

Abstract
Langerhans cells (LCs) serve as epidermal sentinels of the adaptive immune system. Conventional wisdom suggests that LC encounter antigen in the skin, and then migrate to the draining lymph nodes, where the antigen is presented to T cells thus initiating an immune response. Platelet-activation factor (PAF) is a phospholipid mediator with potent biological effects. During inflammation, PAF mediates recruitment of leukocytes to inflammatory sites. Here we tested a hypothesis that PAF induces LC migration. Applying 2,4-dinitrofluorobenzene (DNFB) to wild-type (WT) mice activated LC migration. In contrast, applying DNFB to PAF receptor (PAFR)-deficient mice or mice injected with PAF receptor antagonists failed to induce LC migration. Moreover, after FITC application the appearance of hapten-laden LCs (FITC+, CD11c+, Langerin+) in the lymph nodes of PAF receptor deficient mice was significantly depressed compared with that found in WT mice. LC chimerism indicates that PAF receptor on keratinocytes but not LCs is responsible for LC migration. Contrary to the diminution of LC migration in PAF receptor-deficient mice, we did not observe any difference in the migration of hapten-laden dDCs (FITC+, CD11c+, Langerin−) into the lymph nodes of PAF receptor-deficient mice. In addition, the contact hypersensitivity response generated in WT or PAF receptor-deficient mice was identical. Finally, dDCs, but not LCs isolated from the draining lymph nodes after hapten application activated T cell proliferation. These findings suggest that LC migration may not be responsible for the generation of CHS, and dDCs may play a more important role.
Keywords: Dendritic cells, Lipid mediators, Cell trafficking, Skin, Antigen Presentation/Processing
Dendritic cells (DCs)3 are professional APCs that play a crucial role in activating adaptive immune responses. Langerhans cells (LCs) are a subset of immature DCs that reside in the epidermis. LCs are distinguished from other DCs by the presence of cytoplasmic organelles, known as Birbeck granules (1). LCs are characterized by expression of the transmembrane type-II Ca2+-dependent lectin, langerin/CD207 (2). Because of their specialized location, LCs are thought to constitute the first immunologic barrier to pathogens. LC migration is triggered via a variety of stimuli including, hapten application, TNF-α, and UV irradiation (3-6). LCs are traditionally thought to play a role in the induction of contact hypersensitivity (CHS). However, three different groups reported three distinct results after applying hapten to mice genetically engineered to be deficient in LCs: a diminished reaction (7), a enhanced reaction (8), or an unchanged response (9). Moreover, recent findings suggest that LCs are not required for the induction of humoral and cell-mediated immunity following gene gun immunization (10). Therefore, it remains controversial whether LCs are dispensable for the induction of skin immunity.
Dermal DCs (dDCs) were identified more than 120 years after LCs were discovered (11). There is no exclusive marker for dDCs comparable to langerin/CD207, making dDCs difficult to track in vivo and they have often been overlooked in studies of skin immunity. Recently, Kisssenpfennig et al. demonstrated that CD11c+, CD8α−, CD205intermediate, epidermal growth factor negative, hapten positive, DCs migrating to draining lymph nodes of mice are dDC, and their data suggest dDC are responsible for inducing CHS (9).
Platelet-activating factor (PAF) is a phospholipid mediator with potent biological effects (12). PAF binds to a specific receptor (PAFR), which is found on a wide variety of cells, including platelets, monocytes, mast cells, polymorphonuclear lymphocytes dendritic cells and keratinocytes (13). PAF is produced by physical stress including UV and trauma (14). PAF is involved in UVB, PUVA, and jet fuel-induced immune suppression (13, 15, 16). PAF also plays a role in regulating the immune response to microbial pathogens (14). PAF binding results in the production of numerous cytokines (13, 17, 18). During inflammation, PAF mediates recruitment of leukocytes to inflammatory sites (12, 19), but its role in LC or dDC migration is unclear.
Based primarily on the role of PAF in UV and PUVA-induced immune suppression, we hypothesized that PAF receptor binding induces LC migration. We used PAFR-deficient mice and PAFR antagonists to test the role of PAF in LC migration. We found that PAF is involved in LC migration from the skin to the draining lymph nodes, but does not appear to play a role in dDC migration. Regardless, we did not observe any difference in the CHS response generated in wild type (WT) mice versus PAFR-deficient mice, suggesting the dDC and not LC play an essential role in the induction of CHS.
Materials and Methods
Mice
PAFR-deficient mice, backcrossed onto a C57BL/6 background, originally described by Shimizu and colleagues (20), were provided to us by Dr Jeffrey B. Travers (Indiana University School of Medicine, Indianapolis, IN), with Prof. Shimizu's permission. Wild-type C57BL/6 mice that express the CD45.2 allele were purchased from National Cancer Institute. OT-II mice (Vα2 Vβ5.1/5.2 T cell receptor transgenic) were provided to us by Dr. Yong-Jun Liu (UT MD Anderson Cancer Center). C57BL/6 mice that express the CD45.1 allele (C57BL/6-Ly5.1Peb3b) were purchased from the Jackson Laboratory (Bar Harbor, ME). Within each experiment, all mice were age and sex matched. All procedures were reviewed and approved by the University of Texas M. D. Anderson Cancer Center Animal Care and Use Committee.
