

Abstract
The classic model for neurodegeneration due to mutations in DNA repair genes holds that DNA damage accumulates in the absence of repair, resulting in the death of neurons. This model was originally put forth to explain the dramatic loss of neurons observed in patients with xeroderma pigmentosum neurologic disease, and is likely to be valid for other neurode-generative diseases due to mutations in DNA repair genes. However, in trichiothiodystrophy (TTD), Aicardi–Goutières syndrome (AGS), and Cockayne syndrome (CS), abnormal myelin is the most prominent neuropathological feature. Myelin is synthesized by specific types of glial cells called oligodendrocytes. In this review, we focus on new studies that illustrate two disease mechanisms for myelin defects resulting from mutations in DNA repair genes, both of which are fundamentally different than the classic model described above. First, studies using the TTD mouse model indicate that TFIIH acts as a co-activator for thyroid hormone-dependent gene expression in the brain, and that a causative XPD mutation in TTD results in reduction of this co-activator function and a dysregulation of myelin-related gene expression. Second, in AGS, which is caused by mutations in either TREX1 or RNASEH2, recent evidence indicates that failure to degrade nucleic acids produced during S-phase triggers activation of the innate immune system, resulting in myelin defects and calcification of the brain. Strikingly, both myelin defects and brain calcification are both prominent features of CS neurologic disease. The similar neuropathology in CS and AGS seems unlikely to be due to the loss of a common DNA repair function, and based on the evidence in the literature, we propose that vascular abnormalities may be part of the mechanism that is common to both diseases. In summary, while the classic DNA damage accumulation model is applicable to the neuronal death due to defective DNA repair, the myelination defects and brain calcification seem to be better explained by quite different mechanisms. We discuss the implications of these different disease mechanisms for the rational development of treatments and therapies.
Keywords: Aicardi-Goutières syndrome, Cockayne syndrome, Oligodendrocytes, Myelin, Calcification, Vascular disease, Thyroid hormone, TREX1, RNASEH2, Toll-like receptor, Innate immune system
1. Introduction
It would be reasonable to assume that the major clinical impact of mutations in DNA repair genes would be an increased risk of cancer. Indeed, a greatly elevated risk of different types of cancers is associated with different types of DNA repair deficiencies, as observed in patients with Fanconi anemia, hereditary non-polyposis colon cancer, and xeroderma pigmentosum (XP) [1].
Surprisingly, it is becoming increasingly clear that another common clinical manifestation of DNA repair gene mutations is neurologic disease. As shown in Table 1, there are least 16 hereditary DNA repair diseases in which neurological abnormalities represent a major, and in some cases the only, clinical effect of mutations in DNA repair genes.
Table 1.
List of neurologic diseases resulting from mutations in DNA repair genes
| Disease | Genes | Major CNS features |
|---|---|---|
| Neurodegenerative diseases | ||
| Xeroderma pigmentosum (XP) neurologic disease | XPA,C,D,Fa | Primary neurodegeneration in brain, spinal cord |
| Ataxia telangiectasia (AT) | ATM | Purkinje neuron degeneration, movement disorders |
| AT-like disease (ATLD) | MRE11 | Clinically similar to late-onset AT neurologic disease |
| Spinocerebellar ataxia with axonal neuropathy (SCAN1) | TDP1 | Cerebellar atrophy |
| Ataxia with oculomotor apraxia 1 | APTX (Aprataxin) | Cerebellar atrophy, axonal sensorimotor neuropathy |
| Ataxia with oculomotor apraxia 2 | SETX | Cerebellar atrophy, axonal sensorimotor neuropathy |
| Ataxia with oculomotor apraxia 3 | Unknown | Cerebellar atrophy |
| Diseases of myelin and brain calcification | ||
| Cockayne syndrome (CS) | CSA,B; XPB,D,G | De/dysmyelination, calcification, microcephaly |
| Cerebro-oculo-facio-skeletal (COFS) syndrome | XPD,G; CSB, ERCC1 | De/dysmyelination, calcification, microcephaly |
| Aicardi–Goutières syndrome (AGS) | TREX1, RNASEH2 | De/dysmyelination, calcification, microcephaly, elevated CSF IFN-α, CSF lymphocytosis |
| Trichothiodystrophy (TTD) | XPD, TTDA | Demyelination, microcephaly, ± calcification |
| Microcephaly diseases | ||
| Nijmegen Breakage syndrome (NBS) | NBS1 | Microcephaly, craniofacial abnormalities, mental retardation |
| LIG4 syndrome | LIG4 | Microcephaly, craniofacial abnormalities |
| Seckel syndrome | ATR | Microcephaly, craniofacial abnormalities, mental retardation |
| Immunodeficiency with microcephaly | Cernunnos-XLF | Microcephaly, craniofacial abnormalities, mental retardation |
| Primary microcephaly 1 | MCPH1/BRIT1 | Microcephaly, mental retardation |
The diseases are classified by the brain neuropathology, rather than by DNA repair pathway. References are given in the text. The table is not intended to be a comprehensive list of all DNA repair diseases in which there is nervous system involvement. In many of these diseases, the peripheral nervous system is affected as well. For other caveats, see text.
The neurologic disease in XPF patients is somewhat different than the classic XP neurologic disease in XPA and XPD. In XPC, neurologic disease is asmpytomatic. For further discussion, see [26].
The classic model for neurologic disease due to defective DNA repair holds that the accumulation of DNA damage results in neuronal death. However, in trichiothiodystrophy (TTD), Aicardi–Goutières syndrome (AGS), and Cockayne syndrome (CS), the most prominent neuropathology is not neurodegeneration (i.e. neuronal death), but rather myelin defects. In this mini review, we focus on recent studies of TTD and AGS which indicate that mutations in what we generally think of as “DNA repair” genes can result in myelin defects by mechanisms that are qualitatively different than the accumulation of DNA damage.
A major point of emphasis in this mini review is that there are fundamental qualitative differences in neuropathology amongst the neurological diseases resulting from mutations in DNA repair genes, and that these differences in neuropathology reflect differences in the specific cell types that are affected within the brain. Therefore, we will first give a brief overview of the different types of cells in the nervous system (Fig. 1) to serve as a framework for the discussion that follows.
Fig. 1.
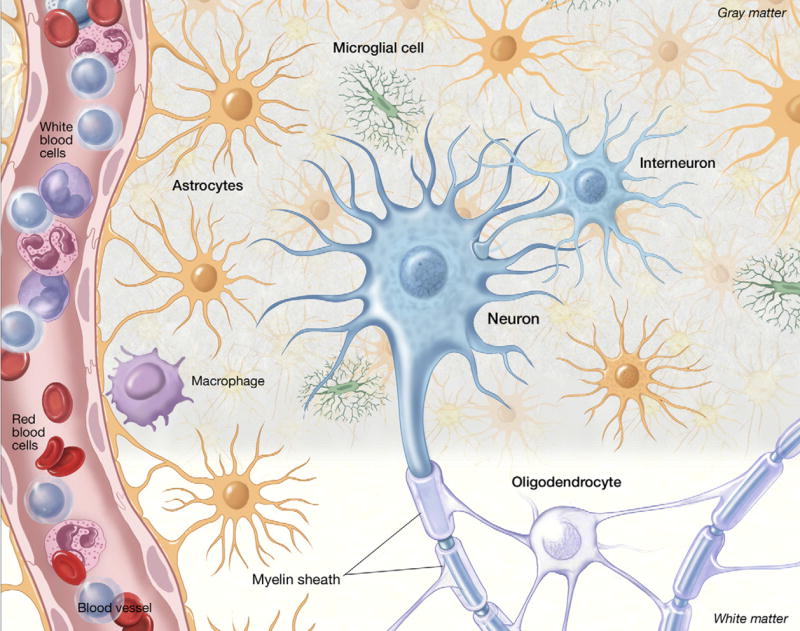
A drawing of the different cell types in the brain to show their anatomical relationships and relative numerical proportions. In addition to the major cell types which are discussed in the text, and marked on the figure, other notable points are the wrapping of myelin produced by oligodendrocytes around the axons of neurons, and the end-feet of the astrocytes wrapping around the blood vessel which in association with vascular endothelial cells, form the blood–brain barrier. The relative sizes of the different cell types are not drawn to scale.
2. The brain is more than neurons: different cell types in the nervous system
In this section, we list the major cell types in the brain, including some relevant functional aspects. For additional information and further references, see [2].
2.1. Neurons
The cell type that characterizes the nervous system and makes it unique is the nerve cell or neuron. Neurons can communicate with each other via synaptic transmission, and it is this communication that underlies all of the activities of the nervous system including cognition, sensation, and the control of movement. Neurons come in many different shapes and sizes. The smallest neurons, called granule neurons are found in the cerebellum, hippocampus and olfactory system, and are only approximately 10 μm in width and length. At the other end of the scale, the motor neurons that connect the cerebral cortex at the top of the brain to the sacral spinal cord in the tailbone can be ≈ 1 m in length.
