

Abstract
Behavioral and electrophysiological studies on mutants defective in the Drosophila inebriated (ine) gene demonstrated increased excitability of the motor neuron. In this paper, we describe the cloning and sequence analysis of ine. Mutations in ine were localized on cloned DNA by restriction mapping and restriction fragment length polymorphism (RFLP) mapping of ine mutants. DNA from the ine region was then used to isolate an ine cDNA. In situ hybridization of ine transcripts to developing embryos revealed expression of this gene in several cell types, including the posterior hindgut, Malpighian tubules, anal plate, garland cells, and a subset of cells in the central nervous system. The ine cDNA contains an open reading frame of 658 amino acids with a high degree of sequence similarity to members of the Na+/Cl−-dependent neurotransmitter transporter family. Members of this family catalyze the rapid reuptake of neurotransmitters released into the synapse and thereby play key roles in controlling neuronal function. We conclude that ine mutations cause increased excitability of the Drosophila motor neuron by causing the defective reuptake of the substrate neurotransmitter of the ine transporter and thus overstimulation of the motor neuron by this neurotransmitter. From this observation comes a unique opportunity to perform a genetic dissection of the regulation of excitability of the Drosophila motor neuron.
Keywords: neuronal excitability, genetic analysis
Neurotransmitters released into synapses can affect the excitability of target neurons by an effect on ion channels. The effects of these neurotransmitters can be attenuated by members of families of proteins called neurotransmitter transporters that catalyze rapid transmitter reuptake (for review, see refs. 1 and 2). The Na+/Cl−-dependent family of transporters catalyze the reuptake of transmitters such as dopamine, serotonin (5HT), norepinephrine (NE), γ-aminobutyric acid (GABA), and others. The importance of members of this family in controlling neuronal function is demonstrated by the observation that they can serve as targets for drugs such as cocaine, amphetamines, and certain classes of antidepressants (refs. 3 and 4; for review, see ref. 5) and by the striking behavioral and neurophysiological abnormalities exhibited by mouse mutants defective in the dopamine transporter (6). Despite the importance of these transporters, their roles at the molecular level in controlling the development and function of their target neurons are incompletely understood.
One useful method to address this question is to isolate and perform a phenotypic analysis of transporter-defective mutants. In Drosophila, mutations in several genes that cause increased neuronal excitability have been isolated (7–11). Several of these genes encode ion channel structural or regulatory proteins (12–15). Mutations in the ine gene confer increased excitability of the larval motor neuron. At the behavioral level, ine mutations enhance the phenotypes of mutations in the Shaker (Sh) and the Hyperkinetic (Hk) genes, which encode K+ channel α (12) and β (14) subunits, respectively. In particular, Sh; ine or Hk; ine double mutants exhibit a “down-turned wings and indented thorax” appearance. This appearance is also exhibited by Sh mutants overexpressing the sodium channel structural gene para or carrying an additional mutation in eag, which encodes a K+ channel α subunit distinct from Sh (13, 16, 17), and is thus an appearance exhibited by Sh mutants when excitability is further increased. In addition, following brief high-frequency nerve stimulation and in the presence of the K+ channel blocking drug quinidine, ine mutants display an abnormal repetitive firing of action potentials and increased transmitter release from the larval motor neuron (7). This phenotype is also exhibited by other mutants exhibiting increased excitability as a result of decreased K+ currents or increased Na+ currents (8, 10, 13–17). In contrast, in an otherwise wild-type background or in the absence of quinidine, ine mutations conferred no obvious behavioral or electrophysiological abnormalities. Thus it was proposed that ine mutations increased excitability, either by increasing Na+ currents or reducing K+ currents, but because of functional redundancy of neuronal ion channels, these effects were revealed phenotypically only when certain K+ channels were blocked by drugs or mutations (7). Other genotypes in which ion currents are affected display a similar functional redundancy (ref. 17 and references therein).