Abs and reagents
Abs (FITC, PE, PerCP-Cy5.5, and/or allophycocyanin) specific for CD45.1, CD45.2, CD8α, CD11c, PE-conjugated mouse anti rat IgG2a, corresponding isotype controls, and secondary reagents (FITC-conjugated streptavidin, PerCP-conjugated streptavidin, and allophycocyaninconjugated streptavidin) were purchased from BD Bioscience. Abs specific (Biotin) for I-A/I-E and Langerin (CD207; clone eBioRMUL.2) were purchased from eBiosciences. Ab specific for the extracellular domain of Langerin (clone 205C1) was purchased from AbCys (Paris, France) (21). Abs specific for PAFR was purchased from Cayman (Ann Arbor, MI). FITC isomer I and 2,4-dinitro-1-fluorobenzene (DNFB) were purchased from Sigma-Aldrich. Alexa Flour 594 goat anti-rat IgG(H+L) Ab was purchased from Invitrogen. The PAFR agonist carbamyl-PAF (c-PAF), the PAFR antagonists PCA4248 and CV3988 were purchased from Biomol. Stock solutions were prepared by dissolving each in a 50% DMSO/PBS buffer and diluted further in PBS before injection. Recombinant murine TNF-α was purchased from PeproTech Inc. The OVA peptide 323−339 (ISQAVHAAHAEINEAGR) was purchased from GenScript Corp.
Preparation of murine epidermal sheets and immunofluorescence analysis
Murine epidermal sheets were prepared as described previously (22). After fixation, the sheets were incubated at room temperature over night with purified anti-mouse Langerin or biotinconjugated anti-mouse I-A/I-E. They were then washed with PBS, were incubated at room temperature for 1 h with Alexa Flour 594 goat anti-rat IgG(H+L) and FITC-conjugated Streptavidin, anti-CD45.1, or anti-CD45.2. After washing with PBS, they were mounted using VECTASHIELD HardSet Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA), respectively. The samples were analyzed using a fluorescence microscope (Olympus, Tokyo, Japan). The number of LCs found in the epidermis was determined by counting at least 10 fields/sample.
DNFB sensitization and in vivo PAF antagonist treatment
WT and PAFR-deficient mice were painted with 10 μl of 0.5% DNFB solubilized in acetone/olive oil (4:1) on both ear surfaces. Twenty-four hours later, the ears were collected and LC numbers enumerated by immunofluorescence. In some experiments, the PAFR antagonists, PCA4248, CV3988 (500 nmol per mouse), were injected into the peritoneal cavity (ip) immediately before DNFB application.
c-PAF or TNF-α-induced LC migration assay
WT mice received c-PAF (150 pmol/ear), murine recombinant TNF-α (50 ng/ear), (intradermal injection) or an equal volume (30 μl) of the vehicle into the pinnae of the ear. The ears were harvested 1 or 24 h after injection.
FITC-bearing LC or dDC migration assay
400 μl of a 7.5 mg/ml solution of FITC (dissolved in 1:1 acetone/dibutylphthalate) was applied on the shaved back skin. Two days later, the draining inguinal lymph nodes were removed, and 5 μm frozen sections were prepared. The sections were stained with purified anti-mouse Langerin at room temperature for 1 hour. Alternatively, lymph nodes were removed and single cell suspensions were analyzed by flow cytometry. For intracellular staining, BD Cytofix/CytopermTM (BD Biosciences) kit was used according to the manufactures' recommendations. Permeabilized fixed cells were incubated with purified with anti-Langerin at 4 °C over night, washed with BD Perm/WashTM, and incubated with PE-conjugated mouse anti-rat IgG2a at 4 °C for 40 min. After washing, flow-cytometric analysis was performed (FACSCalibur, BD Biosciences).
Real-Time quantitative RT-PCR
Murine keratinocytes (PAM 212) were plated and treated with 1 μM of c-PAF. Total RNA was extracted from the PAM 212 cells using Rneasy (Quiagen, Valencia, CA). cDNA was reverse transcribed from total RNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Murine TNF-α (Assay ID: Mm00443258_m1) expression was quantified using Taqman Gene Expression assay (Applied Biosystems) in ABI PRISM 7000 sequence detection system (Applied Biosystems). GAPDH gene expression was measured using the TaqMan rodent GAPDH reagents (Applied Biosystems). Real-time amplifications were analyzed using SDS 7000 1.1 software (Applied Biosystems). Threshold cycle (CT) values for TNF-α were normalized to GAPDH using the following equation; 1.8(GADPH-TNF-α) x10,000, where GADPH is the CT of the GADPH control, TNF-α is the CT of the TNF-α and 10,000 is an arbitrary factor to bring all values above one.
CHS response
WT and PAFR-deficient mice were immunized by applying 50 μl of 0.5% DNFB solution solubilized in acetone/olive oil (4:1) on their shaved abdomens. Six days later, 10 μl of 0.2% DNFB solution was applied to both ear surfaces. On day 7, the mice were sedated, and the thickness of each ear was measured with an engineer's micrometer (Mitutoyo, Tokyo, Japan). The data are expressed as the mean change in ear swelling (left ear + right ear ÷ 2) for each animal (n= 5).
Preparation and analysis of LC chimeric mice
LC chimeric mice were generated as described previously (23). Briefly, 7 to 8-week-old recipient mice (CD45.2+ PAFR-deficient or CD45.1+ congenic) were lethally irradiated with two doses of 550 rads each, 3 h apart, and injected intravenously with bone marrow (BM) cells (5×105) obtained from CD45.1+ or CD45.2+ donors. Seven weeks after BM transplantation, >90% of blood cells were of donor origin. The dorsal hair of the chimeric mice was removed with electric clippers and then exposed to 10kJ/m2 (CD45.1+ mice) or 20 kJ/m2 (PAFR-deficient mice) of UVB (290−320nm, FS-40 sunlamps) radiation as measured with an IL-1700 research radiometer (International Light). The presence of donor- and host-derived LCs in the epidermis was evaluated by flow-cytometric analysis of epidermal cell suspensions and immunofluorescence analysis of epidermal sheets 9 weeks after UVB irradiation (24). Murine dorsal skins were incubated for 16 hours at 4°C in 0.3% trypsin Hank's Balanced Salt Solution (GIBCO) and epidermal sheets were separated from the dermis. The epidermal sheets were incubated with 0.3% trypsin/DNAase I (Sigma) Hank's Balanced Salt Solution for 20 min at 37°C, were shaken vigorously, filtered through 70μm filter, and then re-suspended into 5% FCS/PBS for flow cytometry.
