

Abstract
Synchronized network responses in thalamus depend on phasic inhibition originating in the thalamic reticular nucleus (nRt) and are mediated by the neurotransmitter γ-aminobutyric acid (GABA). A suggested role for intra-nRt connectivity in inhibitory phasing remains controversial. Recently, functional GABA type B (GABAB) receptors were demonstrated on nRt cells, and the slow time course of the GABAB synaptic response seems ideally suited to deinactivate low-threshold calcium channels. This promotes burst firing, a characteristic feature of synchronized responses. Here we investigate GABAB-mediated rebound burst firing in thalamic cells. Whole-cell current-clamp recordings were obtained from nRt cells and somatosensory thalamocortical relay cells in rat brain slices. Synthetic GABAB inhibitory postsynaptic potentials, generated by a hybrid computer–neuron synapse (dynamic clamp), triggered rebound low-threshold calcium spikes in both cell types when peak inhibitory postsynaptic potential hyperpolarization was greater than −92 mV. The threshold inhibitory postsynaptic potential conductance for rebound burst generation was comparable in nRt (7 nS) and thalamocortical (5 nS) cells. However, burst onset in nRt (1 s) was considerably delayed compared with thalamocortical (0.6 s) cells. Thus, GABAB inhibitory postsynaptic potentials can elicit low-threshold calcium spikes in both relay and nRt neurons, but the resultant oscillation frequency would be faster for thalamocortical–nRt networks (3 Hz) than for nRt–nRt networks (1–2 Hz). We conclude, therefore, that fast (>2 Hz) GABAB-dependent thalamic oscillations are maintained primarily by reciprocal connections between excitatory and inhibitory cells. These findings further indicate that when oscillatory neural networks contain both recurrent and reciprocal inhibition, then distinct population frequencies may result when one or the other type of inhibition is favored.
Keywords: rebound bursts, oscillations, synchronization
The thalamus is capable of generating multiple types of oscillations that act as pacemakers of thalamocortical (TC) rhythms. Among those are spindle waves and delta oscillations (reviewed in refs. 1 and 2), which regulate the global attentive states of the forebrain and are responsible for some pathological forms of spike and wave discharges in epilepsy.
The oscillations primarily depend on mutual connectivity of TC relay cells and inhibitory neurons of the adjacent nucleus reticularis thalami (nRt) and the propensity of both cell types to fire bursts of action potentials from a relatively hyperpolarized resting state. Burst firing in thalamic cells is mediated by a low threshold spike (LTS) that is generated by low-voltage-activated (T-type) calcium channels (1, 2). T channels in TC and nRt cells can be distinguished by their biophysical properties as well as by their somatodendritic distribution. In nRt cells, (i) T channels are thought to be located mainly in the dendrites (3, 4); and (ii) they have a slower time course of inactivation and deinactivation and a higher threshold of activation compared with TC cells (5). nRt cells receive excitatory synaptic input from relay cells and, in turn, generate inhibitory postsynaptic potentials (IPSPs) in TC cells. These IPSPs have both γ-aminobutyric acid type A (GABAA) and type B (GABAB) receptor-mediated components and are capable of inducing rebound burst firing in relay cells (6, 7). The GABAA component is critical for spindle generation, whereas the GABAB component mediates slower, more synchronous epileptiform oscillations (6). The slow time course of the GABAB-mediated IPSP is particularly suited to deinactivate T channels in relay cells (reviewed in ref. 8), but a similar action has not yet been shown in nRt cells.
The role of nRt proper in synchronizing and maintaining thalamic oscillations is controversial. The surgically isolated nRt sustains spindle activity, suggesting a pacemaker role of this nucleus (9). This view has recently been supported by computer models of nRt showing that assemblies of interconnected nRt cells segregate into different cell clusters, which maintain oscillations by burst firing out of phase (10). In contrast, disconnection of nRt from the relay nuclei abolished spindle waves in slices of the ferret lateral geniculate nucleus, indicating that the spindles result from network interactions between relay and nRt cells (7).