In this paper, we report the cloning and sequence analysis of ine. We used restriction mapping and restriction fragment length polymorphism (RFLP) mapping to localize three ine mutations to within a 10-kb region of DNA. Sequence analysis of a cDNA isolated from this region revealed similarity to members of the Na+/Cl−-dependent neurotransmitter transporter family. We confirmed that this cDNA encodes Ine protein by sequence and transcription analysis of this gene in ine mutants. Hybridization in situ of the ine cDNA to developing embryos revealed expression in a variety of different cell types, including the posterior hindgut, Malpighian tubules, and a subset of cells in the central nervous system. We conclude that loss of function of the ine transporter and presumed overstimulation by the substrate neurotransmitter increases excitability of the Drosophila motor neuron, possibly by an effect on ion currents.
MATERIALS AND METHODS
Mutations and chromosomes used are described in ref. 18.
Mutant Isolation.
The isolation of the ine1 and ine2 mutants was described previously (7). The ine3 and ine4 mutants were isolated as follows: w f Sh133; bw males (carrying an isogenized second chromosome) were mutagenized with 5000 rads of γ-rays generated from a 137Cs source (Department of Immunology, Baylor College of Medicine; 1 rad = 0.01 Gy). These males were crossed with w f Sh133; CyO/ine1 females carrying a closely linked P element marked with w+ (called Pw25C). F1 progeny exhibiting the “downturned wings” phenotype (7) were selected. The newly induced mutations were distinguished from the ine1 tester allele by the absence of the w+ marker. The ine3 and ine4 mutants were obtained from approximately 50,000 progeny flies.
RFLP Mapping.
Fly lines carrying recombination events around the ine gene were generated as follows: w; CyO/ine1 Pw25C or w; CyO/ine2 Pw25C flies were crossed to line G63 (19): w flies carrying P element marked with w+ at 24E (called Pw24E). Lines carrying recombination events between the P elements were generated by selecting about 500 white-eyed flies from about 40,000 total progeny. Lines were established from these recombinant flies and their genotype at ine was determined by crossing to Sh133; CyO/ine1 tester flies and examining the progeny for the “down-turned wings” phenotype. Of these 500 recombinants, 88 were ine+; in these, recombination had occurred between Pw24E and ine, and these were assayed for RFLPs in the region with Southern blot hybridization.
Nucleic Acid Manipulations.
Nucleic acid manipulations were performed as described (20). RFLPs between the G63 and ine mutant parent chromosomes were identified by digesting genomic DNAs with 15 enzymes and performing Southern blot hybridization using whole phage clones as probes. The ine cDNA was the single positive obtained from a screen of 5 × 105 plaques of a 0- to 24-h embryonic cDNA library (21). This cDNA was subcloned via its SacI and ApaI sites into pBluescript (SK+) and sequenced on both strands by the automated fluorescent dye terminator method (D. Needleman, University of Texas Medical School). RNA preparation and cDNA synthesis for reverse transcription-coupled polymerase chain reaction (PCR) was performed using the GIBCO/BRL Trizol reagent and SuperScript preamplification system kits, respectively. Generation and subcloning of reverse transcription-coupled PCR products for sequencing was performed using the Invitrogen TA or Promega pGEM kits. The Qiagen Qiaquick PCR purification kit was used to prepare PCR products for direct sequencing.
RESULTS
Identification and Characterization of the ine cDNA.
The ine gene was previously localized to the 24E–25A region of chromosome 2 (7). DNA from this region was obtained by “chromosome jumping” from an inversion broken in decapentaplegic and in 24F2 (the inversion stock and DNA flanking the inversion were kindly supplied by W. Gelbart, Harvard University, Cambridge, MA). Additional cloned DNA in phage or cosmids from the region was obtained in collaboration with R. MacIntyre (Cornell University). We used RFLP analysis to localize ine1 and ine2 onto this cloned DNA. We generated 88 lines of flies carrying recombination events between Pw24E and ine1 or ine2 (pooled). Of these, two had undergone recombination between RFLP-1 and RFLP-2 (Fig. 1A), and two had undergone recombination between RFLP-2 and ine. An additional two had undergone recombination between RFLP-1 and ine and were not further localized. None had undergone recombination centromere-proximal to RFLP-3. In addition, a deletion of about 2.4 kb was detected in this region in the ine3 mutant (Fig. 1A).
Figure 1.