A particularly noteworthy type of neuron, located in the cerebellum, is called the Purkinje neuron (Fig. 2) after the Czech neuroanatomist who discovered it. Although from a quantitative standpoint, Purkinje neurons are a minor cell population in the cerebellum compared to granule cells (Fig. 2), they play a crucial role in the output of the cerebellar cortex, a brain region that is essential to the control of balance. Degeneration of cerebellar Purkinje neurons results in ataxia, which as shown in Table 1 is a very common manifestation of mutations in DNA repair genes. Purkinje neurons have been shown to undergo degeneration in the brain of XP, CS, and AT patients, and are likely to be affected in ATLD as well.
Fig. 2.
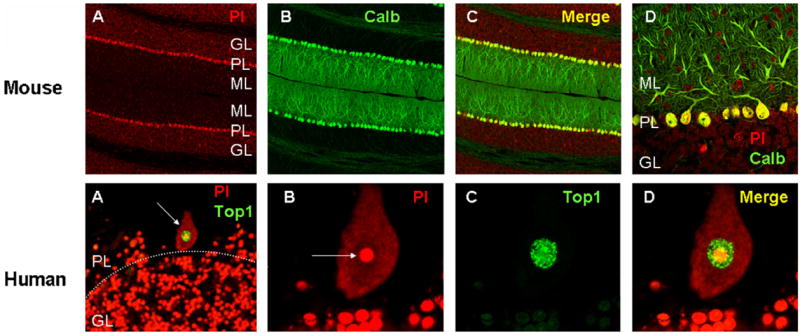
Purkinje neurons in the mouse and human cerebellum. (Top row) (A–C) Sections through the mouse cerebellum stained with propidium iodide (PI), a nucleic acid stain, illustrating the different layers of the cerebellar cortex; ML, molecular layer; PL, Purkinje layer; GL, granule layer. (B) Same section stained with calbindin (Calb), a marker for Purkinje neurons. Panel (C) is a merged image of (A) and (B). Panel (D) is a detail from (C) to illustrate the dendritic trees of the Purkinje neurons. (Bottom row) (A) low-power image of a section through the human cerebellum (case 1465, see [35]) showing a single Purkinje neuron (arrow), adjacent to the granule layer. Note the quantitative difference between the granule neurons (in the GL, below the dotted line) relative to the single Purkinje neuron. Green staining is topoisomerase I (Top1), counterstained with PI. (B–D) Higher magnification of the Purkinje neuron shown in (A), stained with PI (B), Top1 (C) and a merged image (D). In (B), note that the nucleolus of the Purkinje neuron (arrow) is comparable in size to the entire nucleus of the granule neurons shown below. Also note the much greater intensity for Top1 staining in the nucleus of the Purkinje neuron compared to the granule neurons. For further information, see [35]. Images produced by Tracy Gilman (mouse) and Sarah Calkins (human). The conditions used for PI staining and image acquisition were substantially different between the mouse and human material, and therefore the mouse and human images are not directly comparable with each other.
2.2. Glial cells
The majority of cells in the brain are glial cells. The word glia comes from the Latin word meaning glue. This terminology reflects the early idea that neurons are the only important brain cells, and the other cells are just the glue that holds the brain together. As it turns out, there are three functionally distinct types of glial cells, and their functions are much more important than the name would imply. In particular, the important role of glial cells in brain diseases is becoming increasingly appreciated. Also, the different types of glial cells differ in the ability to respond to oxidative stress and in DNA repair capacity [3,4].
2.2.1. Astrocytes
The most common type of glial cell is the astrocyte. Although astrocytes can be thought of as a type of supporting cell for the neuron, they also play an important role in maintaining the extracellular environment, thereby allowing appropriate neurotransmission. Astrocytes surround blood vessels in the brain, thereby contributing to the blood–brain barrier (in addition to endothelial cells, see below).
2.2.2. Oligodendrocytes
The second major type of glial cell is the oligodendrocyte, which forms the myelin sheath that surrounds the axons of multiple neurons, functioning as the insulator to allow rapid synaptic transmission. In the peripheral nervous system, the myelin producing cells are referred to as Schwann cells and insulate a single axon. Myelin is composed of approximately 70% lipid, along with a limited number of proteins. The high-fat content of myelin gives it a white appearance in brain tissue, hence the term “white matter”. A schematic representation of the myelin in relation to white matter and gray matter is given in Fig. 1.
2.2.3. Microglial cells
Although the name implies a similarity with other types of glial cells, it is now generally accepted that microglia are derived from the hematopoetic cell lineage (monocytes) and populate the brain from the blood during early brain development [5]. Consistent with this lineage, microglia are generally considered to be the resident immune cell of the brain [6]. Under basal conditions, microglia are often described as “resting”, but are more accurately described as “alert” or carrying out surveillance [7,8]. In response to brain damage or cell death microglia undergo a graded activation process, ultimately engulfing extracellular debris in a phagocytic process.
In addition to microglia, the brain also contains various types of macrophages [5]. Although not technically considered glial cells, their phagocytic role is more similar to that of the microglia than any other cell type in the brain. In fact, some authors use the term mononuclear phagocytes to include both microglia and macrophages [9].
2.3. Endothelial cells
The brain is highly vascularized and as such also contains endothelial cells within the vasculature. Endothelial cells, along with astrocytes, form the blood–brain barrier. As a result, endothelial cell dysfunction within the brain can ultimately impact brain function, by for example allowing white blood cells to enter the brain, and thereby contribute to neurologic disease.
3. Different brain cells types are affected in different DNA repair diseases
Most reviews of neurologic disease due to DNA repair gene mutations (e.g. [10]) classify the diseases by the repair pathway affected. This tends to obscure some important qualitative differences in neuropathology between diseases considered to result from defects in the same DNA repair pathway (e.g. CS and XP, see [11]). Therefore, in Table 1, we have grouped the diseases not by the repair pathway affected, but by the primary neuropathological feature. Considered from this point of view, the diseases roughly fall into three groups.
3.1. Neurodegenerative disorders
In the first group of diseases, neurons are the primary cell type that is affected. These include XP, AT, ATLD, SCAN1, and the AOAs 1, 2, and 3 [11–15].
3.2. Diseases of myelin and brain calcification
In the second group are those diseases in which the most striking neuropathological feature is abnormal myelination, indicating that oligodendrocytes are primarily affected. Defective myelination includes demyelination (reduced levels of myelin) and also dysmyelination (abnormal myelin). These “white matter” diseases include CS, COFS, TTD, and AGS [16–19]. Notably, calcification of the brain, particularly in the basal ganglia and cerebellum, are also observed in these diseases, most dramatically in CS and AGS.
3.3. Microcephaly
In the last group are those diseases in which microcephaly is the main feature. These include NBS, LIG4 syndrome, Seckel syndrome, immunodeficiency with microcephaly, and MCH1 (reviewed in [10]). As indicated in Table 1, patients with microcephaly diseases may also have craniofacial dysmorphology and mental retardation.
3.4. Caveats
As with any classification of this type, some aspects and distinctions are oversimplified. For example, microcephaly is also observed in CS, TTD, and severe cases of XP. Also, in CS there is degeneration of both granule and Purkinje neurons in the cerebellum [11]. Finally, regarding ATLD, while the clinical description of the original patients [20–22] was similar to a slowly progressing form of AT, some affected members of the most recently described family displayed microcephaly, similar to NBS patients [23]. Despite these caveats, Table 1 reflects the major neurologic features of the different diseases, and serves as a useful framework for considering the mechanisms discussed below.
4. The classic mechanism: accumulation of DNA damage in neurons due to defective DNA repair causes neurodegeneration
Fig. 3 illustrates what we will refer to as the accumulated DNA damage mechanism for neurodegeneration due to DNA repair deficiency. This mechanism was originally proposed by Robbins and co-workers to explain the neurodegeneration observed in XP patients [24]. According to this mechanism, in normal individuals endogenous DNA damage is constantly being produced but also repaired, resulting in a low-steady-state level of damage which is compatible with normal cellular function. However, in patients with DNA repair deficiency, endogenous damage is not repaired and therefore accumulates over time, ultimately leading to neuronal death as a result of impaired transcription.
Fig. 3.
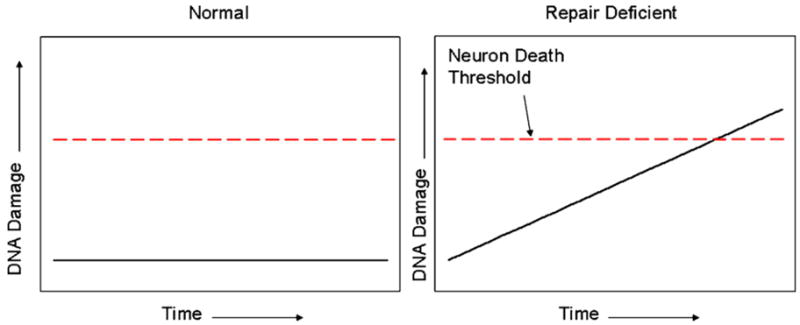
The accumulated damage model of neurodegeneration due to defective DNA repair. For explanation see text. For the neurodegenerative diseases in Table 1, the time necessary for accumulated DNA damage to reach the neuronal death threshold is likely to be several years, if not longer. This may explain why the Atm−/−, Xpa−/− and Aptx−/− mice do not accurately reproduce the progressive neurodegeneration observed in the human patients; the 2-year lifespan of a mouse is simply not long enough to allow sufficient damage to accumulate (see also [10]).