T cell Proliferation assay
4-Ethoxymethylene-2-phenyl-2-oxazoline-5-one (OX) was dissolved in acetone/dibutylphthalate (1:1). Two days after applying 400μl of 2% OX, the draining lymph node were removed, single cells suspension were prepared and the CD11c+ CD8αlow/− Langerin+ (skin-derived LC) and CD11c+ CD8αlow/− Langerin− (skin-derived dDC) were selected by sorting. CD4+ T cells were isolated from OT-II mice by negative selection with magnetic beads cells (Miltenyi Biotec) as described previously (25). To monitor cell proliferation, CD4+ T cells were incubated with CFSE at final concentration of 5μM in 1%FCS/PBS for 10 min at 37°C. CFSE-labeled T cells (1×105), LCs and OVA peptide 323−339 (10μg/ml) and were incubated in 96 well plates in complete RPMI 1640. Proliferation in wells with plate-bound anti-mouse CD3 Abs (10μg/ml; BD Bioscience) and soluble anti-mouse CD28 Abs (5μg/ml; BD Bioscience) served as the positive control. After 4 days, the cells were stained with allophycocyanin-conjugated anti-mouse CD4 (eBioscience) and CFSE content analyzed by FACSCalibur. The division index was calculated using FlowJo software (Tree Star, Ashland, OR). Alternatively 3H-thymidine incorporation was used as a measure of T cell proliferation. Forty-eight hrs after the initiation of culture 1 μCi of radiolabeled thymidine (Amersham) was added to each well. Eight hours later, incorporation of 3H-thymidine into newly synthesized DNA was determined by liquid scintillation counter. Cultures were run in triplicate.
Statistical analysis
Statistical differences between the positive control and the experimental groups were determined by Student's two-tailed t-test. Probabilities less than 0.05 were considered to be significant.
Results
PAF is involved in LC migration from the epidermis
We first tested the hypothesis that PAF plays a role in LC migration. Applying hapten to WT mice had two effects. First, fewer LC were found in the epidermal sheets. Second, there was a noticeable increase in the size of the remaining LC. (Figs. 1A & 1B, & 1C). In contrast, applying DNFB to PAFR-deficient mice failed to induce the migration of LC (Figs. 1A & 1B, & 1C). Next, mice were injected with PCA4248, CV3988, two PAF receptor antagonists, and then treated with hapten. LC migration was measured 24 h later. As expected, applying DNFB induced LC migration (Figs. 1D & 1E). In contrast, applying DNFB immediately after injecting PCA4248 or CV3988, failed to induce LC migration (Figs. 1D, & 1E). To further test the role of PAF in LC migration, carbamyl-PAF (c-PAF) a metabolic stable analogue of PAF, was injected into the skin. Injecting 150 pmol of c-PAF induced LC migration (Fig. 1F). These results indicate that PAF signaling activates LC migration.
FIGURE 1. PAF is involved in LC migration from the epidermis.

A, DNFB was applied to the ears of WT mice and PAFR-deficient mice. Epidermal sheets were collected 1 day after DNFB application and stained with Langerin. B, After staining, the number of LCs found in the epidermis was determined by counting. *, Statistically significant difference (p < 0.001; n=5) from no treatment group of WT mice. C, Epidermal sheets were collected 1 day after DNFB application and stained with I-A. *, Statistically significant difference (p < 0.001; n=5) from no treatment group of WT mice. D, The PAFR antagonists, PCA4248, CV3988 (500 nmol per mouse), or respective vehicle control was injected i.p. immediately before DNFB application. Epidermal sheets were collected 1 day after DNFB application and stained with Langerin. E, After staining, the number of LCs found in the epidermis was determined by counting. *, Statistically significant difference (p < 0.001; n=5) from no treatment group. F, 150 pmol of c-PAF or respective vehicle control was injected into the pinnae of the ear. Epidermal sheets were collected 1 day after injection and stained with Langerin. *, Statistically significant difference (p < 0.02; n=5) from vehicle group. A representative experiment is shown; each experiment was performed at least twice.
PAF is involved in LC migration from the skin to the draining lymph nodes
The previous findings indicate that PAF is involved in inducing LC to leave the skin. To determine if PAF is responsible for inducing LC to migrate to the lymph nodes, the following was done. WT mice were painted with FITC. One to 5 days later, the draining lymph node was removed, a single cell suspension was prepared and the cells were stained with anti-CD11c, anti-CD8α, and anti-Langerin. Flow cytometric analysis indicated that two subsets of CD11c+FITC+ cells were found in the draining lymph nodes (Fig. 2A). The main subset was Langerin−, matching the phenotype expected for migrating hapten-laden dDCs (9). Over 90% of the FITC+ CD11c+ cells exhibited a CD8αlow/− phenotype matching that expected for skin-derived DCs (9, 21) (Fig. 2A right panel). The second subset of FITC+CD11c+ were Langerin+ (Fig. 2A) matching that expected for migrating hapten-laden LCs (9). We observed a time-dependent increase in DC migration and cell number in the lymph node, peaking 2 days to 3days after FITC application and then gradually decreasing (Figs. 2B & 2C). PAFR-deficient mice then were painted with FITC to test a role for PAF in LC migration. The appearance of FITC+CD11c+Langerin+ cells in draining lymph nodes of PAFR-deficient mice was depressed compared to that found in WT mice (Figs. 2D & 2E). These data indicate that PAF is involved in LC migration from the skin to draining lymph nodes.