Organization and sequence analysis of the ine transcription unit. (A) The centromere-proximal and centromere-distal directions were determined by restriction mapping and chromosomal in situ hybridization and are indicated by arrows. The top horizontal line represents genomic DNA, the lower line represents the ine cDNA, and the horizontal bar above genomic DNA represents the location and extent of the ine3 deletion. Positions of EcoRI sites are indicated with vertical lines. The location of the three RFLPs that distinguish the ine+ from the ine1 or ine2 chromosomes and that were used to define the ine transcription unit are indicated by downward arrows. RFLP-1 (to the left) and RFLP-3 (to the right) are EcoRI polymorphisms, whereas RFLP-2 (in the middle) is a NaeI polymorphism; each are either single base-pair changes or very small deletions or insertions. The EcoRI fragment used to isolate the ine cDNA is shown by brackets. From sequence comparison between genomic and the cDNA, we established that the ine cDNA begins 629 nt from the EcoRI site located to the right of the start of the ine cDNA. The location of the seven introns are indicated, but lengths and locations are not drawn to scale. (B) Nucleotide sequence and deduced amino acid sequence of the ine cDNA. The end point of the ine3 deletion is indicated by an arrow beginning at A878. The location of the ine4 mutation (G → A531) is indicated by a #. The 12 transmembrane domains are underlined and labeled with roman numerals. Consensus sequences for CK2 (solid square), PKC (solid circle) and glycosylation (∗) are shown. The intron locations are indicated with bars and numbered 1–7. Intron 1 is about 780 bp long, intron 2 is about 835 bp long, intron 4 is 69 bp long, intron 5 is 56 bp, intron 6 is 65 bp, and intron 7 is 63 bp. The size of intron 3 has not been determined but is greater than 400 bp long.
A cDNA library prepared from RNA from 0-h to 24-h embryos (21) was probed with one EcoRI fragment (depicted in Fig. 1A), and a single cDNA 2.3 kb long was obtained, subcloned, and sequenced on both strands (Fig. 1B). Sequence analysis revealed an open reading frame (ORF) of 658 amino acids with a calculated molecular weight of about 75,000. The putative start codon is preceded by a satisfactory match to the Drosophila consensus sequence for the start of translation (22). There are four in-frame stop codons, and no other methionine codons, in the 140 bp between this putative start codon and the beginning of the cDNA. The cDNA is apparently complete at the 3′ end because it contains a run of 16 adenosines preceded by a consensus poly(A) addition site (23). Sequence comparison between this cDNA and genomic DNA from the region enabled the localization of seven introns (locations shown in Fig. 1B) and the determination of the direction of transcription. Hydropathy analysis of the ine cDNA indicated 12 membrane-spanning domains, shown in Fig. 1B.
Sequence Similarities with Members of the Na+/Cl−-Dependent Neurotransmitter Transporter Family.
Computer searches of the GenBank and GenEMBL data banks demonstrated that ine shares significant sequence similarity with members of the Na+/Cl−-dependent neurotransmitter transporter family. Members of this family have been shown to catalyze the reuptake of neurotransmitters such as glycine, dopamine, 5HT, NE, GABA, as well as metabolites or osmolytes such as taurine, betaine, β-alanine, and creatine. The ine transporter shows the strongest sequence similarity to the rat GABA transporter and human GABA/betaine transporter (40.9% and 40.6% sequence identity, respectively); however, the ine transporter also shows significant (40.4% and 39.9% identity, respectively) homology to the human dopamine and NE transporters. Fig. 2 shows a sequence alignment among several members of this transporter family, including ine. Several structural features common to members of this family are observed in ine. These features include 12 transmembrane domains, a large extracellular domain between the third and fourth transmembrane domains that contains two cysteine residues spaced 9 amino acids apart (residues 173 and 182) and a conserved tryptophan (residue 243) in the fourth transmembrane domain that has been proposed to be important for incorporation of members of this family into the membrane (30). However, the ine transporter is apparently a more distantly related member of this family: ine displays differences in 51 of the 168 amino acids conserved among the five other transporters shown in Fig. 2.
Figure 2.