At present, the accumulated DNA damage mechanism seems to be generally accepted, at least implicitly, by most workers in the field. However, it is worth noting that at the time the mechanism [24] was originally proposed, it was very controversial and not at all well received (J. Robbins, personal communication) in part because at the time there was much less knowledge of the magnitude of endogenous DNA damage than today.
As noted above, the accumulated DNA damage mechanism was originally proposed to explain progressive, primary neurodegeneration that is observed in XP neurologic disease. No subsequent data has emerged to indicate that this mechanism is not applicable, and in fact additional supporting data has been published. Specifically, it has been shown that XPD patients with mutations that specifically affect the NER function of XPD develop “pure” XP, with a progressive neurodegenerative disease similar to XPA patients (though typically with later onset), whereas XPD mutations that either destabilize TFIIH or affect the transcription functions result in TTD or the XP-CS complex ([25]; see also below). Thus with regard to XP neurologic disease, the primary question is which DNA lesions are responsible for neurodegeneration in XP. We have previously proposed that, based on several criteria, the 8,5′-cyclopurine deoxynucleosides represent the best candidates amongst currently known DNA lesions for playing a causative role in XP neurologic disease, but it is certainly possible that other endogenous DNA lesions that are repaired by NER could be involved as well [26].
In addition to XP, the accumulated DNA damage mechanism is also likely to be applicable to several of the other neurologic diseases on the list above, including the ataxias SCAN1 and AOA1, 2, and 3. In the case of AOA1, a good candidate DNA lesion has been proposed [27]. However, because these diseases have been the subject of several comprehensive reviews [15,28,29], they will not be discussed further here.
A complicating factor in applying the accumulated DNA damage mechanism for AT was the published evidence showing that the ATM protein, while localized to the cell nucleus in most cells, was apparently localized to the cytoplasm of Purkinje neurons in the human [30] and rodent [31] brain. Since Purkinje neuron degeneration is the sine qua non of AT, the localization of ATM in this specific cell type is of major clinical significance. Cytoplasmic localization of the ATM protein in these cells would imply a role for this protein other than sensing DNA damage, and therefore a very different pathophysiologic mechanism than a DNA damage sensing or repair defect.
However, several recent studies have now challenged this view. First, work by Shiloh and co-workers showed that in human neuron-like cells, the ATM protein is primarily localized to the nucleus, and responds to DNA damage as it does in other cell types [32,33]. Also, an important study by Barzilai and co-workers showed that ATM is present in the nucleus of cerebellar neurons in the mouse brain, including Purkinje neurons, and can be phosphorylated on a residue equivalent to Ser1981 in the human protein following exposure to ionizing radiation [34]. We have also made similar observations regarding ATM phosphorylation in rodent brain after radiation as well (Gorodetsky and Brooks, unpublished observations).
In addition to these findings in mice, we [35] showed that the ATM protein is predominantly localized to the nucleus of Purkinje neurons in postmortem human brain. Importantly, in contrast to the earlier publication [30], we used postmortem brain tissue from AT patients to demonstrate the specificity of the ATM antibody.
Taken together, these new findings on the localization of the ATM protein are now consistent with the hypothesis that at least one function of the ATM protein in Purkinje neurons is to detect DNA damage. As such, the accumulated DNA damage mechanism of neurodegeneration seems applicable to AT as well.
Having said this, there are important outstanding issues that remain, concerning what type of DNA damage is sensed by ATM in neurons [35], and at what stage of Purkinje cell development the ATM protein functions. Regarding the latter issue, based on their studies using Atm−/− mice [36,37] McKinnon and co-workers proposed a model in which ATM acts at a stage just after terminal differentiation to trigger apoptosis of neurons that have experienced excess DNA damage during brain development. According to this model, in the absence of ATM, such neurons survive and populate the Purkinje neuron layer, only to degenerate later as a result of DNA damage experienced during development [38]. However, it is entirely possible that ATM plays both a proapoptotic role just after terminal differentiation, as in the McKinnon hypothesis, as well as an ongoing role in the detection of DNA damage, and that the final neurological phenotype of AT patients is due to the loss of both pathways.
In summary, the accumulated DNA damage model of neurodegeneration resulting from defective DNA repair holds that DNA damage in neurons accumulates over time due to the lack of repair, ultimately resulting in neuronal death by blocking transcription. Based on current knowledge, this mechanism seems applicable to XP, AT, ATLD, the AOAs, SCAN1, and perhaps also the cerebellar neurodegeneration in CS.
5. A new concept of the transcription syndrome hypothesis: loss of the co-activator function of TFIIH leads to myelination defects
The seminal discovery that the XPB and XPD proteins are part of the transcription factor TFIIH [39] led to the realization that TFIIH plays important roles in both basal transcription and DNA repair. This in turn, led to the concept of transcription syndromes, the idea that some of the clinical manifestation observed in CS and TTD patients with mutations in these genes XPB and XPD patients could be due to transcription defects, as opposed to DNA repair defects [1,40].
As typically described, during NER and transcription, TFIIH functions as a helicase, either opening the DNA during NER, or opening the DNA near the promoter to allow transcription initiation [41]. Clearly, TFIIH is involved in both functions, although it should be noted that regarding the function of XPB, a recent study showed that the ATPase activity, rather than the helicase activity, is important for DNA opening [42].
In addition to the DNA opening roles for TFIIH, it has also been known for some time that TFIIH has another, mechanistically distinct role in transcription regulation, which is that it functions as a kinase that phosphorylates nuclear hormone receptors [41,43]. Here, we focus on a recent study [44] from the Egly laboratory showing that TFIIH has yet another role in transcription regulation. Specifically, TFIIH can function as a co-activator for thyroid hormone-dependent gene regulation, and that the loss of this co-activator function of TFIIH is responsible for some of the major neurological abnormalities observed in TTD patients.
Compe et al. [44] studied the so-called TTD mouse, which carries the same R722W mutation in Xpd as in a human TTD patient [45]. They first showed that total brain weights of the TTD mice at postnatal day 20 were significantly lower than WT controls, and also observed some evidence for reduced myelination. Realizing that several of the neurological features of human TTD patients, notably including demyelination, were similar to what is observed in thyroid hormone deficiency, the authors focused their attention on the expression of known thyroid hormone-dependent genes in the TTD mouse brain, including several involved in myelin synthesis. In general, they observed decreased levels of thyroid-dependent gene expression in most regions of the mouse brain, although there were some exceptions. In addition, ultrastructural studies of the TTD brain showed increased numbers of unmyelinated axons, indicative of a functional myelination defect.
Additional experiments were carried out in which mice were made hypothyroid (by prenatal injections of the drug PTU), and subsequently treated with thyroxine, a physiological thyroid hormone receptor (TR) ligand. In WT mice, hypothyroidism reduced thyroid hormone-dependent gene expression, but this could be restored by hormone replacement. However, in the TTD mice, hormone replacement therapy was ineffective. Taken together, the results indicate a dysregulation of thyroid hormone-dependent gene regulation in the TTD mouse brain.
To address the mechanistic basis for these observations, the authors carried out a detailed and comprehensive analysis of the interaction of the TR isoforms with promoter regions of TR regulated genes. The experiments were carried out in vivo, using a ChIP assay adapted to the mouse brain tissues, and in vitro, using DNAse1 footprinting. The conclusion from these studies was that hormone-dependent recruitment of TRs onto thyroid response elements was deficient in the TTD tissues and cells, reflecting instability of the liganded TRs on thyroid response element (TRE) DNA.
In view of the evidence that TFIIH can phosphorylate nuclear hormone receptors [46], the authors next asked whether the reduced stabilization of the TR in the TTD samples was due to decreased TR phosphorylation. However, while the TR was found to be phosphorylated in vivo, and this phosphorylation affected by TFIIH, phosphorylation of the TR did not affect its ability to bind to the TRE. On the other hand, reducing TFIIH levels by siRNA knockdown of XPD did reduce TR-dependent gene transactivation.
Taken together, these results indicate that TFIIH acts as a co-activator for thyroid hormone-dependent transactivation in the mouse brain, and also in human cells. In contrast, TFIIH does not influence transcriptional repression mediated by unliganded TRs. Direct phosphorylation of TRs by TFIIH is not necessary for this co-activator function, although as the authors noted, the possibility that TFIIH may phosphorylate other proteins in vivo that are involved in thyroid hormone gene regulation cannot be excluded.