In rodents, nRt cells are interconnected by axon collaterals (e.g., ref. 11). These recurrent connections appear to be mediated mainly by GABAA receptors (12–14). As yet, the net effect of this lateral inhibition remains uncertain, although transient hyperpolarizations in nRt cells can trigger rebound bursts of action potentials (15, 16). Direct application of GABA to nRt cells leads to depolarization or weak hyperpolarization (17, 18), leaving some uncertainty about the net effect of a GABAA receptor-mediated increase in chloride conductance in these cells. However, focal application of the GABAA receptor antagonist bicuculline within nRt results in an increased output from this nucleus, suggesting a disinhibitory effect of GABAA receptor blockade (7, 13). In addition, intra-nRt GABAB IPSPs have been investigated by computer simulations, demonstrating their potential role in synchronizing burst firing in this nucleus (19).
In a recent in vitro study, we found a weak GABAB-mediated component in a subpopulation of intra-nRt synapses (20). However, the maximally evokable GABAB synaptic responses in our preparation were quite small (<0.2 nS), which prevented us from systematically examining their role on thalamic cell firing. Therefore, in the present study, we investigate GABAB receptor-mediated burst firing in nRt and relay cells by using a hybrid neuron–computer synapse generated by a dynamic clamp (21). This method allowed us to examine in an in vitro slice preparation the potential function of larger synaptic responses (1–35 nS) that may occur in vivo, where the connectivity within the circuit is probably much higher. The goal of the present study was to determine the threshold and minimal delay of GABAB-mediated rebound burst firing in nRt cells compared with relay neurons in order to demonstrate the potential role of GABAB synapses in synchronizing thalamic network activity.
METHODS
Tissue Preparation.
Sprague Dawley rats of either sex, postnatal days 11–13, were anesthetized with pentobarbital (50 mg/kg, i.p.) and decapitated. The brain was removed, transferred into ice-cold solution containing 234 mM sucrose, 11 mM glucose, 24 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 10 mM MgSO4, and 0.5 mM CaCl2, and equilibrated with 95% O2/5% CO2. Horizontal slices (200 μm) were cut with a Vibratome (TPI, St. Louis), incubated at 32°C in physiological saline containing 126 mM NaCl, 26 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgCl2, 2 mM CaCl2, and 10 mM glucose, and equilibrated with 95% O2/5% CO2 for at least 1 h before recording.
Electrophysiology.
Patch pipettes were pulled from borosilicate glass (Garner Glass, Claremont, CA) and filled with 120 mM K-gluconate, 11 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, and 11 mM EGTA (pH adjusted to 7.3 with KOH, osmolarity 300 mosmol). Slices were transferred into a perfusion chamber and superfused with physiological saline at 30°C. Whole-cell recordings were performed under visual control using a microscope equipped with an infrared-sensitive camera (22). A dynamic clamp based on the method of Sharp et al. (21) was applied through an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA). Bridge balance was monitored throughout the experiment, and unstable recordings were rejected. A personal computer connected to a Labmaster TM-100 A/D converter (Scientific Solutions, Solon, OH) performed the dynamic clamp calculations and updated the injected current at 3 kHz. A liquid junction potential of −12 mV was subtracted on line.
Data Analysis.
Curve fittings were performed in Origin (Microcal, Amherst, MA). Data are presented as mean ± SEM and n designates the number of cells. Nonparametric statistics were determined with the Mann–Whitney U test.
RESULTS
The experiments shown were designed to test the hypothesis that GABAB-mediated IPSPs are capable of generating rebound burst responses in nRt cells and to compare the results quantitatively with data obtained from relay cells, where it is known that GABAB IPSPs can generate a rebound burst under physiological conditions (6). The time course of the GABAB receptor-mediated conductance underlying late IPSPs in thalamic cells has been described previously by a fourth power exponential activation and double exponential inactivation kinetics (20) similar to that in granule cells in the hippocampus (23). The time constants and weights of the fits were left constant while the peak conductance was varied.
Basic Characteristics of Rebound Bursts in Thalamic Cells.