Sequence alignment of the amino acid sequence of the ine transporter with certain other members of the Na+/Cl−-dependent neurotransmitter transporter family. Sequence sources are as follows: human betaine/GABA transporter (hBGT) (24), rat GABA transporter GAT2 (rGABA) (25), human dopamine transporter (hDAT) (26), human NE transporter (hNE) (27), and Drosophila 5HT transporter (dSERT) (28, 29). Boxed areas indicate identical amino acids among sequences. Alignments are based on a Dayhoff matrix using the pileup program of the Genetics Computer Group sequence analysis software pakcage.
The ine transporter does not appear to be more closely related to the Drosophila 5HT transporter dSERT than to other transporters. In fact, there are only 19 amino acids that are the same in ine and dSERT but different in the four other transporters shown. In contrast, there are 31 amino acids that are the same between ine and the two GABA transporters but different in the three monoamine transporters shown.
Assignment of This Transporter as the ine Gene Product.
To determine whether ine encodes this transporter, we used PCR and reverse transcription-coupled PCR to amplify and sequence the transporter gene in ine mutants. We were unable to detect any changes in the transporter coding region in ine1 mutants. The location of the 2.4-kb deletion found in ine3 was determined by amplifying and sequencing DNA from this region with PCR primers flanking the deletion. The deletion removes DNA from codon 293 to about 700 nt past the transcription stop site. Finally, ine4 is a nonsense mutation at codon 177. Both ine3 and ine4 are likely to be null mutants. Thus, the sequence analysis of two independent ine mutations, in addition to the observation that ine1 flies produce no detectable transporter transcripts (see below), constitute our evidence that ine encodes this transporter.
Transcription of ine in Wild-Type and ine Mutants.
Northern blot analysis of poly(A)+-selected RNA prepared from whole flies and probed with the 3′ end of the ine cDNA exhibited two bands, one with a size of about 2.6 kb and a broad band with a size centered around 2.3 kb. Each RNA species is approximately the size of the ine cDNA (Fig. 3). Occasionally a faint band with a size of about 4.9 kb is observed (data not shown); this band could represent unspliced or partially spliced ine transcripts. The 2.3- and 2.6-kb transcripts are undetectable in RNA prepared from ine1 flies; thus, the ine1 mutation might affect the expression, splicing, or stability of the ine message. As expected, these transcripts are absent from RNA prepared from ine3 flies as well. These transcripts appear at normal levels in ine2 flies (Fig. 3).
Figure 3.
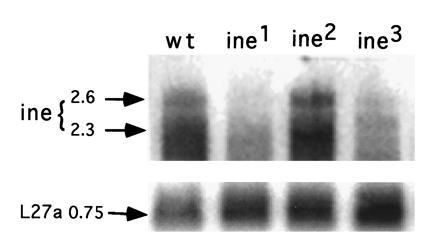
Analysis of ine transcription in wild-type and ine mutant flies by Northern blot analysis. Total RNA was prepared from adult flies. mRNA was isolated using oligo(dT)-cellulose type 77F (Pharmacia), size-separated on a 1% agarose gel that contained 6% formaldelyde, and then transferred to GeneScreen membrane (DuPont). The 3′ end of the ine cDNA (≈800 bp long) was labeled with [32P]dCTP and used as probe. Lanes 1–4 are iso bw (the isogenic wild-type parent to ine mutants), ine1, ine 2, and ine 3, respectively. The relative amounts of mRNA loaded in each lane was deduced from a control probe prepared from ribosomal protein L27a (0.75-kb band). Quantitation of L27a band intensity (arbitrary units): iso bw, 1.0; ine1, 1.5; ine2, 1.6; ine3, 2.1. The image was exposed for 3 days and processed with the FujiX BAS 1000 phosphorimager system.
The Spatial Pattern of ine Transcription in Embryos.
To determine which cell types in the developing embryo express ine, whole mount in situ hybridization was performed with ine antisense RNA as a probe. RNA was present uniformly in the cellular blastoderm, presumably introduced as maternal message. During germ-band extension (stage 9), the primordium of the hindgut shows elevated levels of transcripts. During germ-band retraction (stage 13), the midgut, Malpighian tubules, garland cells, anal plate, and foregut also express transcripts, and specific hybridization to head regions becomes apparent. This central nervous system staining is segmentally repeating in cells flanking the midline of the ventral ganglion. This central nervous system expression pattern is similar to the dSERT expression pattern reported previously (28). In contrast to ine, however, there was no nonneuronal expression reported for dSERT. Fig. 4 shows dorsal and lateral views of ine expressing tissues in stage 15–16 embryos.
