

Abstract
It has long been suspected that proteolytic activity associated with advancing growth cones may be required for axon extension. We have isolated mutations in the kuzbanian (kuz) gene, which is expressed in the nervous system and encodes a putative zinc metalloprotease with a disintegrin domain. Drosophila embryos with loss-of-function mutations in kuz have dramatic defects in the development of central nervous system axon pathways, with many axons stalling and failing to extend through the nerve cord. This phenotype is rescued by panneural expression of kuz mRNA in the embryo. These results show that the Kuz metalloprotease is required for axon extension, suggesting a requirement for proteolytic activity at the growth cone surface.
The growth cone, the leading structure of extending axons, senses guidance cues from the cellular environment and guides axon extension. Ramón y Cajal originally suggested that “the growth cone may be regarded as a sort of club or battering ram … forcing apart cellular interstices until it arrives at its destination” (1). Later workers, however, suggested that proteolytic activity may facilitate growth cone penetration through tissues (e.g., ref. 2). Since these early speculations, several studies have shown that proteases and proteolytic activity are associated with extending neurites in vitro (e.g., refs. 3–5). In addition, application of protease inhibitors to cultured neurons has been reported to affect neurite extension (e.g., refs. 6–8). Moreover, PC12 cell lines that are engineered to constitutively express antisense mRNA for a matrix metalloprotease (stromelysin-1) extend neurites as normal in response to nerve growth factor, but their growth cones show a significantly reduced ability to penetrate a basal lamina as compared with the parental cells (9), suggesting that this metalloprotease confers neurite invasiveness in vitro. These studies have suggested that proteolytic activity plays an important role in the extension process. However, it is not clear to what extent growth cones require proteolytic activity in vivo. Do growth cones need proteolytic activity to extend axons through their complex cellular environments? Or do proteases play a more subtle role— for example, helping to refine cellular connections during learning tasks, as recently suggested for the rat cerebellum (10)?
The Drosophila embryo, with its well-defined anatomy and genetic accessibility, provides an excellent system for asking questions about gene function in vivo. To determine which genes are necessary for proper axon growth and targeting in the Drosophila embryo, we have recently undertaken genetic screens to look for mutations which disrupt the development of the central nervous system (CNS) and motor axon pathways (11, 12). These screens have identified several genes required for axon guidance and targeting (13–15). If proteases play an prominent role in axonal extension, then it is likely that these screens should also recover mutations in protease genes.
The recently discovered a disintegrin and metalloprotease (ADAM) family of proteases (16) has been shown to play a diverse set of roles during development. This family of membrane-bound and secreted zinc metalloproteases is characterized by the presence of a disintegrin domain, which has been implicated in binding integrins. ADAM family members act in sperm–egg binding (17), myoblast fusion (18), and neuronal cell fate determination (19), among other processes.
In this report we show that the kuzbanian (kuz) gene, which encodes a metalloprotease of the ADAM family (19), is expressed in the Drosophila nervous system and is required for axonal extension in the embryonic CNS. During two screens for axon pathway development in Drosophila (11, 12), we isolated loss-of-function mutations in the kuz gene. These null or near-null mutations lead to a large-scale stalling of CNS axons. Later-growing axons seem to be more affected than pioneer axons, suggesting that the need for the Kuz protease increases as the complexity of the cellular environment increases. Panneural expression of kuz from a transgene rescues the loss-of-function axon extension phenotype. This is the first demonstration of the requirement for a specific protease in axonal extension in vivo.
MATERIALS AND METHODS
Histology of the Embryonic Nervous System.
Embryos were prepared and stained with antibodies as previously described (12). Antibodies were used at the following concentrations: mAb 1D4 anti-Fasciclin II, 1:10; mAb 3C10 anti-Even-skipped, 1:2; mAb BP102, 1:5; mAb 7G10 anti-Fasciclin III, 1:20. kuz mutations were introduced in a FasIII mutant background (11, 12), and mAb 7G10 was used to identify homozygous mutant embryos. The phenotypes described in this paper do not depend on the absence of Fasciclin III. The duplication used to generate females carrying two copies of kuz was Dp(2;2)Adh3.