The results of Compe et al. make a convincing case that Xpd mutations in the TTD mouse result in myelination defects due to the loss of the co-activator function of TFIIH in thyroid hormone-dependent gene expression. By extension, to the extent that the mechanisms responsible for aberrant myelin gene expression in the TTD mouse are the same as those responsible for demyelination in the human TTD patients, which seems a reasonable assumption, dysregulation of thyroid dependent gene expression in the brain is a more compelling explanation for the neurologic disease in human TTD patients than either an NER defect or a defect in transcription initiation.
The work by Compe et al. comes on the heels of another important paper, in which Ito et al. [46] showed that XPG could act to stabilize the TFIIH complex. In cells from XPG-CS patients, the CAK complex dissociates from the core of TFIIH, resulting in defects in nuclear hormone stimulated gene expression. In contrast to the TTD work described above, in the XPG-CS cells, reduced phosphorylation of nuclear hormone receptors is the main mechanism for the hormone-dependent transcription defect. Since this work has been recently reviewed by others [47,48], we will not discuss this in any more detail, except to point out that steroid hormones act on the brain as well as in the periphery, and therefore defective phosphorylation of nuclear hormone receptors could also negatively affect brain function in XPG-CS patients.
5.1. Are defects in nuclear hormone phosphorylation also involved in pure CS?
Postmortem examination of the brain of an XPG-CS patient reveals the typical pattern of neuropathologies observed in pure CS patients, including microcephaly, dysmyelination, and basal ganglia calcification [49]. Thus the obvious question arises as to whether defects in nuclear hormone-dependent gene transcription observed in XPG-CS cells are also characteristic of cells from CS patients. However, Ito et al. [46] tested this possibility, but found that nuclear hormone-dependent transcription was normal in CS cells. Thus despite the phenotypic similarity between CS, XPG-CS, and TTD, the underlying molecular defects appear to be different. As the authors noted, it is possible that other transcription defects are involved in CS. The puzzling question of the mechanistic basis of CS neurologic disease will be revisited below.
6. A new model: failure to degrade endogenous DNA leads to neurologic disease by activation of the innate immune system
A fascinating new twist on the accumulated DNA damage model of neurologic disease has recently been provided by a series of studies on another rare genetic disease, AGS. Clinically, AGS patients present in early childhood with psychomotor retardation, microcephaly, and in some cases seizures [50]. In addition, patients also suffer painful inflammatory sores on the skin called chilblains [51]. Neurologic abnormalities observed in the brains of AGS patients include demyelination, calcification of the basal ganglia, vascular abnormalities, and elevated levels of white blood cells and interferon (IFN)-α in the cerebrospinal fluid (CSF) [19]. In many ways, these features of the disease are strikingly similar to a viral infection of the brain.
Given the clinical presentation of AGS patients, it would not have been expected that when the mutant genes responsible for AGS were identified, one would turn out to be encode a protein thought to be involved in DNA repair. Yet, as shown by the laboratories of Crow et al., AGS can be caused by mutations in the gene encoding TREX1, a 3′ → 5′ exonuclease [52]. In other patients, AGS results from mutations in the genes encoding any of the three subunits of RNASEH2 [53]. Genetic studies indicate that there is at least one additional AGS gene to be identified [51].
Two additional points about the phenotypic effects of TREX1 mutations should be mentioned. First, mutations in TREX1 can result in very disparate clinical phenotypes. Specifically, while TREX1 mutations that disable the exonuclease function in humans result in the neurologic features in AGS as described above, heterozygous C-terminal truncation mutations of TREX1, which do not involve the nuclease domain, result in a less severe disease with white matter involvement [54]. In contrast, other heterozygous TREX1 mutations do not result in neurologic disease, but instead cause the autoimmune disease systemic lupus erythromatosus (SLE) [55], and a dominant negative TREX1 mutation results in the related disorder familial chilblain lupus (FCL) [56,57]. Thus mutations in the same gene affect different organs in humans. Second, in contrast to the human diseases, gene-targeted Trex1−/− mice do not develop neurological disease, but instead develop cardiac inflammation [58].
How is it that mutations in TREX1 and RNASH2 can lead to the unusual neurologic symptoms observed in AGS patients and the lupus like symptoms? A compelling answer to this question has been proposed by Crow et al. According to this hypothesis, the failure to degrade some endogenous nucleic acid species, generated as a result of a normal cellular process (see below) results in the activation of the innate immune system, which is in turn responsible for the disease symptoms [52].
In contrast to the well-known adaptive or acquired immune system, which involves the production of immunoglobulin from T and B cells), the innate immune system depends on phagocytic cells, such as macrophages, that engulf foreign substances. This in turn triggers an inflammatory response, and a complex cellular cascade resulting in the synthesis of cytokines such as the IFNs [59,60]. A key step in activation of the innate immune system involves pattern recognition receptors (PRRs), which include the Toll-like receptors (TLRs). The different TLRs, as well as other PRRs, recognize specific ligands which collectively are referred to as pathogen-associated molecular patterns (PAMPs) [60]. Of significance to the current discussion, different nucleic acid species are recognized by different TLRs or other PRRs [61,62].
Thus in AGS, the hypothesis is that as a result of normal cellular function, some type of nucleic acid species is generated that is normally degraded by TREX1 and/or RNASEH2, and is thus non-pathogenic. In the absence of these enzymes due to mutation, the nuclei acid is able to engage a PRR and activate an innate autoimmune response. Stated simply, in AGS, the innate immune system is “tricked” into an antiviral response by the undegraded endogenous nucleic acid.
Late last year, Yang et al. [63] made an important advance in understanding the pathophysiology of AGS. They discovered that a ≈ 60 bp single-stranded DNA species, apparently arising during S-phase of the cell cycle, could be detected within the endoplasmic reticulum (ER) of cells from the Trex1−/− mouse, but not in control mice. Strikingly, the same phenomenon was observed in cells from a human AGS patient as well, despite the different clinical outcomes of TREX1 deficiency in the two species. On the question of how RNASEH2 mutations could lead to AGS, the authors suggested that the initial nucleic acid species released during replication might be from an Okazaki fragment, and thereby composed of both a 5′ RNA primer and DNA. If so, then RNASEH2 could be necessary to degrade the RNA component (see [63] for a more detailed discussion of this point).
In addition to identifying an endogenous nucleic acid substrate for TREX1, these authors also demonstrated chronic ATM activation, as well as defective G1/S transition, in both human and mouse TREX-deficient cells. Based on these and other findings, Yang et al. outlined two possible mechanisms relating the cellular defects to the autoimmune features of the disease. First, the undegraded ssDNA could activate the innate immune system via the toll-like receptor TLR9. This hypothesis is attractive because TLR9 is located within the ER of immune cells, as are TREX1 and the ssDNA that accumulates in the absence of TREX1. The alternative model involves ATM signaling via the NKG2D receptor that is expressed by Granzyme A-secreting immune cells [64]. For a more detailed discussion of two possibilities, as well as other perspectives on this topic, see [65].
Regarding the neurological disease in AGS, the key features to be explained are calcification of the basal ganglia, dysmyelination, and elevated levels of CSF IFN-α. It seems likely that the elevated levels of interferon-α are responsible for the brain calcification, since transgenic mice designed to overexpress IFN-α in the brain (specifically in astrocytes) display a progressive brain calcification that is phenotypically similar to AGS [66]. In addition, of the two models for innate immune activation proposed above (the TLR-mediated versus NKG2D-mediated) the TLR mechanism seems more likely in the brain, since many TLRs, including TLR9, are expressed in the brain [67] whereas NKG2D is believed to be specific to subsets of immune cells.
Based on these considerations, and the hypothesis and proposals made in the original publications [52,63], a speculative model for neurologic disease in AGS due to TREX1 mutations is illustrated in Fig. 4. According to the model, ssDNA is generated as a result of DNA replication in some cell type in the brain. In the absence of TREX1, the undegraded nucleic acid enters the endosome, where it activates TLR9, triggering a signal transduction cascade resulting in increased IFN-α production. The IFN-α is then secreted into the extracellular space of the brain, and therefore into the CSF, where its acts on other cells in the brain, in particular vascular elements and oligodendrocytes, resulting in calcification and dysmyelination. Vascular dysfunction could also explain the elevated levels of white blood cells in the CSF.
Fig. 4.

A speculative working model for AGS neurologic disease, incorporating the proposals [52] and data from AGS cells [66], as well as the neuropathological evidence from AGS patients [19] and the IFN-α transgenic mice [66,68]. In one or more cell types in the normal brain, most likely microglia or macrophages, ssDNA resulting from DNA replication is produced, but is degraded by TREX1. In the AGS brain, ssDNA enters the endosome due to the absence of TREX1, where it activates a TLR. TLR activation results in signal transduction to the nucleus (yellow arrow), increasing the expression of the IFN-α gene (as well as other genes, not shown for clarity). This results in the synthesis and secretion of IFN-α into the CSF, where it can act on the other brain cell types. Effects in IFN-α in the vasculature appear to be of particular importance, and in human patients, dysmyelination is also prominent [19], indicating effects on oligodendrocytes. Activation of astrocytes and functional changes in neurons are also observed in the IFN-α overexpressing mice [66,68].