Figure 4.
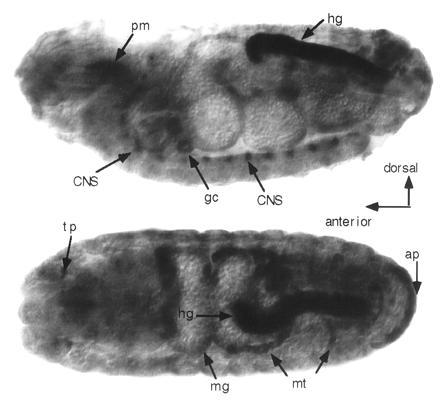
Whole-mount in situ localization of ine transcripts in late-stage embryos. (Upper) Lateral view. (Lower) Dorsal view. Strong staining can be seen in the posterior hindgut (hg), anal plate (ap), Malpighian tubules (mt), midgut (mg), tracheal pits (tp), pharyngeal muscle region (pm), and a subset of segmentally repeating cells in the central nervous system. Additional small patches of specific staining in the head were difficult to identify. Embryo preparation, fixation, hybridization, and staining were conducted as described (31). The probe was prepared from antisense RNA transcribed from the ine cloned cDNA using SP6 RNA polymerase (Boehringer Mannheim). Probe of sense RNA prepared from T7 RNA polymerase (Boehringer Mannheim) was ulilized as a control. The probe contained 200–300 bases from the 5′ end of the ine cDNA. The color reaction was allowed to proceed for 10 h.
DISCUSSION
Previous behavioral and electrophysiological analyses on mutants defective in the Drosophila ine gene revealed increased excitability in the motor neuron (7). In this paper we have described the cloning and sequence analysis of the ine gene. We have found that ine encodes a member of the Na+/Cl−-dependent neurotransmitter transporter family, which in Drosophila and other organisms is responsible for the reuptake of transmitters such as dopamine, 5HT, NE, GABA, and others. Thus, mutations that reduce or eliminate transporter function cause increased excitability of the motor neuron. The mechanism of action by which this increased excitability occurs is unknown; however, the ine-encoded transporter might control the function of the target motor neuron in a manner similar to the action of related transporters studied in other systems. For example, mutations in the mouse dopamine transporter increase the duration of dopamine presence in synapses and thus cause over stimulation of target neurons with dopamine (6). In addition, drugs such as cocaine, amphetamines, and certain classes of antidepressants which block various monoamine transporters are thought to exert their physiological and behavioral effects by similar mechanisms (4). Elimination of the ine transporter might similarly cause overstimulation of the target motor neuron with the substrate neurotransmitter of this transporter.
How could this overstimulation account for the ine mutant phenotypes? As described previously, ine mutants exhibit several features indicative of increased neuronal excitability, which was proposed to result either from increased Na+ currents, or decreases in one (or more) K+ currents (7). Because ion channels are prominent targets of neurotransmitter-mediated regulation of neuronal excitability (for review, see ref. 32), it seems possible that in ine mutants, overstimulation of the motor neuron by the substrate neurotransmitter of ine could then cause overactivation of a signaling pathway leading to increases in Na+ channel activity or decreases in the activity of one (or more) K+ channels. Effects of modulatory neurotransmitters on the opening kinetics, opening probability, and voltage dependence of activation of ion channels in target neurons have been described (for review, see ref. 32). Although the nature of the signaling pathway in the Drosophila motor neuron remains to be elucidated, G-protein-linked second messenger signaling has been implicated in several other systems that respond to modulatory neurotransmitters (32). For example, in classic studies, Kandel and Schwartz (33) reported that 5HT application to the Aplysia sensory neuron caused increased excitability via cAMP-dependent phosphorylation and consequent inhibition of a K+ channel. Further studies will be required to determine if a similar phenomenon is occurring in the Drosophila motor neuron.