In Situ Hybridization.
Digoxigenin-UTP (Boehringer Mannheim)-labeled RNA probes for whole mount in situ hybridization were synthesized from a kuz cDNA inserted into the pBluescript vector (Stratagene) using the T3 and T7 RNA polymerases (Pharmacia). Hybridizations were performed as described in ref. 20 with minor modifications (C. Kopczynski and C.S.G., unpublished data).
Generation of the kuz Rescue Construct.
A 4.9-kb BamHI/EcoRI fragment, blunt-ended with the Klenow fragment of DNA polymerase, containing the complete kuz coding region (19), was inserted into the pUAST vector (21), which had been digested with XbaI and blunt-ended with the Klenow fragment. The resulting construct was used for P element-mediated transformation.
RESULTS
In analyzing mutations generated in two screens for mutations affecting the CNS and motor axon pathways in the Drosophila embryonic nervous system (11, 12), several lethal mutations were recovered that result in the large-scale stalling of CNS axons (see Fig. 3). Mapping and complementation analysis showed that these mutations map to the l(2)34Da locus, recently cloned and renamed kuzbanian (19). kuz encodes a transmembrane protein with extracellular zinc-metalloprotease and disintegrin domains (Fig. 1).
Figure 3.
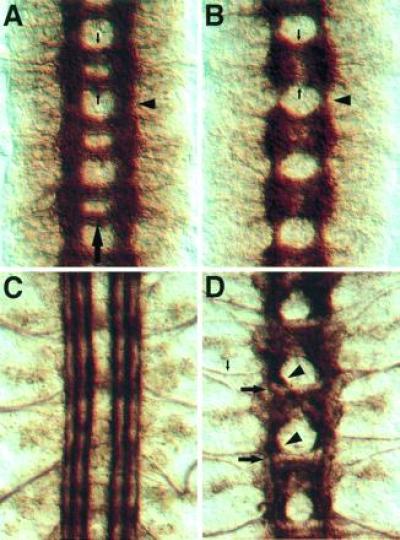
Phenotype of the kuz mutations. Each panel shows three segments of the CNS, and the large arrow in A marks the midline. (A) A wild-type stage 16 embryo stained with mAb BP102, which stains all CNS axon pathways. The arrowhead shows a longitudinal connective in the intercommissural region. The small downward arrow marks the anterior commissure, and the upward arrow indicates the posterior commissure, in one of the segments. (B) A kuz mutant stage 16 embryo stained with mAb BP102. Note the reduction in the longitudinal connectives (arrowhead) in contrast to the intense staining in the commissural region. The commissures (small arrows) are less distinct. (C) A wild-type stage 17 embryo stained with mAb 1D4, which recognizes the Fas II protein. At this stage Fas II is expressed on three parallel axon bundles on either side of the midline. (D) A kuz mutant stage 17 embryo stained with mAb 1D4. The three parallel bundles fail to form. Instead, most axons stall as large clumps in the commissural region (arrowheads). Only a small number of the Fas II-positive axons make it through the longitudinal connective (large arrows). In contrast to the CNS, the axons of the aCC neuron-pioneered intersegmental nerve (small arrow) extend large distances into the periphery, indicating that the CNS axon stalls are not due to a failure in the axon growth machinery.
Figure 1.
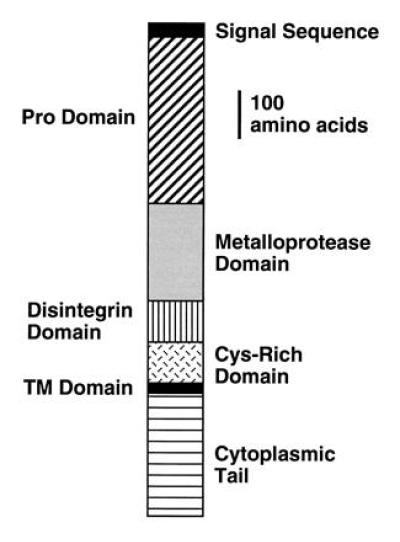
Schematic of the Kuz protein. The protein contains a number of different domains typical of the ADAM family of metalloproteases (16, 19), including a pro-, metalloprotease, disintegrin, cysteine-rich, transmembrane (TM), and cytoplasmic domain.