In a first set of experiments, the basic features of LTS responses in thalamic cells were determined by ramp and step current injections. As described previously (e.g., ref. 5), relay and nRt cells had similar basic electrophysiological features. The mean resting potentials of both cell types were similar (TC: −77 ± 2.6 mV, n = 10; nRt: −78 ± 2.3 mV, n = 18). The mean input resistance in relay and nRt neurons was 282 ± 48 MΩ and 222 ± 15 MΩ, respectively; thus, they were not significantly different. The membrane time constant (τm) was not different in the two cell types (TC, 64 ± 6.9 ms; nRt, 53 ± 6.2 ms). Fig. 1 A and B shows examples of current-clamp recordings in a relay cell (Fig. 1A) and an nRt neuron (Fig. 1B). In all cells, robust rebound burst firing was generated upon relaxation of the membrane potential from step hyperpolarizations (postanodal exaltation, Fig. 1 A and B). The burst consisted of a depolarizing envelope, mediated by a low-threshold calcium spike, which triggered high-frequency action potentials (Fig. 1 A and B). Relay cells were clearly distinguishable from nRt cells not only by their characteristic localization within the slice (6, 14), but also by the presence of a hyperpolarization-activated cation current (Ih; ref. 24) that produced a pronounced sag in the hyperpolarizing voltage response (e.g., Fig. 1A, arrow). In addition, bursts in relay cells normally contained fewer action potentials compared with nRt cells, and the underlying calcium spike had a faster rise and decay time course (5).
Figure 1.
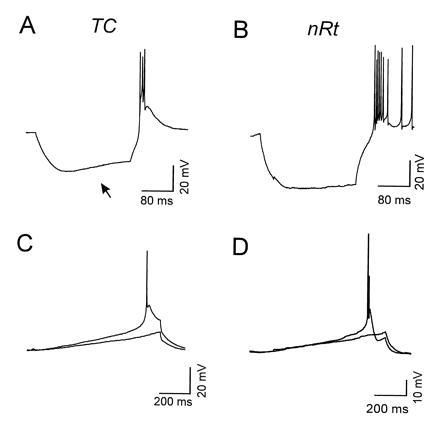
Rebound burst firing in thalamic neurons. (A and B) Hyperpolarizing current injections from the resting membrane potential (A, −72 mV; B, −70 mV) induced rebound bursts of action potentials overriding a low threshold calcium spike in a relay cell (A) and an nRt neuron (B). (A) Note the depolarizing sag in the relay cell response resulting from activation of a hyperpolarization-activated cation current (arrow). (C and D) Bursts of action potentials in a relay cell (C) and an nRt neuron (D) triggered by ramp currents from a hyperpolarized resting state of −85 mV (C) and −75 mV (D). Two traces are shown around threshold for each cell type. The slope of the threshold ramp was 0.026 mV/ms (C) and 0.022 mV/ms (D).
The threshold stimulus for burst initiation was determined by injecting depolarizing ramp currents (25, 26) with a slope of 10–100 pA/s from a relatively hyperpolarized steady-state membrane potential of approximately −80 mV, at which T channels are deinactivated to a significant (≈50%) degree (Fig. 1 C and D; ref. 5). Fig. 1 C and D shows examples of ramp-induced membrane depolarizations in a relay cell (Fig. 1C) and an nRt neuron (Fig. 1D) with slopes sub- and suprathreshold for burst firing. The minimal rate of rise of the membrane potential that was capable of initiating a low-threshold spike was not different between relay and reticular neurons (TC, 23 ± 2 mV/s; nRt, 21 ± 2 mV/s). In addition, the voltage threshold for burst firing was similar in both cell types (TC, −73 ± 2.8 mV; nRt, −74 ± 1.5 mV).
In summary, the present experiments confirm that nRt and relay cells both are capable of generating rebound burst responses after step hyperpolarizations, and that under steady-state conditions the threshold stimulus for burst generation is similar in the two cell types.
Slow IPSP-Mediated Burst Firing.