The substrate neurotransmitter of the ine transporter has not yet been identified. It should be noted that reuptake of glutamate, the excitatory transmitter at insect neuromuscular junctions, is accomplished in mammals by proteins in a family that is structurally and functionally distinct from the Na+/Cl−-dependent family described herein (34, 35). Identification will most likely require expression in a heterologous system such as Xenopus oocytes followed by appropriate uptake assays. The sequence of the ine transporter, however, provides clues as to the nature of the substrate. The ine transporter contains a tryptophan residue (Trp-235) that is conserved among GABA transporters and has been proposed to participate in substrate binding (30). Furthermore, the ine, GABA, and creatine transporters each contain a glycine residue (Gly-71 for ine) where the 5HT, dopamine, and NE transporters contain an aspartate residue. This aspartate has been proposed to be required for the binding of the amino group present in 5HT, dopamine, and NE (36). These data raise the possibility that the ine transporter may transport GABA but will fail to transport monoamine neurotransmitters.
Furthermore, the expression pattern of ine is consistent with that expected for a GABA transporter but not a monoamine transporter. Expression of monoamine transporters occurs in restricted subsets of neurons or glia, usually in the same neurons that produce the substrate transmitter. For example, the expression of the previously characterized Drosophila 5HT transporter dSERT (28, 29) is restricted to the subset of cells in the nervous system that produce 5HT. In contrast, certain GABA transporters are expressed in nonneuronal tissues, such as the kidney and liver, as well (25, 37). This nonneuronal expression is apparently the result of overlapping substrate specificity exhibited by GABA transporters. For example, the GABA transporter GAT3 can also catalyze the reuptake of the osmolyte betaine (38), which is taken up by medulla cells in the kidney to neutralize the high ion concentrations that enable fluid reuptake to occur. As is found for GABA transporters, the ine gene is expressed in several nonneuronal cell types in developing embryos. These tissues include the hindgut, Malpighian tubules, and anal plate, which perform fluid resorption in insects (39), and the garland cells, which might also perform a similar function. Thus it is possible that the ine transporter accomplishes uptake of an osmolyte in addition to uptake of a classical neurotransmitter.
There are several possible mechanisms to account for the increased excitability of the motor neuron observed in ine mutants. One possibility is that the signaling system is entirely autonomous to the motor neuron (Fig. 5A). In this view, the neurotransmitter substrate for the ine transporter is produced, released by the motor nerve terminal, and then acts on receptors at the motor nerve terminal to cause increased excitability. In this case, the ine transporter is also expressed in the motor neuron and localized to the motor nerve terminal. Several transmitters have been reported at the motor nerve terminal that could be potential substrates for the ine transporter. These transmitters include glutamate itself and octopamine (40, 41). Alternatively, the signaling system could result from intercellular communication between an interneuron and its target motor neuron (Fig. 5B). In this view, the neurotransmitter substrate of the ine transporter is produced and released by an interneuron that synapses onto the motor neuron and acts on receptors in the motor neuron to cause increased excitability. In this case, the ine transporter is expressed in the interneuron. In either view, loss of the ine transporter would lead to reduced reuptake of the neurotransmitter and thus overstimulation of the target motor neuron by the transmitter. The consequent increase in excitability of the motor neuron could occur via G-protein-based signals acting on ion channels in the motor neuron, as described above. These possibilities might be distinguishable with experiments on the location of the ine protein with immunocytochemical methods.
Figure 5.
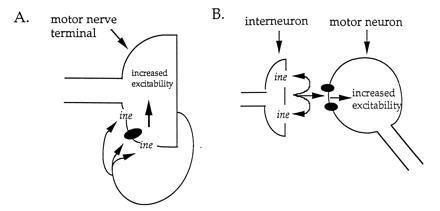
Two possible mechanisms for the role of the ine transporter and its substrate neurotransmitter in controlling excitability of the motor neuron. Neurotransmitter release is indicated by arrows; neurotransmitter receptors are indicated by solid ellipses; ine-encoded transporter is indicated by ine. (A) The motor neuron synthesizes and releases the substrate neurotransmitter, which then can act on receptors located in the motor nerve terminal membrane. Binding of neurotransmitter to receptor causes an increase in nerve terminal excitability. The duration of neurotransmitter action is attenuated by the ine transporter, which is expressed by the motor neuron and localized to the nerve terminal. (B) The substrate neurotransmitter is synthesized and released by an interneuron that synapses onto the motor neuron. The synapse is drawn onto the motor neuron cell body for convenience. Binding of neurotransmitter to receptors in the motor neuron causes increase in motor neuron excitability. The duration of neurotransmitter action is attenuated by the ine transporter, which is expressed by the interneuron.