Analysis of kuz mRNA distribution in the embryo reveals that kuz is broadly expressed in the embryonic CNS, probably in all neurons. (Fig. 2). kuz mRNA is also expressed at lower levels in the epidermis. The distribution of kuz mRNA in the nervous system is consistent with the phenotypes observed in the kuz mutation.
Figure 2.
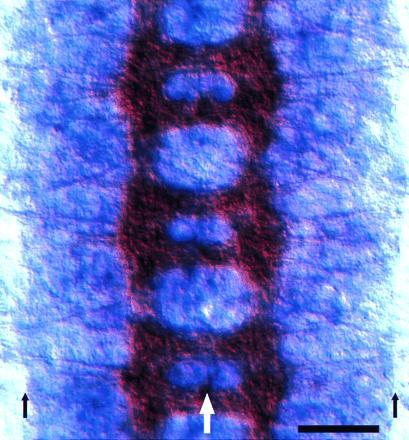
Expression of kuz mRNA in the embryonic nervous system. Three segments of the embryonic CNS from a stage 15 Drosophila embryo are shown. The CNS axon tracts are stained brown with mAb BP102, while kuz mRNA appears purple. kuz appears to be expressed in all neurons at a uniform level. This expression remains constant through the axon extension period. The large white arrow marks the midline and the small black arrows mark the lateral borders of the CNS. (Bar = 20 μm.)
Mutant embryos from heterozygous parents show major defects in the embryonic axon pattern, such as a reduction in the thickness of the longitudinal axon tracts (Fig. 3 A and B). The alleles are likely null or near null, as the phenotypes do not appear stronger when transheterozygous with Df(2L)b88, which removes the kuz gene. This reduction in staining of the longitudinal bundles is accompanied by the accumulation of a mass of axonal material in the commissural region. Staining with mAb 1D4, which recognizes the Fasciclin II protein expressed on three parallel axon bundles on either side of the CNS midline (22, 23), shows that this decrease in longitudinal staining and increase in commissural region staining results from a failure of longitudinal axon bundles to extend through the intercommissural region (Fig. 3 C and D). Instead the bundles stall before crossing the segment boundary and form masses of axon material in the commissural region.
Although the late pattern of axon bundles is highly aberrant in kuz mutants, at least some initial axon projections are normal. For example, the pCC neuron, which pioneers a longitudinal axon bundle (24), extends its axon normally through the intercommissural region (Fig. 4 A and B). Other early-extending neurons also show wild-type trajectories at the outset of axon extension.
Figure 4.
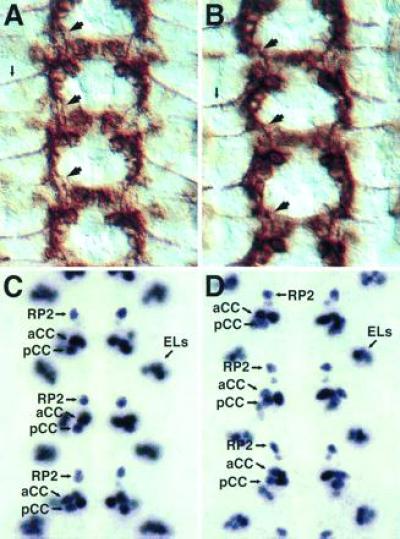
Early-extending axons and cell fates are normal in kuz mutants. Each panel shows three segments of the CNS. (A) A wild-type stage 13 embryo stained with mAb 1D4. Fas II is expressed on a subset of neurons, including the longitudinal connective pioneer neuron pCC and the aCC motoneuron. The pCC axon navigates anteriorly through the connective (large arrows). The aCC axon exits the CNS laterally (small arrow). (B) A kuz mutant stage 13 embryo stained with mAb 1D4. The normal pattern of cells express Fas II. The pCC axon extends its axon through the longitudinal connective (large arrows) and the aCC axon exits the CNS laterally (small arrow), as in wild-type embryos. Overall the CNS is very close to wild type at this stage. (C) A wild-type stage 15 embryo stained with mAb 3C10, which recognizes the Even-skipped nuclear protein. Even-skipped is expressed in a small subset of CNS cells, including the RP2 and aCC motoneurons and the pCC interneuron, as well as a lateral cluster of cells known as the ELs (25). (D) A kuz mutant stage 15 embryo stained with mAb 3C10. The Even-skipped-positive cells are present and in their correct locations, indicting that the cell fates of these neurons are not altered in the kuz mutant.