Although step and ramp currents are capable of initiating burst firing in thalamic neurons, they are not particularly physiological stimuli. Therefore, we applied a dynamic clamp to relay and nRt cells, which allowed for the injection of currents with a time course mimicking synaptic events. The time constants of the GABAB kinetics in nRt and relay cells were, respectively, 46 ms and 36 ms for activation, 134 ms (relative weight, 87%) and 104 ms (92%) for the fast inactivation, and 495 ms (13%) and 642 ms (8%) for the slow inactivation. Thalamic cells were held at −60 to −65 mV by dc injection. A GABAB-like current of varying amplitude was injected into the neurons according to the precalculated conductance and the measured driving force. The potassium reversal potential was set to −105 mV (6, 20). Fig. 2 shows examples of computer-generated IPSPs that were near threshold for burst firing in a relay cell (Fig. 2A) and a reticular neuron (Fig. 2B). Dynamic clamp IPSPs with a peak conductance in the range of 1–20 nS were capable of generating rebound bursts in 13 of 15 nRt and in 8 of 10 relay cells with stable resting membrane potentials. The mean peak amplitude of the threshold IPSP was −92 mV in both cell types. The current injected into the cells by the dynamic clamp to generate the IPSP is shown in Fig. 2C for the relay cell and in Fig. 2D for the reticular neuron, and the underlying conductances are shown in Fig. 2 E and F. The mean threshold peak conductance for burst firing was found not to be significantly different in relay compared with nRt cells (TC, 5.0 ± 0.9 nS; nRt, 7.2 ± 1.1 nS).
Figure 2.
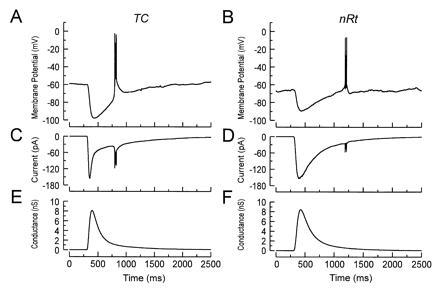
GABAB-mediated rebound burst firing in thalamic cells. (A and B) Examples of IPSPs generated by the dynamic clamp, which were capable of generating rebound bursts of sodium spikes in a relay cell (A) and a reticular neuron (B). Note the delayed onset of the low-threshold spike in the reticular neuron compared with the relay cell. (C and D) Time course of the current injected by the dynamic clamp into the relay (C) and the nRt (D) cell. Note that, as expected for a K+-mediated synaptic conductance, the magnitude of the current varied with the membrane potential. The underlying conductance in the relay (E) and reticular (F) neuron had a time course typical for GABAB receptor-mediated synaptic events in these cells.
However, a prominent difference between nRt and relay cells was the delay of the burst, i.e., the time between IPSP onset and the occurrence of the first spike, which was significantly longer in nRt compared with relay neurons. The mean (threshold) burst delay in relay and nRt cells was 580 ± 80 and 970 ± 70 ms, respectively (P < 0.002). The corresponding minimum delay values were 380 and 800 ms (Fig. 3 C and D). A longer delay to burst in nRt cells could be partially explained by the finding that the voltage threshold for rebound burst firing induced by GABAB IPSPs was depolarized by 4.8 ± 0.7 mV compared with the ramp-triggered LTS (P < 0.0001, paired t test). By contrast, no stimulus-dependent differences in voltage threshold were found in relay cells. Given the rate of GABAB IPSP decay, to reach the increased voltage threshold in nRt cells, the membrane potential had to rise for approximately an additional 250 ms. This accounts in large part for the observed difference in LTS delay between the two cell types. Another possible factor that may contribute to the shorter delay of the burst in relay cells is the activation of Ih during the GABAB response, which would accelerate membrane depolarization leading to an earlier response. However, it was found that Ih is not critical in determining the LTS delay in relay cells, because bath application of 1 mM Cs+, which inhibits Ih (24) and blocked the hyperpolarization-induced sag, had no consistent effect on the delay of the threshold burst (n = 4; data not shown).
Figure 3.