FIGURE 2. PAF is involved in LC migration from the skin to the draining lymph nodes.

A, FITC was applied onto the backs of WT mice. Two days after FITC application, draining lymph nodes were collected and were stained with CD11c and Langerin. A. We gated on the FITC+ CD11c+ population, and counted the Langerin+ and CD8α+ cells. In Langerin panel, blue line indicates Langerin staining, red line indicate isotype control. In CD8a panel, red line indicates CD8α expression in whole cells DLN (positive control), blue line indicates CD8α expression in FITC+ CD11c+ population, and green line indicates isotype control. B, The number of FITC+ CD11c+ Langerin+ cells (LCs) and FITC+ CD11c+ Langerin− cells (dDCs) found in draining lymph nodes was counted at various times after FITC application. C, Cell numbers of the lymph nodes was increased after hapten application. D, FITC was applied onto the backs of WT mice and PAFR-deficient mice. Two days later, the number of FITC+ CD11c+ Langerin+ cells (LCs) in the draining lymph nodes were counted. *, Statistically significant difference (p < 0.05; n=5) from WT mice. E, FITC was applied onto the backs of WT mice and PAFR-deficient mice. Two days later, the inguinal lymph nodes were collected and stained with Langerin (red). In PAFR-deficient mice, fewer FITC+ Langerin+ cells (yellow; LCs) were found compared to those in WT mice. Bar = 200 μm. A representative experiment is shown; each experiment was performed at least twice.
PAFR on keratinocytes but not LCs plays an important role in LC migration
LC migration is regulated by keratinocyte-derived cytokines, such as TNF-α or IL-1β produced by LCs (26). PAF binds to a specific receptor found on a wide variety of cells, including, DCs and keratinocytes (13, 27). To determine whether PAF binding to its receptor on keratinocytes drives LC migration, we made LC chimeric mice. The first group (PAF receptor −/− CD45.2 BM cells into CD45.1 recipients) is characterized by PAF receptor positive keratinocytes and PAF receptor negative LCs. The second group (CD45.1 BM cells into CD45.2 PAFR −/− mice) is characterized by PAF receptor negative keratinocytes and PAF receptor positive LCs. We observed that high dose UV irradiation (10−20 kJ/m2) of both groups depleted the host LCs and resulted in LC chimeric mice. We found Langerin+ LCs in mice exposed to 10kJ/m2 of UV express CD45.1 (host phenotype) 9 weeks after UV irradiation (Figs. 3A & 3B). Similarly, when PAFR −/− BM cells (CD45.2) were used to reconstitute x-irradiated CD45.1 mice, and these mice were subsequently exposed to 10 kJ/m2 of UVB, 9 weeks post-UV irradiation, all the LCs in the skin were of donor origin (CD45.2) (Figs. 3A & 3B). We also could produce LC chimerism in PAFR-deficient recipient mice, however, higher dose of UVB radiation were required (Figs. 3C & 3D). We then applied DNFB to the ears of the LC chimeric mice to activate LC migration. There was no significant difference in LC migration between CD45.1 control mice and group 1 (PAFR −/− CD45.2 bone marrow into CD45.1 recipients) mice (Fig. 3E). However, LC migration was totally abrogated when group 2 mice (CD45.1 bone marrow cells into CD45.2 PAFR −/− mice) were treated with DNFB (Fig. 3F). These results indicate that PAFR on keratinocytes, but not LCs, plays an important role in LC migration after hapten application.
FIGURE 3. PAFR on keratinocytes but not LCs plays an important role in LC migration.

Two groups of chimeric mice were prepared: Group 1 (PAFR −/− CD45.2 bone marrow cells into CD45.1 recipients; LC are PAFR−/−); Group 2 (CD45.1 bone marrow into CD45.2 PAFR −/−mice; keratinocytes are PAFR−/−). A, Representative experiment showing the expression of CD45.1 and CD45.2 on gated I-A+ CD11c+ LCs isolated from CD45.1 recipients reconstituted with PAFR−/− cells. B, Epidermal sheets obtained from CD45.1 mice, reconstituted with BM from PAFR −/− mice and then irradiated with 10kJ/m2 of UVB. Epidermal sheets were stained with CD45.1 or CD45.2 and Langerin. C, Representative experiment showing the expression of CD45.1 and CD45.2 on gated I-A+ CD11c+ LCs isolated from PAFR−/− mice reconstituted with CD45.1 BM. D, Epidermal sheets obtained from PAFR−/− mice reconstituted with BM from CD45.1 mice and then irradiated with 20 kJ/m2 of UVB. E, DNFB was applied onto the ear of CD45.1+ control mice and CD45.1+ mice reconstituted with PAFR−/− BM (Group 1). One day after application, epidermal sheets were stained with Langerin. The cell density of the no treatment group was adjusted to 100%. *, Statistically significant difference (p < 0.001; n=5) from no treatment group. F, DNFB was applied onto the ear of CD45.2+ PAFR−/− mice and CD45.2+ PAFR−/− mice reconstituted with BM from CD45.1 mice (Group 2). One day later epidermal sheets were stained with Langerin. n=5. A representative experiment is shown; each experiment was performed at least twice.