Analysis of the aCC motoneuron shows that the failure of axon extension observed in the CNS is not due to a failure in the general axon growth machinery. In both wild-type embryos and most segments of kuz mutant embryos the aCC motoneuron extends its axon out of the CNS and around the body wall to the dorsal-most body wall musculature, a distance almost equal to the full length of the CNS at this stage of development (data not shown). Upon reaching its target, aCC shows a normal embryonic synaptic morphology.
The defects observed in the axon bundles also do not appear to be due to a failure of proper cell fate determination. Given the severity of the axon extension phenotype, one would expect fairly broad changes in neural cell fate if that were the cause of the extension defect. Analysis of CNS cell fate markers, such as the Even-skipped (Fig. 4 C and D) and Engrailed proteins (data not shown), reveals no major changes in the patterns of staining cells. Instead, the normal patterns of staining cell nuclei are observed. In the case of Even-skipped, the nuclei of the aCC, pCC, and RP2 neurons are easily observed in approximately their normal positions (the nuclear positions later become more distorted, presumably due to the malformation of adjacent axon tracts). In addition, the nuclei of clusters of neurons of unknown function, such as the EL neurons (25), appear in the correct positions and numbers.
To demonstrate that the defects we observe are due to the loss of the kuz gene, we made transgenic flies carrying the kuz cDNA (19) cloned in the pUAST vector (21). The pUAST vector contains the transcription factor GAL4-responsive upstream activation sequence (UAS) and drives expression only in the presence of GAL4. The C155-GAL4 enhancer trap line drives GAL4 expression panneurally and is located in or near the elav gene (26). Embryos carrying the pUAST-kuz construct and the C155-GAL4 enhancer trap line express high levels of kuz mRNA in all neurons. Embryos chromosomally mutant for kuz, and carrying the pUAST-kuz and C155-GAL4 constructs, were analyzed for CNS axon tract formation (Fig. 5). Panneural expression of kuz mRNA rescues the mutant phenotype, indicating that the kuz gene is responsible for the axon extension defects observed in the kuz mutants.
Figure 5.
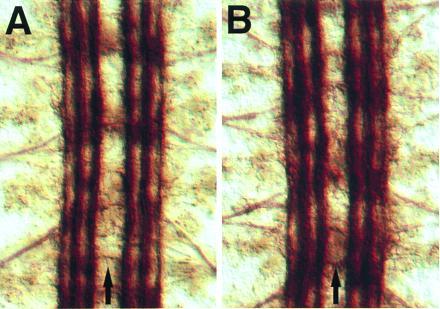
Rescue of the kuz phenotype with panneural expression of a kuz transgene. Each panel shows three segments of the CNS from a stage 17 embryo stained with mAb 1D4. (A) A wild-type embryo, showing three parallel axon tracts on either side of the CNS midline (arrows; same panel as Fig. 3C). (B) An embryo chromosomally mutant for kuz but carrying the C155-GAL4 and pUAST-kuz constructs. These constructs drive expression of kuz from the transgene in all neurons, rescuing the wild-type pattern of axon pathways in the CNS (compare Fig. 3D).