A model for AGS due to RNASEH2 mutations would be similar, except that the immunostimulatory nucleic acid would be an RNA [63] or possibly a chimeric RNA-DNA molecule. Such a molecule might act at a different TLR or other PRR, but ultimately result in IFN-α synthesis and secretion as shown in Fig. 4.
In terms of the downstream targets of IFN-α, some insights might be gained from analysis of the IFN-α transgenic mice, which as noted above display brain calcification similar to human AGS patients [66,68]. In fact, these mice may be a better animal model for AGS neurologic disease than the Trex1−/− mice, since as noted above, the Trex1−/− mice develop cardiac inflammation, but no reported neurologic abnormalities [58]. The curious lack of neuropathology in the mice might be due to species differences in either the cellular localization of the relevant PRRs, or in the complex signaling pathway between PRR activation and downstream transcriptional response.
In summary, the studies of patients with AGS have provided evidence for a fundamentally different model of neurologic disease due to a DNA repair defect. In the accumulated DNA damage model, the accumulation of unrepaired DNA damage is proposed to result in a cell-autonomous pathophysiologic process. In this new model, the failure to degrade naturally occurring nucleic acids results in neurologic disease via a non cell-autonomous mechanism involving activation of the innate immune system and secretion of IFN-α.
7. Dysmyelination and brain calcification in AGS and CS neurologic disease
Surprisingly, of the 16 DNA repair diseases listed in Table 1, the neurological manifestations of AGS are most similar to those in patients with CS and COFS resulting from a CSB mutation [17,69]. The similar neuropathologies in COFS, CS, and AGS have been noted previously by others [19,70]. COFS can also result from mutations in XPD [71], XPG [72], or ERCC1 [73], but neuropathological information on these cases is limited.
Considered from the standpoint of DNA repair pathways involved, it is not at all obvious why this should be the case. There are at least two other possibilities. First, vascular degeneration, particularly affecting small vessels (“microvessels”) in the brain appears to be particularly important in AGS neurologic disease [19]. Likewise, vascular damage is a prominent neuropathological feature in CS as well [16]. Specifically, Rapin et al. [74] described the presence of obliterated microvessels in the brain of a CSA patient. In both diseases, it has been suggested that at least some of the myelination defect could be secondary to vascular damage, though this has not been conclusively proven [19,74].
In AGS, the vascular changes and calcification may be due to increased IFN-α, based on similar observation in the IFN-α transgenic mouse [66]. While there is no data on IFN-α levels in the CS brain, it is possible that the loss of the CS proteins results in a pattern of gene expression changes that is somehow similar to what takes place in the AGS brain. Supporting this idea, Weiner and co-workers [75] showed that under basal conditions, i.e. in the absence of exposure to exogenous DNA damage, CSB deficiency results in a substantial number of gene expression changes. Interestingly, based on their results, they concluded that CSB-deficient cells appear to be under chronic inflammatory stress [75].
So, does this mean that there is no role for defective DNA repair in the dysmyelination and calcification in the CS brain? Not necessarily. Examination of CS brain tissue has observed evidence of neuroinflammation, including activated microglia [49], markers of oxidative stress and lipid peroxidation products [76]. Since myelin is primarily composed of lipid, oxidative stress from an inflammatory response in white matter would be expected to generate lipid peroxidation products. DNA adducts from malondialdehyde and other lipid peroxidation products have been shown to block transcription [77], and are therefore likely to be subject to TC-NER. The hallmark of CS cells is their defect in TC-NER. Thus, it is possible that a “vicious cycle” of neuroinflammation and cell death could be established in the brain of CS patients, facilitated by the loss of TC-NER (see Fig. 5). The defective transcriptional responses to DNA damage in CS cells [78,79] may also contribute to this cycle.
Fig. 5.
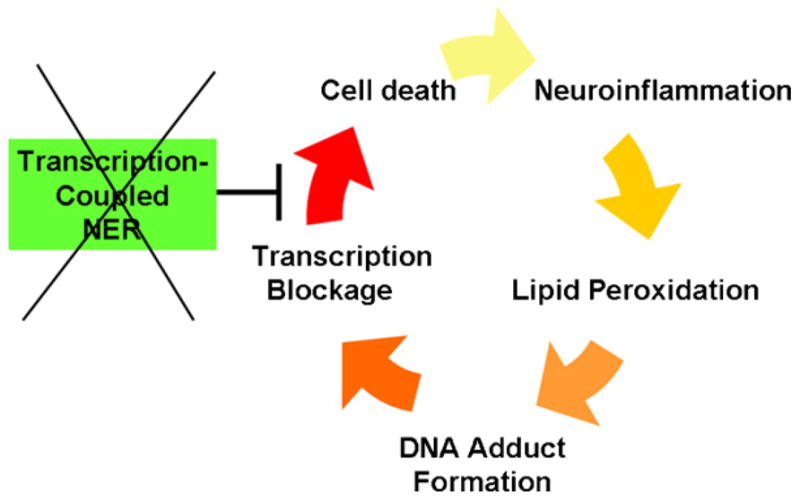
A speculative role for TC-NER in preventing a neuroinflammatory cycle of dysmyelination and calcification of the brain in CS. Inflammation results in lipid peroxidation and transcription-blocking DNA damage, which is repaired by TC-NER. CS gene mutations inactivate TC-NER, resulting in cell death, and in turn more inflammation from phagocytic cells. Under these conditions, the transcriptional defects in CS cells under conditions of DNA damage [78,79] may also play a role.
It should be stressed that the model in Fig. 5 specifically applies to the myelination defects and brain calcification in CS. As noted above, CS patients experience degeneration of neurons in the cerebellum, and this particular neurologic feature has also been observed in Csb−/− Xpc−/−mice [80]. The neuronal loss is unlikely to be due to defective TC-NER, since neuronal cells and other terminally differentiated cells in humans employ a different form of NER, called differentiation-associated repair (DAR) [81], and CSB is not necessary for DAR [82]. For other possible explanations for the cerebellar degeneration in the mice, see [80]. The model in Fig. 5 assumes that glial cells, which do retain the capacity for cell division, use TC-NER.
In summary, the surprising overlap in neurological symptoms between CS and AGS suggests that vascular changes, and perhaps alterations in gene expression may play a role in the white matter phenotypes and brain calcification in both diseases.
8. Does the complete absence of CSB result in CS neurologic disease?
A complicating factor in any discussion of CS and the role of defective TC-NER in disease is the observation of a UV-sensitive syndrome (UVsS) patient, 1KOSV, who was reported to lack any detectable CSB protein and was defective in TC-NER [83]. However, other UVsS patients have normal CSB expression and normal TC-NER [84].
Two very recent reports provide additional insight into genotype–phenotype correlations in CSB. Laugel et al. [85] recently showed that complete absence of CSB expression due to a promoter mutation leads to the classic features of CS. Also, Hashimoto et al. [86] reported a Japanese patient, KPSX6 with late-onset CS neurologic disease. Strikingly, the mutation in KPSX6 is a premature termination near the N-terminus of CSB (codon 82), similar to that in 1KOSV (codon 77). Based on these results, the authors raised the possibility that 1KOSV and another patient with the same mutation may develop neurologic disease later in life, similar to KPSX6.
Extending the proposal of Laugel et al. [85], it may be that in cells from 1KOSV (and KPSX6 as well), there is a small amount of functional CSB protein made, either by read-through of the stop codon during translation or translational re-initiation downstream, which is too little to be detected by Western blotting but sufficient to carry out the function(s) of CSB in brain cells, and thereby prevent (or delay) the onset of neurologic disease. Of the two mechanisms, downstream translational re-initiation seems more likely, since this would explain why stop codon mutations farther downstream in the protein coding sequence are more deleterious that those near the N-terminus. Also, there is a precedent for such a re-initiation mechanism in another hereditary DNA repair disease, NBS [87].
To further complicate matters, cells from a different UVsS patient, Kps3, which has a normal CSB gene, can be distinguished from CSB cells by differences in host-cell reactivation of plasmids containing oxidative DNA lesions such as thymine glycol [88]. However, it is not clear whether this difference reflects a difference in TC-BER of oxidative DNA damage, a global BER deficiency, or some other difference in the way cells from patients with the two diseases cells handle the transfected oxidized DNA. Also, CSB cells show reduced survival after exposure to hydrogen peroxide compared to Kps3 cells [88]. While it is not immediately obvious how this difference could be related to the dysmyelination and calcification in CS neurologic disease, it is possible that this differential susceptibility to oxidative stress could be involved in other aspects of the CS phenotype, such as dwarfism. However, a full discussion of the role of the CS proteins in repair of oxidative DNA damage is beyond the scope the current article; see [89] for a recent review.