Injecting TNF-α overcomes the defect in LC migration in PAFR−/− mice
Cumberbatch et al. demonstrated that injecting TNF-α into the dermis, or TNF-α produced by keratinocytes, induces LC migration (26, 28). Because others have noted that treating PAFR-expressing human epidermal cells with the c-PAF resulted in increased TNF-α mRNA and protein secretion (18), we determined if treating PAM 212 cells induces TNF-α expression. Similar to human epidermal cells, c-PAF treatment increased expression of TNF-α RNA in PAM212 cells in a time-dependent manner (Fig. 4A). Whereas a subcutaneous injection of c-PAF induced significant LC migration from the epidermis in WT mice, it did not induce LC migration in PAFR-deficient mice (Fig. 4B). In contrast, injecting recombinant TNF-α induced equivalent LC migration from the epidermis of WT mice and PAFR-deficient mice (Fig. 4C). These data indicate that PAF may be activating LC migration via a TNF-α dependent mechanism, and suggest PAF is up-stream of TNF-α.
FIGURE 4. TNF-α activates LC migration in PAFR−/− mice.
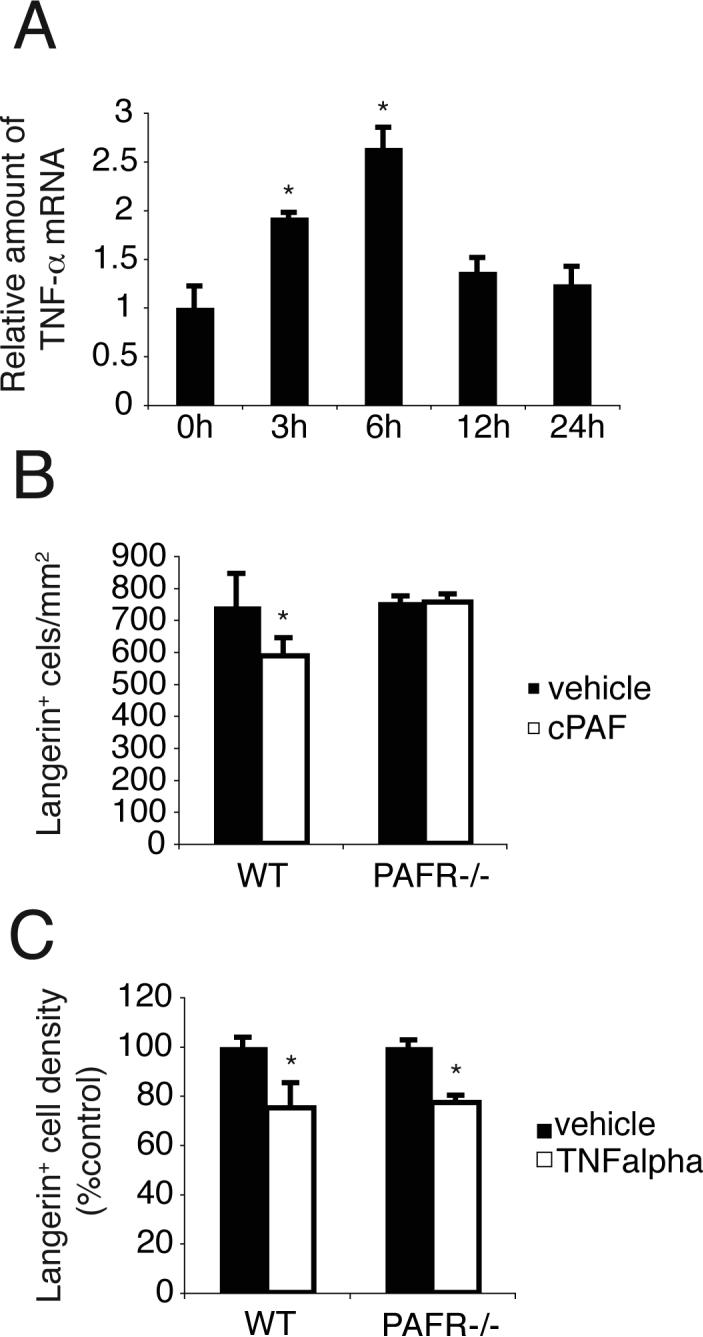
A, PAM 212 cells were treated with 1μM of c-PAF and TNF-α expression determined by real-time PCR. The levels of TNF-α mRNA were normalized to GAPDH expression. *, Statistically significant difference (p < 0.02; n=4) from non-treated (0 h) group. B, 150 pmol of c-PAF was injected into the ears of WT or PAFR −/− mice. Epidermal sheets were collected 1 day after injection and stained with Langerin. *, Statistically significant difference (p < 0.02; n=5) from vehicle treated WT mice. C, 50 ng of recombinant TNF-α was injected into the ears of WT or PAFR −/− mice. Epidermal sheets were collected 1 h after injection and stained with Langerin. *, Statistically significant difference (p < 0.001; n=5) from vehicle treated mice. A representative experiment is shown; each experiment was performed at least twice.
CHS and dDC migration in WT and PAFR-deficient mice
Although traditional evidence suggests that LCs play a critical role in induction of CHS (29), more recent studies have led to questions regarding the role of LC in CHS (30). Based on the data presented above, we tested the CHS response in PAFR-deficient mice. We did not observe any difference in the magnitude of the CHS response generated in WT or PAFR-deficient mice (Fig. 5A). These data indicate that PAF signaling is not essential for inducing CHS.
FIGURE 5. CHS and dDC migration in WT and PAFR −/− mice.

A, WT and PAFR −/− mice were sensitized by applying 50 μl of 0.5% DNFB solution on their shaved abdomens. On day 6, 10 μl of 0.2% DNFB solution was applied to both ear surfaces. On day 7, the thickness of each ear was measured. Irritation controls were not sensitized, but were challenged. *, Statistically significant difference (p < 0.001; n=5) from respective irritation control. B, FITC was applied onto the back skin of WT mice and PAFR −/− mice. Two days after FITC application, FITC+ CD11c+ Langerin− cells (dDCs) in the draining lymph nodes were counted. n=5. C, Increase in inguinal lymph node cell number was identified 2 days after FITC application in both mice (n=5). Each experiment was performed at least twice. A representative experiment is shown; each experiment was performed at least twice.