To determine if the axon extension phenotype is a purely zygotic function of kuz, we generated female flies carrying a duplication of the kuz gene. These females should lay eggs containing the wild-type amount of kuz mRNA. If lack of maternally deposited kuz contributes to the axon extension phenotype, then the phenotype of mutant embryos laid by females carrying two copies of kuz should be less severe than the phenotype of mutant embryos laid by females carrying only a single copy of kuz. We observed that kuz mutant embryos laid by both classes of females have an equally severe axon extension phenotype, indicating that lack of maternal kuz does not contribute to the axon extension phenotype.
DISCUSSION
In this paper we show that the panneurally expressed Kuzbanian extracellular metalloprotease is required for embryonic axon extension in Drosophila. In kuz mutant embryos most CNS axons appear to stall during extension, resulting in incomplete lateral axon tracts through the intercommissural region and the accumulation of axonal material in the commissural region of the embryo. Interestingly, analysis of individual pioneer axons, such as the pCC axon, show that these axons can successfully extend through the intercommissural region. Later-extending follower axons are more severely affected. This result is not due to residual maternal kuz in early-extending axons; embryos from mothers carrying a duplication of kuz have a phenotype identical to that of embryos from mothers carrying only a single copy of kuz. If maternal kuz rescues the early-extending axons, then additional maternal kuz should lead to a more complete rescue of axon extension, which was not observed. Later axons may have additional need for the Kuz metalloprotease because they extend their axons through a more complex, denser cellular environment than do pioneer axons.
The Kuz protein contains both a zinc metalloprotease and a disintegrin domain in its extracellular portion, suggesting a requirement for one or both of these domains in the extension process. Previous studies have indicated that proteases are present on axons and suggested a role for them in axon extension and neural remodeling during learning (e.g., refs. 3–10). Moreover, it seems logical that proteases may be required—growth cones must extend through a complex and dense environment, squeezing between apposed cell membranes and penetrating through extracellular matrix. The presence of proteases to degrade matrix and cell surface molecules would presumably facilitate growth cone penetration through the complex cellular environment of the nervous system. Indeed, the ability of growth cones to cause the proteolytic degradation of extracellular matrix components has been well demonstrated (e.g., refs. 4 and 5).
Disintegrin domains bind to integrins and thus can act as adhesion molecules. For example, the disintegrin domain of the fertilin protein, expressed by sperm, binds to the egg α6β1 integrin, mediating the crucial sperm–egg binding step in the fertilization process (17, 27). How disintegrin-mediated adhesion might be required for axon extension is unclear. Drosophila neurons express a wide variety of adhesion molecules (28). Mutations disrupting various Drosophila adhesion molecules, for example the panneurally expressed Neuroglian molecule (29) or the more specifically expressed Fas II molecule (23), do not give rise to axon stall phenotypes like that observed for kuz. The possibility exists that the disintegrin could act as a receptor for specific signaling, but this would be a novel function for a disintegrin.
Additional experiments will be required to distinguish a requirement for proteolytic activity versus a requirement for disintegrin-mediated adhesion in axon extension. The phenotypic rescue experiment, where expression of kuz by the elav promoter (by means of the C155-GAL4 intermediate; see ref. 26) rescues the kuz mutant phenotype (Fig. 5), suggests a way to distinguish the roles of the different domains. It should be possible to express engineered forms of the Kuz protein, lacking either the metalloprotease domain or the disintegrin domain, using the same expression system, and assay the effectiveness of the phenotypic rescue. It will also be interesting to see if different proteases can substitute for kuz function in this system.