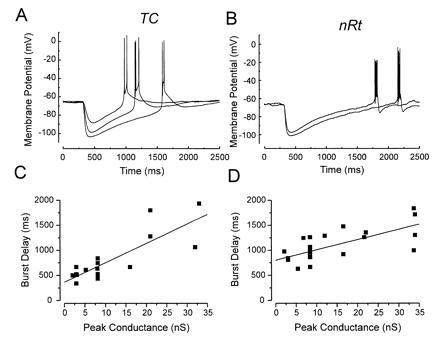
(A) Relationship between IPSP amplitude and delay of LTS onset in relay (A and C) and nRt (B and D) neurons. (A and B) Examples of IPSPs in thalamic cells that were suprathreshold for LTS generation. (C and D) Scatter plots of burst delay versus peak conductance of all IPSPs recorded. Straight lines are linear regression fits (C, r = 0.71; D, r = 0.83). The slope of the fit was 39 ms/nS in C and 20 ms/nS in D. The y-intercepts in C and D were 366 ms and 805 ms, respectively.
When threshold was reached, further increases of the IPSP peak conductance prolonged the delay to burst onset, as shown in Fig. 3 A and B for a relay and an nRt cell, respectively. Fig. 3 C and D shows the linear relationship between burst delay and peak conductance of all IPSPs recorded from relay (Fig. 3C) and nRt (Fig. 3D) neurons. These data suggest that the frequency of intrathalamic oscillations can be regulated within a wide range by changes in the strength of GABAB synapses.
DISCUSSION
A dynamic clamp was used in the present study to assess GABAB IPSP-mediated rebound burst firing in thalamic cells. The experiments have been carried out at a developmental stage, when GABAB receptor density was shown to be highest in thalamus (27). Although T channel density may still be increasing (28), the robust thalamic oscillations obtained from the same preparation indicate that rebound burst firing in thalamic cells is already prominent at this age (6, 14).
GABAB Receptor-Mediated LTS in Thalamic Cells and Its Functional Significance.
By applying a dynamic clamp to thalamic cells, we could experimentally demonstrate that a synaptic conductance with the kinetics characteristic of a slow GABAB IPSP is capable of inducing rebound burst firing in reticular and relay neurons. Whereas GABAB-mediated LTS generation in relay cells has been experimentally demonstrated before (reviewed in ref. 8), comparable data for nRt cells are not yet available. The dynamic clamp allows for adjustment of the peak conductance of the IPSP gradually and across a wide dynamic range in a way that would be difficult, if not impossible, to obtain by extracellular stimulation in vitro. In the present study, we found that the mean threshold IPSP peak conductance for LTS generation was not significantly different in nRt compared with relay cells.
The dynamic clamp GABAB conductance necessary to evoke an LTS in thalamic reticular cells (7 nS) was more than one order of magnitude higher than the mean peak conductance of GABAB-mediated IPSPs obtained by focal stimulation in nRt (0.14 nS; ref. 20). The critical point here is to know how much GABAB conductance is activated during thalamic oscillations in which many neurons fire in synchrony, therefore coreleasing large amounts of neurotransmitter that would theoretically lead to significant GABAB receptor activation (29). Huguenard and Prince (6) estimated the conductance underlying GABAB receptor-mediated inhibitory postsynaptic currents in relay cells between 0.4 and 7.4 nS during delta-like oscillations in thalamic slices. This is well in the range necessary for LTS generation in relay cells, as shown in the present study. Unfortunately, comparable data are not available for nRt cells at present. Baclofen application to thalamic cells activated a potassium conductance that was four times smaller in nRt cells compared with relay neurons (20). These data lead to the suggestion that the number of available GABAB receptors on nRt cells is smaller, and that the slow IPSPs that occur in only a subpopulation of nRt cells may remain subthreshold for LTS generation.
If GABAB receptors in nRt cells were concentrated in the dendrites, then our burst threshold conductances (≈7 nS) deduced from dynamic clamp experiments with “somatic” IPSPs might be an overestimate. Furthermore, because T channels in nRt cells are thought to be predominantly dendritic (e.g., ref. 4), colocalization of dendritic GABAB receptors with T channels might be an interesting mechanism that would promote distal LTS generation in reticular neurons (e.g., ref. 30). However, simulation studies suggest that, because of the slow time course of the GABAB IPSP, the site of inhibitory synapses in nRt cells does not strongly influence rebound burst generation (unpublished observations).