We have shown that excitability of the Drosophila motor neuron is regulated by a neurotransmitter transporter encoded by ine. This observation should enable a genetic dissection of this signaling pathway that controls excitability of the motor neuron.
Note Added in Proof.
A neurotransmitter transporter with properties similar to ine can rescue the photoreceptor potential defects of mutants defective in the receptor oscillation A. (rosA) gene, which is allelic to ine, following introduction into flies by germ-line transformation (42).
Acknowledgments
We are grateful to Ross MacIntyre, in whose lab some of this work was accomplished; to William Gelbart, Judy Kassis, and Bob Levis for providing fly stocks; to W. Gelbart for cloned DNA; to Nigel Atkinson for providing cDNA libraries; to Sylvia Lee and Tarun Mahajan for technical assistance; to Bernie Andruss for his generous help and advice with embryo in situ hybridization; and to Mike Gustin for comments on the manuscript. This work was supported by Grant GM 46566 from the National Institutes of Health and a Grant-in-Aid from the American Heart Association to M.S.
Footnotes
Abbreviations: RFLP, restriction fragment length polymorphism; 5HT, serotonin; NE, norepinephrine; GABA, γ-aminobutyric acid.
Data deposition: The sequence reported in this paper has been deposited in the GenBank data base (accession no. U6640).
References
- 1.Amara S, Kuhar M J. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 2.Schloss P, Puschel A W, Betz H. Curr Opin Cell Biol. 1994;6:595–599. doi: 10.1016/0955-0674(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 3.Self D W, Nestler E J. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 4.Ritz M C, Lamb R J, Goldberg S R, Kuhar M J. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 5.Kuhar M J, Ritz M C, Boja J W. Trends Neurosci. 1991;14:299–302. doi: 10.1016/0166-2236(91)90141-g. [DOI] [PubMed] [Google Scholar]
- 6.Giros B, Jaber M, Jones S R, Wightman R M, Caron M G. Nature (London) 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 7.Stern M, Ganetzky B. J Neurogenet. 1992;8:157–172. doi: 10.3109/01677069209083445. [DOI] [PubMed] [Google Scholar]
- 8.Stern M, Blake N, Zondlo N, Walters K. J Neurogenet. 1995;10:103–118. doi: 10.3109/01677069509083458. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan W D, Trout W E., III Genetics. 1969;61:399–409. doi: 10.1093/genetics/61.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jan Y N, Jan L Y. Proc Natl Acad Sci USA. 1978;75:515–519. doi: 10.1073/pnas.75.1.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallart A, Angautpetit D, Bourretpoulain C, Ferrus A. J Neurogenet. 1991;7:75–84. doi: 10.3109/01677069109066212. [DOI] [PubMed] [Google Scholar]
- 12.Tempel B L, Papazian D M, Schwarz T L, Jan Y N, Jan L Y. Science. 1987;237:770–775. doi: 10.1126/science.2441471. [DOI] [PubMed] [Google Scholar]
- 13.Warmke J, Drysdale R, Ganetzky B. Science. 1991;252:1560–1562. doi: 10.1126/science.1840699. [DOI] [PubMed] [Google Scholar]
- 14.Chouinard S W, Wilson G F, Schlimgen A K, Ganetzky B. Proc Natl Acad Sci USA. 1995;92:6763–6767. doi: 10.1073/pnas.92.15.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulain C, Ferrus A, Mallart A. Pfluegers Arch. 1994;427:71–79. doi: 10.1007/BF00585944. [DOI] [PubMed] [Google Scholar]