kuz appears to play two distinct roles during neural development in the Drosophila embryo. Maternally loaded kuz (i.e., mRNA supplied in the oocyte by the mother) is required for the epidermal versus neural cell fate choice, suggesting that kuz is involved in the lateral inhibition process by which the number of neurons generated is controlled (19). In this paper we have shown that zygotic kuz is required for axon extension. The question arises whether these two functions are truly distinct or rather stem from the same root cause. Specifically, might the axon extension phenotype be related to the neural cell fate specification function of kuz? Three lines of evidence suggest that these are in fact distinct functions of kuz. First, in kuz mutants the expression patterns of various cell surface and nuclear markers are normal (e.g., Fas II and Even-skipped; Fig. 4). If zygotic loss of kuz were affecting cell fate, one would expect these cell markers to show aberrant expression patterns. Second, the initial axon extensions of various cells, such as pCC (Fig. 4), are normal, again suggesting that their cell fates have been correctly specified. Third, the phenotypic rescue is consistent with the axon extension phenotype being unrelated to cell fate defects; C155-GAL4 is an enhancer trap insert in the elav gene, which is expressed in maturing and mature neurons but not in neuroblasts (30, 31). In addition, the GAL4-UAS system introduces an additional delay (approximately 1 hr) before kuz is expressed in this rescue experiment. Thus expression of kuz in neurons is sufficient for rescue—there is no need for kuz expression in neural progenitors to rescue the axon extension phenotype. Maternal kuz, it seems, is sufficient to correctly specify cell fates, while neurons show an additional requirement for zygotic kuz during axon extension.
One alternative hypothesis is that kuz is required to maintain the differentiated neural state. In this scenario loss of kuz expression results in a shutdown of axon extension because neurons revert to a pre-outgrowth state. However, this hypothesis is not consistent with the development of the motoneurons in kuz mutant embryos. The aCC motoneuron, which during development grows the greatest distance of any motoneuron, often successfully reaches its dorsal target muscle and appears to differentiate a synapse. If neurons were reverting to a pre-outgrowth undifferentiated state, then presumably aCC would not show this behavior.
In conclusion, we have identified a Drosophila extracellular metalloprotease that is required for CNS axon extension in vivo. While it is possible that domains of the protein other than the metalloprotease domain may be required for axon extension, it seems likely that the proteolytic domain is responsible for the extension function.
Acknowledgments
We thank Nicholas Seeds and Marc Tessier-Lavigne for thoughtful comments on the manuscript. D.P. is supported by a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund for Medical Research, D.F. was a Predoctoral Fellow and C.S.G. and G.M.R. are Investigators with the Howard Hughes Medical Institute.
Footnotes
Abbreviations: ADAM, a disintegrin and metalloprotease; CNS, central nervous system.
References
- 1.Ramón y Cajal S. Trab Lab Invest Biol. 1906;4:219–284. [Google Scholar]
- 2.Weiss P. J Exp Zool. 1934;68:398–448. [Google Scholar]
- 3.Krystosek A, Seeds N W. Science. 1981;213:1532–1534. doi: 10.1126/science.7197054. [DOI] [PubMed] [Google Scholar]
- 4.Pittman R N. Dev Biol. 1985;110:91–101. doi: 10.1016/0012-1606(85)90067-3. [DOI] [PubMed] [Google Scholar]
- 5.McGuire P G, Seeds N W. Neuron. 1990;4:633–642. doi: 10.1016/0896-6273(90)90121-u. [DOI] [PubMed] [Google Scholar]
