 100 pM had no effect on Aβ toxicity, in the presence of IGFBP-3, a dramatic increase in viability was observed. Ten nanomolar IGFBP-3 together with HNG exerted substantial protection at HNG concentrations of 100 pM (where none was seen without IGFBP-3), and full protection was seen at 1 nM, whereas 10 nM HNG was required without coaddition of IGFBP-3. Representative photomicrographs with calcein-specific fluorescence are shown in Fig. 6 (Fig. 5 Upper). Viability was also quantified by measuring calcein-specific fluorescence in a 96-well format by using a spectrofluorometer. The addition of 10 nM IGFBP-3 potentiated HNG rescue at 100 pM by 10-fold, with viability increasing from 6% to 70%, as measured with calcein staining (P < 0.0001) (Fig. 5 Lower).
Fig. 5.

IGFBP-3 enhances HN protection against Aβ1–43 toxicity. Mouse cortical neurons were plated on poly-l-lysine-coated 96-well plates (5 × 104 cells per well). Neurons were treated for 72 h with 25 μM Aβ1–43 with or without IGFBP-3 (10 nM) and with or without HN (10 pM to 10 nM). Representative photomicrographs are shown from calcein-stained cells at excitation 485 nm and emission 535 nm. Quantification of calcein fluorescence intensity is shown (Lower) (n = 3; **, P < 0.001; ***, P < 0.0001).
Discussion
HN is a recently described small molecular-weight cell survival-promoting agent that has been shown to potently function against AD-related insults in vitro. HN has been suggested to also have a broader survival-promoting activity: it protects human cerebrovascular smooth muscle cells from Aβ-induced toxicity (22). In addition, the activity of HN, or HN-like peptides, may involve protection against other types of cell stress as well, suggested by protection against serum withdrawal (23), and N-methyl-d-aspartate-induced excitotoxic cell death in primary cortical/glial cocultures by the recently cloned rat homologue of HN, rattin (18). The origin of HN and how it is expressed in vivo are still largely unknown (14, 15). HN peptide has a distinct expression pattern during mouse development and in humans is specifically expressed in the AD brain. It is found in large intact neurons of the occipital lobe and in reactive glial cells in the hippocampus (17). Intracellular expression of HN is regulated in part by tripartite motif protein, TRIM11, through a ubiquitin-mediated protein degradation pathway (24). The in vivo functions of HN and the implication of its expression in AD remain to be clarified.
Our findings indicate an interaction between the proapoptotic IGFBP-3 and HN. Moreover, we show that HN can specifically block IGFBP-3-induced cell death in glioblastoma cells. This protection depends on the IGFBP-3-binding capability of HN, as shown by the lack of protection by F6A-HN, a nonbinding mutant of IGFBP-3.
HN, when expressed in mammalian cells, needs to be secreted to be protective (14). The L9R-HN mutant blocks the secretion of the peptide into the culture medium and also makes the peptide ineffective against cell death when transfected to cells. However, when administered exogenously, L9R-HN protects against cell death (14), suggesting an extracellular action for HN and demonstrating its potential functional regulation by IGFBP-3. Indeed, reminiscent of its carrier function for the IGFs, IGFBP-3 could act as a carrier protein for HN both in the circulation and in tissues and thus could be an important regulator of extracellular HN levels as well as transport of HN to the CNS.
Our results demonstrate an association between the IGF system and AD involving IGFBP-3 and HN. HN binds to IGFBP-3 with high affinity and specificity in vitro, with Phe-6 being essential for the binding and also the antiapoptotic function in IGFBP-3-treated cells. Substitution of a critical cysteine residue at position 8 with alanine (HNA) completely destroys the protective activity of HN in AD-related in vitro systems (14, 25). Notably, the residues important for IGFBP-3 binding are different, suggesting that HN acts in both IGFBP-3-dependent and independent fashion. Importantly, Phe-6 is conserved in the recently cloned rat homologue of HN, rattin, indicating the importance of IGFBP-3 binding (18). Guo et al. (15) predicted similar HN homologs by BLAST search in plants and nematodes as well, with Phe-6 being conserved (15).
Recently, HN was shown to interfere with Bax activation and thereby protect from apoptosis (15). HN prevents the mitochondrial translocation of Bax and subsequent release of cytochrome c. Reduced HN levels were shown to predispose cells to apoptotic stimuli in a Bax-dependent manner (15). However, it cannot be ruled out that IGFBP-3 can act as a regulator of HN–Bax interaction. Because IGFBP-3 is present in extracellular, cytoplasmic, and nuclear compartments, this regulation can exist at multiple levels (6). Thus, the exact mechanisms and consequences of IGFBP-3–HN–Bax interactions remain to be clarified.