- 16.Loughney K, Kreber R, Ganetzky B. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- 17.Stern M, Kreber R, Ganetzky B. Genetics. 1990;124:133–143. doi: 10.1093/genetics/124.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindsley D L, Zimm G G. The Genome of Drosophila melanogaster. London: Academic; 1992. [Google Scholar]
- 19.Kassis J A, VanSickle E P, Sensabaugh S M. Genetics. 1991;128:751–761. doi: 10.1093/genetics/128.4.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 21.Palazzolo M J, Hamilton B A, Ding D, Marin C H, Mead D A, Mieredorf R C, VijayRaghavan K, Meyerowitz E M, Lipshitz H D. Gene. 1990;88:25–36. doi: 10.1016/0378-1119(90)90056-w. [DOI] [PubMed] [Google Scholar]
- 22.Cavener D. Nucleic Acids Res. 1991;19:3185–3192. doi: 10.1093/nar/19.12.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Proudfoot N J, Brownlee G G. Nature (London) 1976;263:211–214. doi: 10.1038/263211a0. [DOI] [PubMed] [Google Scholar]
- 24.Borden L A, Smith K E, Gustafson E L, Branchek T A, Weinshank R L. J Neurochem. 1995;64:977–984. doi: 10.1046/j.1471-4159.1995.64030977.x. [DOI] [PubMed] [Google Scholar]
- 25.Borden L A, Smith K E, Hartig P R, Branchek T A, Weinshank R L. J Biol Chem. 1992;267:21098–21104. [PubMed] [Google Scholar]
- 26.Vandenbergh D J, Persico A M. Mol Brain Res. 1992;15:161–166. doi: 10.1016/0169-328x(92)90165-8. [DOI] [PubMed] [Google Scholar]
- 27.Pacholczyk T, Blakely R D, Amara S G. Nature (London) 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- 28.Demchyshyn L L, Pristupa Z B, Sugamori K S, Barker E L, Blakely R D, Wolfgang W J, Forte M A, Niznik H B. Proc Natl Acad Sci USA. 1994;91:5158–5162. doi: 10.1073/pnas.91.11.5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corey J L, Quick M W, Davidson N, Lester H A, Guastella J. Proc Natl Acad Sci USA. 1994;91:1188–1192. doi: 10.1073/pnas.91.3.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinberger-Doron N, Kanner B I. J Biol Chem. 1994;269:3063–3067. [PubMed] [Google Scholar]
- 31.Tautz D, Pfeifle C. Chromosoma. 1989;89:81–85. doi: 10.1007/BF00291041. [DOI] [PubMed] [Google Scholar]
- 32.Hille B. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 33.Kandel E R, Schwartz J H. Science. 1982;218:433–443. doi: 10.1126/science.6289442. [DOI] [PubMed] [Google Scholar]
- 34.Pines G, Danbolt N C, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner B I. Nature (London) 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- 35.Kanai Y, Hediger M A. Nature (London) 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 36.Kitayama S, Shimada S, Xu H, Markham L, Donovan D M, Uhl G R. Proc Natl Acad Sci USA. 1992;89:7782–7785. doi: 10.1073/pnas.89.16.7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu R-R, Lopez-Corcuera B, Mandiyan S, Nelson H, Nelson N. J Biol Chem. 1993;268:2106–2112. [PubMed] [Google Scholar]
- 38.Yamauchi A, Uchida S, Kwon H M, Preston A S, Robey R B, Garcia-Perez A, Burg M G, Handler J S. J Biol Chem. 1992;267:649–652. [PubMed] [Google Scholar]
- 39.Wharton G W. In: Comprehensive Insect Physiology, Biochemistry, and Pharmacology. Kerkut G A, Gilbert L I, editors. Oxford: Pergamon; 1985. pp. 565–603. [Google Scholar]
- 40.Jan L Y, Jan Y N. J Physiol (London) 1976;262:215–236. doi: 10.1113/jphysiol.1976.sp011593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halpern M E, Anderson M S, Johansen J, Keshishian K. Soc Neurosci Abstr. 1991;14:383. [Google Scholar]
- 42.Geng C, Burg M G, Koliantz G, Guan Y, Pak W L. Soc Neurosci Abstr. 1996;22:295.3. [Google Scholar]