The time interval between successive bursts in relay neurons during thalamic network activity is occupied primarily by the time course of the IPSP (31). The burst delay estimated in relay cells (400–1700 ms; Fig. 3C) extrapolated to a temperature of 37°C with a Q10 of 2.1 (23) would result in an interburst frequency of 1–4 Hz, which is well within the frequency range of delta oscillations in this preparation (14). By contrast, a slower interburst frequency of 1–2 Hz would result from intra-nRt GABAB-mediated burst firing.
The difference in the delay of rebound burst firing in TC and nRt cells could be explained by the higher LTS threshold in nRt cells after the GABAB IPSP compared with the ramp input. This difference presumably results from the slow rate of deinactivation or repriming of T channels in nRt cells (5). However, IPSP and ramp thresholds were identical in relay cells. It appears then that the threshold for burst firing, especially in nRt cells, is a dynamic variable depending on the preceding trajectory of the membrane voltage (cf. ref. 32). For example, with the current ramps obtained from a steady-state resting potential of −80 mV, ≈50% of the total T channels would be available in both cell types, and their voltage thresholds for LTS generation are similar. By contrast, because the time constant of T channel deinactivation is about twice as long in nRt cells (≈300 ms at −80 mV and 30°C; ref. 5) compared with relay cells (≈150 ms), then during GABAB IPSPs, we estimate that T channels in nRt cells would have recovered to a level ≈30% less than those in relay cells. This lower post-IPSP channel availability in nRt cells presumably results in a depolarizing shift in threshold for regenerative LTS generation.
Formally, the intrathalamic circuitry can be considered as a recurrent inhibitory network with reciprocal inhibition. It is interesting that during GABAB-dependent thalamic oscillations, it seems that the excitatory–inhibitory loop dictates the frequency of the rhythm. In other words, recurrent excitation from TC cells most likely reactivates nRt cells in a pre-emptive fashion such that IPSP-triggered bursts do not occur. This type of recurrent network interaction is supported by numerical computer models (33) and data obtained from in vitro thalamic network preparations (6, 7). It is interesting to speculate that in such a network, where postinhibitory rebound is a feature of both the inhibitory and excitatory elements, that differential regulation of the strength of recurrent versus reciprocal inhibition may result in a shift in the network oscillatory activity from one mode to another. Overall, these findings reinforce the view that intrinsic cellular properties and intercellular interactions must both be taken into account when interpreting neural network behavior (34).
We conclude that GABAB receptor-mediated IPSPs are capable of inducing LTS in relay cells with a time delay compatible with synchronous network activity in the delta frequency range, but intra-nRt GABAB-mediated LTS generation seems to play little, if any, role for phasing these thalamic oscillations.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke (NS 06477, NS34774), the Pimley Research Fund, the Roche Research Foundation, and the Schweizerische Stiftung für medizinisch-biologische Stipendien.
Footnotes
Abbreviations: GABA, γ-aminobutyric acid; GABAA and GABAB, GABA types A and B, respectively; IPSP, inhibitory postsynaptic potential; LTS, low threshold spike; nRt, nucleus reticularis thalami; TC, thalamocortical.
References
- 1.Steriade M, Llinás R R. Physiol Rev. 1988;68:649–742. doi: 10.1152/physrev.1988.68.3.649. [DOI] [PubMed] [Google Scholar]