Failure of LC to migrate from the skin of PAFR-deficient mice, coupled with the observation that CHS was equal in these mice, suggests that LCs are not the relevant APC for CHS. So we decided to focus on the role of dDC in this reaction. First we measured dDC migration after hapten application. In contrast to LC migration, the migration of dDC (FITC+ CD11c+ Langerin−) into the draining lymph nodes of PAFR-deficient mice was equivalent to that found in WT mice (Fig. 5B). In addition, the increase in lymph node cell numbers found after dermal hapten application of PAFR-deficient mice was equivalent to that found in WT mice (Fig. 5C). These results indicate that PAF does not appear to play a role in dDC migration.
Lymph node dDCs but not LCs have an essential role in CD4+ T cell proliferation
Our data imply that dDC and not LC play an essential role in the generation of CHS in vivo. To directly test the ability of each cell type to present antigen, we performed the following experiment. An antibody specific for the extracellular domain of Langerin (clone 205C1) was used to sort LC from the draining lymph nodes of hapten sensitized mice. We observed that both LC (CD11c+CD8αlow/−Langerin+) and dDCs (CD11c+CD8αlow/−Langerin−) were located in the draining lymph nodes 2 days after hapten application (Fig. 6A). Moreover, we noted the dDC outnumbered the LC by a ratio of 10:1 (Fig. 6A). This physiological ratio of LCs to dDCs was used in a functional assay. CFSE-labeled OT-II CD4+ T cells and either LCs or dDCs were co-cultured with OVA peptide 323−339 for 6 days (Fig. 6B). As a positive control some T cells were stimulated with anti-CD3 + anti-CD28, which induced significant T cell proliferation. Using physiological conditions, migrating dDCs induced a stronger proliferative response than migrating LCs (Fig. 6B; right two panels). Even when the number of dDC was reduced to one-half that seen in physiological conditions, they induced stronger T cell proliferation that LCs (Fig. 6C). We also asked if the LC could suppress the stimulation of T cells by dDC in a mixing experiment. Co-culture of dDCs and LCs at physiological concentration did not affect T cell proliferation as the response of this group was similar to the response found when an equal number of dDC was used (Fig. 6C). To confirm these observations, we repeated these experiments using the classical technique of 3H-thymidine incorporation to measure T cell proliferation.. Note that when non-physiological concentrations of dDC and LC were used, and the cells were added to the wells in equal numbers, dDC presented antigen whereas the by LC did not (Fig 6D). These data indicate that dDCs but not LCs activates T cell proliferation.
FIGURE 6. dDCs but not LCs present Ag to T cells.

A, Two days after hapten application, CD11c+ CD8αlow/− Langerin+ (skin-derived LCs) and CD11c+ CD8αlow/− Langerin− (skin-derived dDCs) were isolated by cell sorting. In the Langerin stained panel, white histograms show the expression of Langerin on CD11c+ CD8αlow/− population and gray histogram indicates isotype control. B, CD4+ T cells were isolated from OT-II mice and were labeled with CFSE. CD4+ T cells and DC were co-cultured with OVA peptide for 4 days and proliferation measured by flow cytometry. Representative data indicating the intensity of CFSE on CD4+ T cells are shown. C, Division index was calculated using the proliferation platform of FlowJo software. Mean values and SDs of triplicates are shown. Arrows indicate physiological conditions (i.e., ratio of dDC to LC found in lymph node). *, Statistically significant difference (p < 0.001) from T cells alone. **, Statistically significant difference (p < 0.002) from LC 2x103 group. ***, Statistically significant difference (p < 0.001) from LC 1x103 and 2x103 group. D, Equal numbers of dDC and LC were cultured with OT-II T cells and proliferation was measured by 3H-Thymidine incorporation. Mean values and SDs of triplicates are shown. *, Statistically significant difference (p < 0.0001) from T cells alone. **, Statistically significant difference (p < 0.005) from LC 1x103 group. ***, Statistically significant difference (p < 0.0002) from LC 1x104 group. A representative experiment is shown; each experiment was performed at least twice.
Discussion
Conventional wisdom suggests that upon encountering antigen in the skin, epidermal LCs carry that antigen to the draining lymph nodes where they initiate an immune reaction. Generally, the signal that induces LC to migrate from the skin to the lymph node is physiological stress. Because stress induces keratinocytes to release PAF (31), a lipid mediator of inflammation that plays a role in leukocyte migration (32), we wanted to determine whether PAF contributes to LC migration in vivo. We observed the following: First, we found that in PAF-deficient mice, hapten-induced LC migration was significantly impaired. Second, we observed that hapten-induced LC migration from the epidermis was blocked by pretreatment with selective PAFR antagonists. Third, subcutaneous local injection of c-PAF, a structural analog of PAF, induced LC migration from the epidermis. Finally, studies with chimeric mice indicate that PAF receptor on the keratinocytes, and not the LC is more important for the induction of migration. These results suggest that PAF is involved in hapten-induced LC migration. On the other hand, we noted no difference in the magnitude of the CHS reaction generated in PAFR-deficient and WT mice. This last finding suggested that LC are not involved in initiating an immune reaction. Using sorted LC and dDC from the lymph nodes of hapten-sensitized mice, we observed that only the dDC and not the LC, could present antigen to CD4+ T cells.