The interaction between IGFBP-3 and HN appears to involve the HBD of IGFBP-3. Furthermore, we observed in cross-linking and ligand blot studies that HN did not bind IGF-I (data not shown). Consequently, HN may not act as a displacing molecule, liberating IGF-I from IGFBP-3. Because a growing body of evidence indicates changes in the expression of IGFs and IGFBPs in the CNS in response to injury, it is plausible that even though the IGFs are expressed at higher levels, the adjoining expression of the IGFBPs may abate their protective effects (11). Because IGFBP-3 appears to be elevated in AD brain (12), part of HN activity may be to block the apoptotic activity of IGFBP-3 in the CNS, having both direct and indirect survival effects.
Increased IGFBP-3 immunoreactivity in AD brain has been detected both in senile plaques and neurofibrillary tangles, strongly suggesting a role for IGFBP-3 in AD-related cell death (12). If IGFBP-3 contributes to neuronal death during AD, our finding that HN inhibits IGFBP-3-induced caspase activation is of major significance. IGFBP-3 proapoptotic effects are transduced via at least two different cell death pathways. In SH-SY5Y neuroblastoma cells, the slow increase in caspase activity points to transcription-dependent mechanisms that have been proposed to be mediated by RXR binding (6). On the other hand, the rapid increase in caspase activation in A172 glioblastoma cells could be transcription-independent, involving possibly a putative IGFBP-3 receptor, which has been suggested to lead to inhibition of growth as observed in human breast cancer cells (26).
An interesting twist is brought about by our results in primary neuronal cells, where the well established protection of HN against Aβ-induced cell toxicity (14, 18, 20, 24) is potentiated by IGFBP-3. It is recognized in other systems that IGFBP-3 can also be proliferative and synergistic with IGF-I (27). Our results clearly enhance the recently emerging multimechanistic role of IGFBP-3 in the regulation of cell proliferation, survival, and apoptosis and also reveal that the interaction between IGFBP-3 and HN is of biological significance.
In summary, we cloned HN, an AD-related survival factor, as an interacting protein of IGFBP-3. HN is a recently described potent survival-promoting molecule, originally found to be expressed in the occipital lobe of Alzheimer brains. Our results not only show a physical association between IGFBP-3 and HN but also point to a functional interaction with cell type-specific effects, including opposing effects in some models and cooperative effects in others. That HN is expressed in surviving and unaffected areas of AD brain suggests it may be involved in antagonism of IGFBP-3 activity and cell death. In combination with recent studies in other cell culture models, HN represents a promising small-molecular-weight neuroprotective factor with therapeutic potential in AD. Our results validate further investigation of IGFBP-3–HN interaction in the CNS and reinforce the importance of the IGF system in relation to neuronal death and AD.
Supplementary Material
Acknowledgments
We thank Mina Chang and Dr. Heju Li for technical support and Dr. Sami Ikonen for help with the manuscript. This work was supported in part by National Institutes of Health Grants AG20954, UO1 CA 84128, and CA 84128; Pharmacia Grants P50 CA 92131 and P30 DK 063491 (to P.C.); a fellowship award from the Giannini Foundation (to K.-W.L.); and the Academy of Finland Grant 102913 (to M.I.).
Abbreviations: AD, Alzheimer's disease; IGF, insulin-like growth factor; Aβ, amyloid-β; IGFBP, IGF-binding protein; HN, humanin; HBD, heparin-binding domain; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling; PVDF, poly(vinylidene difluoride); TPA, phorbol 12-tetradecanoate 13-acetate.