- 2.Steriade M, McCormick D A, Sejnowski T J. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 3.Mulle C, Madariaga A, Deschênes M. J Neurosci. 1986;6:2134–2145. doi: 10.1523/JNEUROSCI.06-08-02134.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Destexhe A, Contreras D, Steriade M, Sejnowski T J, Huguenard J R. J Neurosci. 1996;16:169–185. doi: 10.1523/JNEUROSCI.16-01-00169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huguenard J R, Prince D A. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huguenard J R, Prince D A. J Neurosci. 1994;14:5485–5502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Krosigk M, Bal T, McCormick D A. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- 8.Crunelli V, Leresche N. Trends Neurosci. 1991;14:16–21. doi: 10.1016/0166-2236(91)90178-w. [DOI] [PubMed] [Google Scholar]
- 9.Steriade M, Domich L, Oakson G, Deschênes M. J Neurophysiol. 1987;57:260–273. doi: 10.1152/jn.1987.57.1.260. [DOI] [PubMed] [Google Scholar]
- 10.Destexhe A, Contreras D, Sejnowski T J, Steriade M. J Neurophysiol. 1994;72:803–818. doi: 10.1152/jn.1994.72.2.803. [DOI] [PubMed] [Google Scholar]
- 11.Cox C L, Huguenard J R, Prince D A. J Comp Neurol. 1996;366:416–430. doi: 10.1002/(SICI)1096-9861(19960311)366:3<416::AID-CNE4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Bal T, von Krosigk M, McCormick D A. J Physiol (London) 1995;483:665–685. doi: 10.1113/jphysiol.1995.sp020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huguenard J R, Prince D A. J Neurophysiol. 1994;71:2576–2581. doi: 10.1152/jn.1994.71.6.2576. [DOI] [PubMed] [Google Scholar]
- 14.Ulrich D, Huguenard J R. Neuron. 1995;15:909–918. doi: 10.1016/0896-6273(95)90181-7. [DOI] [PubMed] [Google Scholar]
- 15.Avanzini G, DeCurtis M, Panzica F, Spreafico R. J Physiol (London) 1989;416:111–122. doi: 10.1113/jphysiol.1989.sp017752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bal T, McCormick D A. J Physiol (London) 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCormick D A, Prince D A. Nature (London) 1986;319:402–405. doi: 10.1038/319402a0. [DOI] [PubMed] [Google Scholar]
- 18.Spreafico R, DeCurtis M, Frassoni M, Avanzini G. Neuroscience. 1988;27:629–638. doi: 10.1016/0306-4522(88)90294-1. [DOI] [PubMed] [Google Scholar]
- 19.Wang X-J, Rinzel J. Neuroscience. 1993;53:899–904. doi: 10.1016/0306-4522(93)90474-t. [DOI] [PubMed] [Google Scholar]
- 20.Ulrich D, Huguenard J R. J Physiol (London) 1996;493:845–854. doi: 10.1113/jphysiol.1996.sp021427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp A A, O’Neil M B, Abbot L F, Marder Y. J Neurophysiol. 1993;69:992–995. doi: 10.1152/jn.1993.69.3.992. [DOI] [PubMed] [Google Scholar]
- 22.Stuart G J, Dodt H-U, Sakmann B. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 23.Otis T S, DeKoninck Y, Mody Y. J Physiol (London) 1993;463:391–407. doi: 10.1113/jphysiol.1993.sp019600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCormick D A, Pape H-C. J Physiol (London) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jahnsen H, Llinás R. J Physiol (London) 1984;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crunelli V, Lightowler S, Pollard C E. J Physiol (London) 1989;413:543–561. doi: 10.1113/jphysiol.1989.sp017668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turgeon S M, Albin R L. Neuroscience. 1994;62:601–613. doi: 10.1016/0306-4522(94)90392-1. [DOI] [PubMed] [Google Scholar]
- 28.Pirchio M, Lightowler S, Crunelli V. Neuroscience. 1990;38:39–45. doi: 10.1016/0306-4522(90)90372-b. [DOI] [PubMed] [Google Scholar]
- 29.Destexhe A, Sejnowski T J. Proc Natl Acad Sci USA. 1995;92:9515–9519. doi: 10.1073/pnas.92.21.9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deschênes M, Madariaga-Domich A, Steriade M. Brain Res. 1985;334:165–168. doi: 10.1016/0006-8993(85)90580-3. [DOI] [PubMed] [Google Scholar]
- 31.Bal T, von Krosigk M, McCormick D A. J Physiol (London) 1995;483:641–663. doi: 10.1113/jphysiol.1995.sp020612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch C, Bernander Ö, Douglas R J. J Comput Neurosci. 1995;2:63–82. doi: 10.1007/BF00962708. [DOI] [PubMed] [Google Scholar]
- 33.Golomb D, Wang X-J, Rinzel J. J Neurophysiol. 1994;72:1109–1126. doi: 10.1152/jn.1994.72.3.1109. [DOI] [PubMed] [Google Scholar]
- 34.Getting P A. Annu Rev Neurosci. 1989;12:185–204. doi: 10.1146/annurev.ne.12.030189.001153. [DOI] [PubMed] [Google Scholar]