CHS is a classic delayed-type hypersensitivity reaction to topically applied haptens. Conventional wisdom concerning LC function would predict that the defect in LC migration we noted in PAFR-deficient mice would result in a diminished CHS response (7, 33). We observed, however, no difference in the magnitude of the CHS response generated in WT or PAFR-deficient mice. This observation is consistent with data reported by Kissenpfennig et al. that the absence of LCs at the time of sensitization does not affect CHS in genetically engineered LC knockout mice (9). Also our data is consistent with the conclusion arrived at by others (7-9) that the dDC play a critical role in initiating CHS and migrate to DLNs in greater numbers and more quickly in response to hapten application when compared to LC. Our findings extent on these observations in two important ways. First, we used FACS to isolate LC and dDC from the lymph nodes of hapten-sensitized mice and asked directly whether they could present antigen to T cells. As shown in Figure 6, only the dDC and not the LC could present antigen. Second, we used un-manipulated WT mice. Although gene knockout mice are the most rigorous way to measure the function of any particular gene, it must be kept in mind, as pointed out by Olson and colleagues nearly 10 years ago that, “such manipulations (necessary to create a knockout) may disrupt the expression of other genes located near the intended target and therefore confound the interpretation of phenotypes” (34). This may contribute to the fact that mice genetically engineered to be LC deficient show diminished (7), or enhanced (8), or an unchanged (9) CHS reaction. Our findings confirm that the dDC and not the LC is the primary antigen-presenting cell in CHS. Finally Kaplan et al. proposed that LCs served to regulate the CHS response (8). In our co-mixing experiment, we could not find any evidence that LC regulate the function of dDC. However, we may not have used the proper conditions to activate immune modulatory LC. For example, one well-known way to activate LC to become immune regulatory is through the use of UV radiation (35). In this study we simply used UV as a tool to remove the LC from the skin and then allow reconstitution of the skin 9 weeks later by donor-derived LC. We did not ask if LC found in the lymph nodes of UV-irradiated mice 2 to 3 days after UV induce tolerance. Studies are currently in progress, using sorted lymph node LC to address this question.
Our findings also illustrate the importance of PAFR on keratinocytes in LC migration. The literature indicates that both keratinocytes and dendritic cells express functional PAFR (27). Although our initial findings (Fig. 1) indicated the importance of PAFR signaling in LC migration, we did not know whether PAFR on either LCs or keratinocytes, or both, was more important. To test this, we produced LC chimeric mice whose keratinocytes were PAFR−/− or whose LCs were PAFR−/−. In these mice, we observed that PAFR on keratinocytes, but not LCs, plays an important role in hapten induced LC migration. We assume that cytokines from keratinocytes affect LC migration. This is supported by the fact that PAF is a known regulator of transcription, and has been shown to up-regulate the secretion of a variety of cytokines (32), including TNF-α (Fig. 4). These results imply that PAF produced by keratinocytes after hapten application binds to keratinocytes in an autocrine fashion, and activates them to produce TNF-α, which then induces LC migration.
In summary, our results reveal that PAF is involved in LC migration. However because PAFR−/− mice generate a CHS reaction no different from controls, our findings support the hypothesis that LC are not involved in activating CHS. By demonstrating that dDC, but not LC, sorted from the lymph nodes of hapten sensitized mice, can present antigen to T cells we confirm the critical, and not well appreciated role for these cells in skin immunity. These findings suggest that further studies into the role of dDC in other cutaneous immune diseases such as atopic dermatitis, psoriasis, and UV-induced carcinogenesis (37) have merit.
Acknowledgments
We thank Nasser Kazimi for assistance with the animal experiments, Karen Ramirez and David Z. He for help with sorting and flow cytometry, and Tomohide Yamazaki for help with measuring T cell proliferation. We also thank Prof. Shimizu and Dr. Ishii for providing PAFR-deficient mice.
Footnotes
This work was supported by grants from the National Cancer Institute (CA75575, CA88943 & CA112660). SNB was supported by a C. J. Martin Fellowship (# 307726) from the National Health & Medical Research Council of Australia. The animal, histology, and flow cytometry facilities at the MDACC are supporting in part by a core grant from the NCI (CA16672).
Abbreviations used in this paper: DC, Dendritic cell; LC, Langerhans cell; CHS, contact hypersensitivity; dDC, dermal dendritic cell; PAF, platelet-activating factor; PAFR, PAF receptor; DNFB, 2,4-dinitro-1-fluorobenzene; c-PAF, carbamyl-PAF; WT, wild type; BM, bone marrow; OX, 4-Ethoxymethylene-2-phenyl-2-oxazoline-5-one
“This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.”
Disclosures: The authors declare no conflict of interests.