9. Implications for treatment and therapy
A clear understanding of disease mechanisms is of obvious relevance to the development of rational treatments and therapies for diseases. The only potential exception to this would be in the case of gene therapy, in that the delivery of a normal copy of the mutant gene into cells of the patients would be expected to have a therapeutic effect even if the normal function of the gene in cells is poorly understood. While gene transfer to the brain has in fact been carried out for some other rare genetic brain diseases [90,91], the mutant genes in these diseases encode enzymes that can act extracellularly to degrade toxic metabolites. In such cases, active enzyme secreted from a small number of transduced cells could have a therapeutic effect. In contrast, DNA repair proteins must obviously act inside specific cells, and specifically within the cell nucleus. Thus gene therapy for the neurological aspect of genetic DNA repair diseases seems an unlikely prospect in the near term.
Aside from gene therapy, the other therapeutic strategy based on the accumulated DNA damage mechanism would be to prevent the accumulation of the DNA lesion or lesions that are normally repaired by the compromised repair pathway. In the case of XP neurologic disease, the best candidate DNA lesions are the cyclopurine deoxynucleosides, as reviewed elsewhere [26]. To the extent that these lesions are involved in XP neurologic disease, then antioxidants or other drugs to prevent their formation might be useful. However, a role for these lesions in XP neurologic the disease has not been conclusively proven, and it remains entirely possible that other lesions normally repaired by NER may be involved in XP neurologic disease as well. Unfortunately, Xpa−/− mice do not develop neurodegeneration, perhaps because they do not live long enough for sufficient damage to accumulate.
Atm−/− mice do not develop neurologic disease to nearly the extent that the human patients do, though some Atm−/− mice do show some mild behavioral defects consistent with progressive Purkinje neuron dysfunction [92]. A recent study by Lavin and co-workers showed that treating the Atm−/− mice with an antioxidant produced dramatic improvements on some behavioral tests of balance and motor coordination [93]. For additional discussion of this work, as well as other therapeutic strategies for AT that might be applicable to other related disease see [94].
In contrast to the accumulated DNA damage model, if the primary cause of the neurologic defects in TTD or CS involve defects in nuclear hormone receptor dependent gene transcription, potential therapeutic strategies are very different. Since the defect in TTD is in the manner in which the target cell responds to hormone, rather than in hormone levels per se, simply treating the patients with hormone replacement therapy is not sufficient. At least in the case of TTD, since some TFIIH is present in the patient cells but unstable, it might be possible to develop a drug to stabilize the protein and thereby increase cellular concentrations. The availability of a the TTD mouse model, well-characterized patient cells, as well as the reconstituted biochemical systems [44] would likely be very useful in this regard.
Finally, in the case of AGS, additional knowledge about the signaling pathway between the undigested nucleic acid and activation of the innate immune system should help to identify therapeutic targets. For example, to the extent that a specific TLR pathway is involved, it might be possible to develop or utilize a drug that interferes with that pathway (see e.g. [95]). In light of evidence that TLRs and neuroinflammation play a role in more common brain diseases [96,97] research in these areas has increased dramatically in recent years. This increases the likelihood that effective drugs targeting these pathways in the brain will be developed for more common diseases [98], but which might benefit AGS patients as well.
10. Conclusions and future perspectives
We began this mini review by asking the question “Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage?” Based on the information available at present, the answer is that while degeneration of neurons in patients with DNA repair gene mutations is due to the accumulation of DNA damage, other neurologic abnormalities are better explained by other mechanisms. Specifically, dysregulation of thyroid hormone-dependent myelin gene expression is a more compelling explanation for the demyelination in TTD patients than accumulation of DNA damage. In AGS, it is not the accumulation of DNA damage that results in neurologic disease, but rather a pathological activation of the innate immune system in the brain due to undegraded normal nucleic acid that appears to be responsible. Finally, the overlapping neuropathologies in AGS and CS suggest that the dysmyelination and brain calcification in both diseases may be due in part to vascular changes, and perhaps reflecting common alterations in gene expression patterns. In view of the evidence for inflammation in the brain of CS patients, it is possible that lack of TC-NER or other DNA repair pathways may contribute to cell loss resulting from neuroinflammation.
Regarding the microcephalies, it is not clear that any of these three mechanisms outlined here are directly applicable. Instead, the key problem in these diseases is dealing with strand breaks occurring during S-phase, perhaps in the rapidly dividing cells in those regions of the embryo that give rise to the brain and craniofacial system. As such, ATR-dependent checkpoints and related responses seem to be most important [10,99]. Also, the development of the nervous system is an exceedingly complex process, and the specific DNA repair mechanisms involved vary depending on the specific cell type and stage of brain development [100]. Thus, the three mechanisms we have outlined here are not intended to cover all forms of neurologic disease due to DNA repair gene mutations.
On a final note, there can be no doubt that studies of microorganisms (bacteria and yeast), as well as readily accessible cells (e.g. lymphoblasts and fibroblasts) from human disease patients have been, and continue to be, of tremendous importance for understanding the cell-autonomous functions of DNA repair proteins. However, the complex phenotypes of human diseases resulting from DNA repair gene mutations may involve interactions between different cell types in the body. Therefore, mouse models are clearly valuable, particularly to the extent that they can recapitulate the human neurologic disease and be used to test therapies [93]. On the other hand, as illustrated by the absence of a neurologic phenotype in the Trex−/− mouse, some disease mechanisms in humans may be species specific. Therefore, for a complete understanding of the mechanisms underlying the neurologic diseases resulting from DNA repair gene mutations in humans, it will be important to focus future research efforts on the specific brain cell types affected in human diseases, as well as the interaction of these cells with others. Such a complete understanding of disease mechanisms is crucial for the rational development of treatments and therapies for the patients.
Acknowledgments
We thank Lydia Kibiuk for artistic assistance with Fig. 1, and Cheryl Marietta for helpful discussions and comments on the manuscript.
Footnotes
Note added in proof
After this manuscript as accepted for publication, a report appeared demonstrating that astrocytes are responsible for the production of IFN-α in the brain of AGS patients: J.T. Van Heteren, F. Rozenberg, E. Aronica, D. Troost, P. Lebon, T.W. Kuijpers, Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi-Goutières syndrome. Glia. 56 (2008) 568–578.