References
- 1.Bredesen, D. E. (1995) Ann. Neurol. 38 839-851. [DOI] [PubMed] [Google Scholar]
- 2.Conway, K. A., Baxter, E. W., Felsenstein, K. M. & Reitz, A. B. (2003) Curr. Pharm. Des. 9 427-447. [DOI] [PubMed] [Google Scholar]
- 3.Vincent, A. M. & Feldman, E. L. (2002) Growth Horm. IGF Res. 12 193-197. [DOI] [PubMed] [Google Scholar]
- 4.Carro, E., Trejo, J. L., Gomez-Isla, T., LeRoith, D. & Torres-Aleman, I. (2002) Nat. Med. 8 1390-1397. [DOI] [PubMed] [Google Scholar]
- 5.Niikura, T., Hashimoto, Y., Okamoto, T., Abe, Y., Yasukawa, T., Kawasumi, M., Hiraki, T., Kita, Y., Terashita, K., Kouyama, K., et al. (2001) J. Neurosci. 21 1902-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu, B., Lee, H. Y., Weinzimer, S. A., Powell, D. R., Clifford, J. L., Kurie, J. M. & Cohen, P. (2000) J. Biol. Chem. 275 33607-33613. [DOI] [PubMed] [Google Scholar]
- 7.Rajah, R., Valentinis, B. & Cohen, P. (1997) J. Biol. Chem. 272 12181-12188. [DOI] [PubMed] [Google Scholar]
- 8.Grimberg, A. & Cohen, P. (2000) J. Cell Physiol. 183 1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckbinder, L., Talbott, R., Velasco-Miguel, S., Takenaka, I., Faha, B., Seizinger, B. R. & Kley, N. (1995) Nature 377 646-649. [DOI] [PubMed] [Google Scholar]
- 10.Beilharz, E. J., Klempt, N. D., Klempt, M., Sirimanne, E., Dragunow, M. & Gluckman, P. D. (1993) Brain Res. Mol. Brain Res. 18 209-215. [DOI] [PubMed] [Google Scholar]
- 11.Roschier, M., Kuusisto, E., Suuronen, T., Korhonen, P., Kyrylenko, S. & Salminen, A. (2001) J. Neurochem. 76 11-20. [DOI] [PubMed] [Google Scholar]
- 12.Rensink, A. A., Gellekink, H., Otte-Holler, I., ten Donkelaar, H. J., de Waal, R. M., Verbeek, M. M. & Kremer, B. (2002) Acta Neuropathol. 104 525-533. [DOI] [PubMed] [Google Scholar]
- 13.Hashimoto, Y., Tsuji, O., Niikura, T., Yamagishi, Y., Ishizaka, M., Kawasumi, M., Chiba, T., Kanekura, K., Yamada, M., Tsukamoto, E., et al. (2003) J. Neurochem. 84 864-877. [DOI] [PubMed] [Google Scholar]
- 14.Hashimoto, Y., Niikura, T., Tajima, H., Yasukawa, T., Sudo, H., Ito, Y., Kita, Y., Kawasumi, M., Kouyama, K., Doyu, M., et al. (2001) Proc. Natl. Acad. Sci. USA 98 6336-6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo, B., Zhai, D., Cabezas, E., Welsh, K., Nouraini, S., Satterthwait, A. C. & Reed, J. C. (2003) Nature 423 456-461. [DOI] [PubMed] [Google Scholar]
- 16.Shen, L., Dean, N. M. & Glazer, R. I. (1999) Mol. Pharmacol. 55 396-402. [DOI] [PubMed] [Google Scholar]
- 17.Tajima, H., Niikura, T., Hashimoto, Y., Ito, Y., Kita, Y., Terashita, K., Yamazaki, K., Koto, A., Aiso, S. & Nishimoto, I. (2002) Neurosci. Lett. 324 227-231. [DOI] [PubMed] [Google Scholar]
- 18.Caricasole, A., Bruno, V., Cappuccio, I., Melchiorri, D., Copani, A. & Nicoletti, F. (2002) FASEB J. 16 1331-1333. [DOI] [PubMed] [Google Scholar]
- 19.Yamagishi, Y., Hashimoto, Y., Niikura, T. & Nishimoto, I. (2003) Peptides 24 585-595. [DOI] [PubMed] [Google Scholar]
- 20.Terashita, K., Hashimoto, Y., Niikura, T., Tajima, H., Yamagishi, Y., Ishizaka, M., Kawasumi, M., Chiba, T., Kanekura, K., Yamada, M., et al. (2003) J. Neurochem. 85 1521-1538. [DOI] [PubMed] [Google Scholar]
- 21.Campbell, P. G., Durham, S. K., Suwanichkul, A., Hayes, J. D. & Powell, D. R. (1998) Am. J. Physiol. 275 E321-E331. [DOI] [PubMed] [Google Scholar]
- 22.Jung, S. S. & Van Nostrand, W. E. (2003) J. Neurochem. 84 266-272. [DOI] [PubMed] [Google Scholar]
- 23.Kariya, S., Takahashi, N., Ooba, N., Kawahara, M., Nakayama, H. & Ueno, S. (2002) NeuroReport 13 903-907. [DOI] [PubMed] [Google Scholar]
- 24.Niikura, T., Hashimoto, Y., Tajima, H., Ishizaka, M., Yamagishi, Y., Kawasumi, M., Nawa, M., Terashita, K., Aiso, S. & Nishimoto, I. (2003) Eur. J. Neurosci. 17 1150-1158. [DOI] [PubMed] [Google Scholar]
- 25.Niikura, T., Hashimoto, Y., Tajima, H. & Nishimoto, I. (2002) J. Neurosci. Res. 70 380-391. [DOI] [PubMed] [Google Scholar]
- 26.Yamanaka, Y., Fowlkes, J. L., Wilson, E. M., Rosenfeld, R. G. & Oh, Y. (1999) Endocrinology 140 1319-1328. [DOI] [PubMed] [Google Scholar]
- 27.Weinzimer, S. A., Gibson, T. B., Collett-Solberg, P. F., Khare, A., Liu, B. & Cohen, P. (2001) J. Clin. Endocrinol. Metab. 86 1806-1813. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}