- 6.Hawkins R L, Seeds N W. Dev Brain Res. 1989;45:203–209. doi: 10.1016/0165-3806(89)90039-4. [DOI] [PubMed] [Google Scholar]
- 7.Schlosshauer B, Walter J, Bonhoeffer F. Neurosci Lett. 1990;113:333–338. doi: 10.1016/0304-3940(90)90607-b. [DOI] [PubMed] [Google Scholar]
- 8.Sheffield J B, Krasnopolsky V, Dehlinger E. Dev Dyn. 1994;200:79–88. doi: 10.1002/aja.1002000108. [DOI] [PubMed] [Google Scholar]
- 9.Nordström L A, Lochner J, Yeung W, Ciment G. Mol Cell Neurosci. 1995;6:56–68. doi: 10.1006/mcne.1995.1006. [DOI] [PubMed] [Google Scholar]
- 10.Seeds N W, Williams B L, Bickford P C. Science. 1995;270:1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- 11.Seeger M A, Tear G, Ferres-Marco D, Goodman C S. Neuron. 1993;10:409–426. doi: 10.1016/0896-6273(93)90330-t. [DOI] [PubMed] [Google Scholar]
- 12.Van Vactor D, Sink H, Fambrough D, Tsoo R, Goodman C S. Cell. 1993;73:1137–1153. doi: 10.1016/0092-8674(93)90643-5. [DOI] [PubMed] [Google Scholar]
- 13.Hu S, Fambrough D, Atashi J R, Goodman C S, Crews S T. Genes Dev. 1995;9:2936–2948. doi: 10.1101/gad.9.23.2936. [DOI] [PubMed] [Google Scholar]
- 14.Tear G, Harris R, Sutaria S, Kilomanski K, Goodman C S, Seeger M A. Neuron. 1996;16:501–514. doi: 10.1016/s0896-6273(00)80070-7. [DOI] [PubMed] [Google Scholar]
- 15.Fambrough, D. & Goodman, C. S. (1996) Cell, in press. [DOI] [PubMed]
- 16.Wolfsberg T G, Primakoff P, Myles D G, White J M. J Cell Biol. 1995;131:275–278. doi: 10.1083/jcb.131.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almeida E A C, Huovila A-P J, Sutherland A E, Stephens L E, Calarco P G, Shaw L M, Mercurio A M, Sonnenberg A, Primakoff P, Myles D G, White J M. Cell. 1995;81:1095–1104. doi: 10.1016/s0092-8674(05)80014-5. [DOI] [PubMed] [Google Scholar]
- 18.Yagami-Hiromasa T, Sato T, Kurisaki T, Kamijo K, Nabeshima Y-I, Fujisawa-Sehara A. Nature (London) 1995;377:652–656. doi: 10.1038/377652a0. [DOI] [PubMed] [Google Scholar]
- 19.Rooke, J., Pan, D., Xu, T. & Rubin, G. M. (1996) Science, in press. [DOI] [PubMed]
- 20.Kopczynski C, Muskavitch M A. J Cell Biol. 1992;119:503–512. doi: 10.1083/jcb.119.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brand A H, Perrimon N. Development (Cambridge, UK) 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 22.Grenningloh G, Rehm E J, Goodman C S. Cell. 1991;67:45–57. doi: 10.1016/0092-8674(91)90571-f. [DOI] [PubMed] [Google Scholar]
- 23.Lin D M, Fetter R D, Kopczynski C, Grenningloh G, Goodman C S. Neuron. 1994;13:1055–1069. doi: 10.1016/0896-6273(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs J R, Goodman C S. J Neurosci. 1989;9:2412–2422. doi: 10.1523/JNEUROSCI.09-07-02412.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel N H, Schafer B, Goodman C S, Holmgren R. Genes Dev. 1989;3:890–904. doi: 10.1101/gad.3.6.890. [DOI] [PubMed] [Google Scholar]
- 26.Lin D M, Goodman C S. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 27.Myles D G, Kimmel L H, Blobel C P, White J M, Primakoff P. Proc Natl Acad Sci USA. 1994;91:4195–4198. doi: 10.1073/pnas.91.10.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hortsch M, Goodman C S. Annu Rev Cell Biol. 1991;7:505–557. doi: 10.1146/annurev.cb.07.110191.002445. [DOI] [PubMed] [Google Scholar]
- 29.Bieber A J, Snow P M, Hortsch M, Patel N H, Jacobs J R, Traquina Z, Schilling J, Goodman C S. Cell. 1989;59:447–460. doi: 10.1016/0092-8674(89)90029-9. [DOI] [PubMed] [Google Scholar]
- 30.Bier E, Ackerman L, Barbel S, Jan L, Jan Y N. Science. 1988;240:913–916. doi: 10.1126/science.3129785. [DOI] [PubMed] [Google Scholar]
- 31.Rabinow S, White K. J Neurobiol. 1991;22:443–461. doi: 10.1002/neu.480220503. [DOI] [PubMed] [Google Scholar]