References
- 1.Friedberg E, Walker G, Siede W, Wood R, Schultz R, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, DC: 2005. [Google Scholar]
- 2.Kandel E, Schwatrz J, Jessel T. Principles of Neural Science. Elsevier; New York: 1991. [Google Scholar]
- 3.Ledoux SP, Shen CC, Grishko VI, Fields PA, Gard AL, Wilson GL. Glial cell-specific differences in response to alkylation damage. Glia. 1998;24:304–312. [PubMed] [Google Scholar]
- 4.Hollensworth SB, Shen C, Sim JE, Spitz DR, Wilson GL, LeDoux SP. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med. 2000;28:1161–1174. doi: 10.1016/s0891-5849(00)00214-8. [DOI] [PubMed] [Google Scholar]
- 5.Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- 6.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 7.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 8.Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Kadiu I, Glanzer JG, Kipnis J, Gendelman HE, Thomas MP. Mononuclear phagocytes in the pathogenesis of neurodegenerative diseases. Neurotox Res. 2005;8:25–50. doi: 10.1007/BF03033818. [DOI] [PubMed] [Google Scholar]
- 10.Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:991–1004. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 11.Itoh M, Hayashi M, Shioda K, Minagawa M, Isa F, Tamagawa K, Morimatsu Y, Oda M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326–333. doi: 10.1016/s0387-7604(99)00033-9. [DOI] [PubMed] [Google Scholar]
- 12.Crawford TO. Ataxia telangiectasia. Semin Pediatr Neurol. 1998;5:287–294. doi: 10.1016/s1071-9091(98)80007-7. [DOI] [PubMed] [Google Scholar]
- 13.Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)—its clinical presentation and molecular basis. DNA Repair (Amst) 2004;3:1219–1225. doi: 10.1016/j.dnarep.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 14.Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia. Cell. 2003;112:7–10. doi: 10.1016/s0092-8674(02)01247-3. [DOI] [PubMed] [Google Scholar]
- 15.Gueven N, Chen P, Nakamura J, Becherel OJ, Kijas AW, Grattan-Smith P, Lavin MF. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience. 2007;145:1418–1425. doi: 10.1016/j.neuroscience.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 16.Brumback RA, Brooks PJ, Leech R. Cockayne syndrome. In: Golden J, Harding B, editors. Pediatric Neuropathology. International Society of Neuropathology Press; Pegnitz: 2004. [Google Scholar]
- 17.Del Bigio MR, Greenberg CR, Rorke LB, Schnur R, McDonald-McGinn DM, Zackai EH. Neuropathological findings in eight children with cerebro-oculo-facio-skeletal (COFS) syndrome. J Neuropathol Exp Neurol. 1997;56:1147–1157. doi: 10.1097/00005072-199710000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype–phenotype relationship. Neuroscience. 2007;145:1388–1396. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barth PG. The neuropathology of Aicardi–Goutières syndrome. Eur J Paediatr Neurol. 2002;6(Suppl A):A27–A31. doi: 10.1053/ejpn.2002.0570. discussion A37-29, A77–86. [DOI] [PubMed] [Google Scholar]
- 20.Hernandez D, McConville CM, Stacey M, Woods CG, Brown MM, Shutt P, Rysiecki G, Taylor AM. A family showing no evidence of linkage between the ataxia telangiectasia gene and chromosome 11q22–23. J Med Genet. 1993;30:135–140. doi: 10.1136/jmg.30.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein C, Wenning GK, Quinn NP, Marsden CD. Ataxia without telangiectasia masquerading as benign hereditary chorea. Mov Disord. 1996;11:217–220. doi: 10.1002/mds.870110217. [DOI] [PubMed] [Google Scholar]
- 22.Delia D, Piane M, Buscemi G, Savio C, Palmeri S, Lulli P, Carlessi L, Fontanella E, Chessa L. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum Mol Genet. 2004;13:2155–2163. doi: 10.1093/hmg/ddh221. [DOI] [PubMed] [Google Scholar]
- 23.Fernet M, Gribaa M, Salih MA, Seidahmed MZ, Hall J, Koenig M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet. 2005;14:307–318. doi: 10.1093/hmg/ddi027. [DOI] [PubMed] [Google Scholar]
- 24.Andrews A, Barrett S, Robbins J. Xeroderma pigmentosun neurological abnormalities correlate with the colony forming ability after ultraviolet irradiation. Proc Natl Acad Sci USA. 1978;75:1984–1988. doi: 10.1073/pnas.75.4.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev. 2001;15:15–23. doi: 10.1101/gad.859501. [DOI] [PubMed] [Google Scholar]
- 26.Brooks PJ. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443:713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- 28.el-Khamisy SF, Caldecott KW. DNA single-strand break repair and spinocerebellar ataxia with axonal neuropathy-1. Neuroscience. 2007;145:1260–1266. doi: 10.1016/j.neuroscience.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 29.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genom Hum Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 30.Oka A, Takashima S. Expression of the ataxia-telangiectasia gene (ATM) product in human cerebellar neurons during development. Neurosci Lett. 1998;252:195–198. doi: 10.1016/s0304-3940(98)00576-x. [DOI] [PubMed] [Google Scholar]
- 31.Barlow C, Ribaut-Barassin C, Zwingman TA, Pope AJ, Brown KD, Owens JW, Larson D, Harrington EA, Haeberle AM, Mariani J, Eckhaus M, Herrup K, Bailly Y, Wynshaw-Boris A. ATM is a cytoplasmic protein in mouse brain required to prevent lysosomal accumulation. Proc Natl Acad Sci USA. 2000;97:871–876. doi: 10.1073/pnas.97.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biton S, Dar I, Mittelman L, Pereg Y, Barzilai A, Shiloh Y. Nuclear ataxia-telangiectasia mutated (ATM) mediates the cellular response to DNA double strand breaks in human neuron-like cells. J Biol Chem. 2006;281:17482–17491. doi: 10.1074/jbc.M601895200. [DOI] [PubMed] [Google Scholar]
- 33.Biton S, Gropp M, Itsykson P, Pereg Y, Mittelman L, Johe K, Reubinoff B, Shiloh Y. ATM-mediated response to DNA double strand breaks in human neurons derived from stem cells. DNA Repair (Amst) 2007;6:128–134. doi: 10.1016/j.dnarep.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 34.Dar I, Biton S, Shiloh Y, Barzilai A. Analysis of the ataxia telangiectasia mutated-mediated DNA damage response in murine cerebellar neurons. J Neurosci. 2006;26:7767–7774. doi: 10.1523/JNEUROSCI.2055-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorodetsky E, Calkins S, Ahn J, Brooks PJ. ATM, the Mre11/Rad50/Nbs1 complex, and topoisomerase I are concentrated in the nucleus of Purkinje neurons in the juvenile human brain. DNA Repair (Amst) 2007;6:1698–1707. doi: 10.1016/j.dnarep.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y, Barnes DE, Lindahl T, McKinnon PJ. Defective neurogenesis resulting from DNA ligase IV deficiency requires Atm. Genes Dev. 2000;14:2576–2580. doi: 10.1101/gad.837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y, Chong MJ, McKinnon PJ. Ataxia telangiectasia mutated-dependent apoptosis after genotoxic stress in the developing nervous system is determined by cellular differentiation status. J Neurosci. 2001;21:6687–6693. doi: 10.1523/JNEUROSCI.21-17-06687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abner CW, McKinnon PJ. The DNA double-strand break response in the nervous system. DNA Repair (Amst) 2004;3:1141–1147. doi: 10.1016/j.dnarep.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 39.Schaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, Chambon P, Egly JM. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 40.Vermeulen W, van Vuuren AJ, Chipoulet M, Schaeffer L, Appeldoorn E, Weeda G, Jaspers NG, Priestley A, Arlett CF, Lehmann AR, et al. Three unusual repair deficiencies associated with transcription factor BTF2(TFIIH): evidence for the existence of a transcription syndrome. Cold Spring Harb Symp Quant Biol. 1994;59:317–329. doi: 10.1101/sqb.1994.059.01.036. [DOI] [PubMed] [Google Scholar]
- 41.Egly JM. The 14th Datta lecture. TFIIH: from transcription to clinic. FEBS Lett. 2001;498:124–128. doi: 10.1016/s0014-5793(01)02458-9. [DOI] [PubMed] [Google Scholar]
- 42.Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell. 2007;26:245–256. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 43.Rochette-Egly C, Adam S, Rossignol M, Egly JM, Chambon P. Stimulation of RAR alpha activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell. 1997;90:97–107. doi: 10.1016/s0092-8674(00)80317-7. [DOI] [PubMed] [Google Scholar]
- 44.Compe E, Malerba M, Soler L, Marescaux J, Borrelli E, Egly JM. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat Neurosci. 2007;10:1414–1422. doi: 10.1038/nn1990. [DOI] [PubMed] [Google Scholar]
- 45.de Boer J, de Wit J, van Steeg H, Berg RJ, Morreau H, Visser P, Lehmann AR, Duran M, Hoeijmakers JH, Weeda G. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell. 1998;1:981–990. doi: 10.1016/s1097-2765(00)80098-2. [DOI] [PubMed] [Google Scholar]
- 46.Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell. 2007;26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 47.Friedberg EC, Wood RD. New insights into the combined Cockayne/xeroderma pigmentosum complex: human XPG protein can function in transcription factor stability. Mol Cell. 2007;26:162–164. doi: 10.1016/j.molcel.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Scharer OD. The molecular basis for different disease states caused by mutations in TFIIH and XPG. DNA Repair (Amst) 2008;7:339–344. doi: 10.1016/j.dnarep.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindenbaum Y, Dickson D, Rosenbaum P, Kraemer K, Robbins I, Rapin I. Xeroderma pigmentosum/Cockayne syndrome complex: first neuropathological study and review of eight other cases. Eur J Paediatr Neurol. 2001;5:225–242. doi: 10.1053/ejpn.2001.0523. [DOI] [PubMed] [Google Scholar]
- 50.Aicardi J. Aicardi–Goutières syndrome: special type early-onset encephalopathy. Eur J Paediatr Neurol. 2002;6(Suppl A):A1–A7. doi: 10.1053/ejpn.2002.0567. discussion A23–25, A77–86. [DOI] [PubMed] [Google Scholar]
- 51.Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen HJ, Corry PC, Cowan FM, Cox H, D’Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SG, Garcia-Cazorla A, Gener B, Goizet C, Goutières F, Green AJ, Guet A, Hamel BC, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang YH, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EG, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard ML, Nischal KK, Ostergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Burgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JB, Tacke U, Tan TY, Till M, Tolmie JL, Tomlin P, Vagnarelli F, Valente EM, Van Coster RN, Van der Aa N, Vanderver A, Vles JS, Voit T, Wassmer E, Weschke B, Whiteford ML, Willemsen MA, Zankl A, Zuberi SM, Orcesi S, Fazzi E, Lebon P, Crow YJ. Clinical and molecular phenotype of Aicardi–Goutières syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi–Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 53.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martinez-Frias ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JB, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi–Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 54.Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068–1070. doi: 10.1038/ng2082. [DOI] [PubMed] [Google Scholar]
- 55.Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 56.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, Crow YJ. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi–Goutières syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, Pfeiffer C, Hollis T, Gahr M, Perrino FW, Lieberman J, Hubner N. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007;85:531–537. doi: 10.1007/s00109-007-0199-9. [DOI] [PubMed] [Google Scholar]
- 58.Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE. Gene-targeted mice lacking the Trex1 (DNase III) 3′ → 5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 60.Unterholzner L, Bowie AG. The interplay between viruses and innate immune signaling: recent insights and therapeutic opportunities. Biochem Pharmacol. 2007;75:589–602. doi: 10.1016/j.bcp.2007.07.043. [DOI] [PubMed] [Google Scholar]
- 61.Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol. 2006;27:525–532. doi: 10.1016/j.it.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 62.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 63.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 64.Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coscoy L, Raulet DH. DNA mismanagement leads to immune system oversight. Cell. 2007;131:836–838. doi: 10.1016/j.cell.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161:5016–5026. [PubMed] [Google Scholar]
- 67.Crack PJ, Bray PJ. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol Cell Biol. 2007;85:476–480. doi: 10.1038/sj.icb.7100103. [DOI] [PubMed] [Google Scholar]
- 68.Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T, Carr DJ, Gold LH, Henriksen SJ, Siggins GR. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-alpha. Brain Res. 1999;835:46–61. doi: 10.1016/s0006-8993(99)01328-1. [DOI] [PubMed] [Google Scholar]
- 69.Meira LB, Graham JM, Jr, Greenberg CR, Busch DB, Doughty AT, Ziffer DW, Coleman DM, Savre-Train I, Friedberg EC. Manitoba aboriginal kindred with original cerebro-oculo-facio-skeletal syndrome has a mutation in the Cockayne syndrome group B (CSB) gene. Am J Hum Genet. 2000;66:1221–1228. doi: 10.1086/302867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lanzi G, D’Arrigo S, Drumbl G, Uggetti C, Fazzi E. Aicardi–Goutières syndrome: differential diagnosis and aetiopathogenesis. Funct Neurol. 2003;18:71–75. [PubMed] [Google Scholar]
- 71.Graham JM, Jr, Anyane-Yeboa K, Raams A, Appeldoorn E, Kleijer WJ, Garritsen VH, Busch D, Edersheim TG, Jaspers NG. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet. 2001;69:291–300. doi: 10.1086/321295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hamel BC, Raams A, Schuitema-Dijkstra AR, Simons P, van der Burgt I, Jaspers NG, Kleijer WJ. Xeroderma pigmentosum–Cockayne syndrome complex: a further case. J Med Genet. 1996;33:607–610. doi: 10.1136/jmg.33.7.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jaspers NG, Raams A, Silengo MC, Wijgers N, Niedernhofer LJ, Robinson AR, Giglia-Mari G, Hoogstraten D, Kleijer WJ, Hoeijmakers JH, Vermeulen W. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80:457–466. doi: 10.1086/512486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rapin I, Weidenheim K, Lindenbaum Y, Rosenbaum P, Merchant SN, Krishna S, Dickson DW. Cockayne syndrome in adults: review with clinical and pathologic study of a new case. J Child Neurol. 2006;21:991–1006. doi: 10.1177/08830738060210110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci USA. 2006;103:9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hayashi M, Itoh M, Araki S, Kumada S, Shioda K, Tamagawa K, Mizutani T, Morimatsu Y, Minagawa M, Oda M. Oxidative stress and disturbed glutamate transport in hereditary nucleotide repair disorders. J Neuropathol Exp Neurol. 2001;60:350–356. doi: 10.1093/jnen/60.4.350. [DOI] [PubMed] [Google Scholar]
- 77.Cline SD, Riggins JN, Tornaletti S, Marnett LJ, Hanawalt PC. Malondialdehyde adducts in DNA arrest transcription by T7 RNA polymerase and mammalian RNA polymerase II. Proc Natl Acad Sci USA. 2004;101:7275–7280. doi: 10.1073/pnas.0402252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kyng KJ, May A, Brosh RM, Jr, Cheng WH, Chen C, Becker KG, Bohr VA. The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene. 2003;22:1135–1149. doi: 10.1038/sj.onc.1206187. [DOI] [PubMed] [Google Scholar]
- 79.Proietti-De-Santis L, Drane P, Egly JM. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J. 2006;25:1915–1923. doi: 10.1038/sj.emboj.7601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci USA. 2007;104:1389–1394. doi: 10.1073/pnas.0610619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nouspikel T. DNA repair in differentiated cells: some new answers to old questions. Neuroscience. 2007;145:1213–1221. doi: 10.1016/j.neuroscience.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 82.Nouspikel TP, Hyka-Nouspikel N, Hanawalt PC. Transcription domain-associated repair in human cells. Mol Cell Biol. 2006;26:8722–8730. doi: 10.1128/MCB.01263-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci USA. 2004;101:15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Spivak G, Itoh T, Matsunaga T, Nikaido O, Hanawalt P, Yamaizumi M. Ultraviolet-sensitive syndrome cells are defective in transcription-coupled repair of cyclobutane pyrimidine dimers. DNA Repair (Amst) 2002;1:629–643. doi: 10.1016/s1568-7864(02)00056-3. [DOI] [PubMed] [Google Scholar]
- 85.Laugel V, Dalloz C, Stary A, Cormier-Daire V, Desguerre I, Renouil M, Fourmaintraux A, Velez-Cruz R, Egly JM, Sarasin A, Dollfus H. Deletion of 5′-sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. Eur J Hum Genet. 2008;16:320–327. doi: 10.1038/sj.ejhg.5201991. [DOI] [PubMed] [Google Scholar]
- 86.Hashimoto S, Suga T, Kudo E, Ihn H, Uchino M, Tateishi S. Adult-onset neurological degeneration in a patient with Cockayne syndrome and a null mutation in the CSB gene. J Invest Dermatol. 2008 Jan 10; doi: 10.1038/sj.jid.5701210. [Epub ahead of print] PMID: 18185538. [DOI] [PubMed] [Google Scholar]
- 87.Maser RS, Zinkel R, Petrini JH. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet. 2001;27:417–421. doi: 10.1038/86920. [DOI] [PubMed] [Google Scholar]
- 88.Spivak G, Hanawalt PC. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 2006;5:13–22. doi: 10.1016/j.dnarep.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 89.Frosina G. The current evidence for defective repair of oxidatively damaged DNA in Cockayne syndrome. Free Radic Biol Med. 2007;43:165–177. doi: 10.1016/j.freeradbiomed.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 90.Janson C, McPhee S, Bilaniuk L, Haselgrove J, Testaiuti M, Freese A, Wang DJ, Shera D, Hurh P, Rupin J, Saslow E, Goldfarb O, Goldberg M, Larijani G, Sharrar W, Liouterman L, Camp A, Kolodny E, Samulski J, Leone P. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther. 2002;13:1391–1412. doi: 10.1089/104303402760128612. [DOI] [PubMed] [Google Scholar]
- 91.Crystal RG, Sondhi D, Hackett NR, Kaminsky SM, Worgall S, Stieg P, Souweidane M, Hosain S, Heier L, Ballon D, Dinner M, Wisniewski K, Kaplitt M, Greenwald BM, Howell JD, Strybing K, Dyke J, Voss H. Clinical protocol. Administration of a replication-deficient adeno-associated virus gene transfer vector expressing the human CLN2 cDNA to the brain of children with late infantile neuronal ceroid lipofuscinosis. Hum Gene Ther. 2004;15:1131–1154. doi: 10.1089/hum.2004.15.1131. [DOI] [PubMed] [Google Scholar]
- 92.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 93.Gueven N, Luff J, Peng C, Hosokawa K, Bottle SE, Lavin MF. Dramatic extension of tumor latency and correction of neurobehavioral phenotype in Atm-mutant mice with a nitroxide antioxidant. Free Radic Biol Med. 2006;41:992–1000. doi: 10.1016/j.freeradbiomed.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 94.Lavin MF, Gueven N, Bottle S, Gatti RA. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br Med Bull. 2007;81–82:129–147. doi: 10.1093/bmb/ldm012. [DOI] [PubMed] [Google Scholar]
- 95.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 96.Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis. 2008;29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 97.Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, oxidative stress and the pathogenesis of Parkinson’s disease. Clin Neurosci Res. 2006;6:261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Infante-Duarte C, Waiczies S, Wuerfel J, Zipp F. New developments in understanding and treating neuroinflammation. J Mol Med. 2008 Jan 15; doi: 10.1007/s00109-007-0292-0. [Epub ahead of print] PMID: 18196212. [DOI] [PubMed] [Google Scholar]
- 99.Frappart PO, McKinnon PJ. BRCA2 function and the central nervous system. Cell Cycle. 2007;6:2453–2457. doi: 10.4161/cc.6.20.4785. [DOI] [PubMed] [Google Scholar]
- 100.Lee Y, McKinnon PJ. Responding to DNA double strand breaks in the nervous system. Neuroscience. 2007;145:1365–1374. doi: 10.1016/j.neuroscience.2006.07.026. [DOI] [PubMed] [Google Scholar]