References
- 1.Wolff K. The fine structure of the Langerhans cell granule. J Cell Biol. 1967;35:468–473. doi: 10.1083/jcb.35.2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, Duvert-Frances V, Vincent C, Schmitt D, Davoust J, Caux C, Lebecque S, Saeland S. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12:71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- 3.Kripke ML, Munn CG, Jeevan A, Tang JM, Bucana C. Evidence that cutaneous antigen-presenting cells migrate to regional lymph nodes during contact sensitization. J Immunol. 1990;145:2833–2838. [PubMed] [Google Scholar]
- 4.Cumberbatch M, Gould SJ, Peters SW, Kimber I. MHC class II expression by Langerhans' cells and lymph node dendritic cells: possible evidence for maturation of Langerhans' cells following contact sensitization. Immunology. 1991;74:414–419. [PMC free article] [PubMed] [Google Scholar]
- 5.Moodycliffe AM, Kimber I, Norval M. The effect of ultraviolet B irradiation and urocanic acid isomers on dendritic cell migration. Immunology. 1992;77:394–399. [PMC free article] [PubMed] [Google Scholar]
- 6.Fukunaga A, Nagai H, Noguchi T, Okazawa H, Matozaki T, Yu X, Lagenaur CF, Honma N, Ichihashi M, Kasuga M, Nishigori C, Horikawa T. Src homology 2 domain-containing protein tyrosine phosphatase substrate 1 regulates the migration of Langerhans cells from the epidermis to draining lymph nodes. J Immunol. 2004;172:4091–4099. doi: 10.4049/jimmunol.172.7.4091. [DOI] [PubMed] [Google Scholar]
- 7.Bennett CL, van Rijn E, Jung S, Inaba K, Steinman RM, Kapsenberg ML, Clausen BE. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J Cell Biol. 2005;169:569–576. doi: 10.1083/jcb.200501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23:611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 10.Stoecklinger A, Grieshuber I, Scheiblhofer S, Weiss R, Ritter U, Kissenpfennig A, Malissen B, Romani N, Koch F, Ferreira F, Thalhamer J, Hammerl P. Epidermal langerhans cells are dispensable for humoral and cell-mediated immunity elicited by gene gun immunization. J Immunol. 2007;179:886–893. doi: 10.4049/jimmunol.179.2.886. [DOI] [PubMed] [Google Scholar]
- 11.Lenz A, Heine M, Schuler G, Romani N. Human and murine dermis contain dendritic cells. Isolation by means of a novel method and phenotypical and functional characterization. J Clin Invest. 1993;92:2587–2596. doi: 10.1172/JCI116873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishii S, Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res. 2000;39:41–82. doi: 10.1016/s0163-7827(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 13.Walterscheid JP, Ullrich SE, Nghiem DX. Platelet-activating factor, a molecular sensor for cellular damage, activates systemic immune suppression. J Exp Med. 2002;195:171–179. doi: 10.1084/jem.20011450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Mousdicas N, Yi Q, Al-Hassani M, Billings SD, Perkins SM, Howard KM, Ishii S, Shimizu T, Travers JB. Staphylococcal lipoteichoic acid inhibits delayed-type hypersensitivity reactions via the platelet-activating factor receptor. J Clin Invest. 2005;115:2855–2861. doi: 10.1172/JCI25429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolf P, Nghiem DX, Walterscheid JP, Byrne S, Matsumura Y, Matsumura Y, Bucana C, Ananthaswamy HN, Ullrich SE. Platelet-activating factor is crucial in psoralen and ultraviolet A-induced immune suppression, inflammation, and apoptosis. Am J Pathol. 2006;169:795–805. doi: 10.2353/ajpath.2006.060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramos G, Kazimi N, Nghiem DX, Walterscheid JP, Ullrich SE. Platelet activating factor receptor binding plays a critical role in jet fuel-induced immune suppression. Toxicol Appl Pharmacol. 2004;195:331–338. doi: 10.1016/j.taap.2003.07.014. [DOI] [PubMed] [Google Scholar]
- 17.Pei Y, Barber LA, Murphy RC, Johnson CA, Kelley SW, Dy LC, Fertel RH, Nguyen TM, Williams DA, Travers JB. Activation of the epidermal platelet-activating factor receptor results in cytokine and cyclooxygenase-2 biosynthesis. J Immunol. 1998;161:1954–1961. [PubMed] [Google Scholar]
- 18.Dy LC, Pei Y, Travers JB. Augmentation of ultraviolet B radiation-induced tumor necrosis factor production by the epidermal platelet-activating factor receptor. J Biol Chem. 1999;274:26917–26921. doi: 10.1074/jbc.274.38.26917. [DOI] [PubMed] [Google Scholar]
- 19.Lou YP, Takeyama K, Grattan KM, Lausier JA, Ueki IF, Agusti C, Nadel JA. Platelet-activating factor induces goblet cell hyperplasia and mucin gene expression in airways. Am J Respir Crit Care Med. 1998;157:1927–1934. doi: 10.1164/ajrccm.157.6.9709113. [DOI] [PubMed] [Google Scholar]
- 20.Ishii S, Kuwaki T, Nagase T, Maki K, Tashiro F, Sunaga S, Cao WH, Kume K, Fukuchi Y, Ikuta K, Miyazaki J, Kumada M, Shimizu T. Impaired anaphylactic responses with intact sensitivity to endotoxin in mice lacking a platelet-activating factor receptor. J Exp Med. 1998;187:1779–1788. doi: 10.1084/jem.187.11.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Douillard P, Stoitzner P, Tripp CH, Clair-Moninot V, Ait-Yahia S, McLellan AD, Eggert A, Romani N, Saeland S. Mouse lymphoid tissue contains distinct subsets of langerin/CD207 dendritic cells, only one of which represents epidermal-derived Langerhans cells. J Invest Dermatol. 2005;125:983–994. doi: 10.1111/j.0022-202X.2005.23951.x. [DOI] [PubMed] [Google Scholar]
- 22.Fukunaga A, Nagai H, Yu X, Oniki S, Okazawa H, Motegi S, Suzuki R, Honma N, Matozaki T, Nishigori C, Horikawa T. Src homology 2 domain-containing protein tyrosine phosphatase substrate 1 regulates the induction of Langerhans cell maturation. Eur J Immunol. 2006;36:3216–3226. doi: 10.1002/eji.200635864. [DOI] [PubMed] [Google Scholar]
- 23.Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, Weissman IL, Cyster JG, Engleman EG. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002;3:1135–1141. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Fukunaga A, Nagai H, Oniki S, Honma N, Ichihashi M, Matozaki T, Nishigori C, Horikawa T. Engagement of CD47 inhibits the contact hypersensitivity response via the suppression of motility and B7 expression by Langerhans cells. J Invest Dermatol. 2006;126:797–807. doi: 10.1038/sj.jid.5700176. [DOI] [PubMed] [Google Scholar]
- 25.Moodycliffe AM, Nghiem D, Clydesdale G, Ullrich SE. Immune suppression and skin cancer development: regulation by NKT cells. Nat Immunol. 2000;1:521–525. doi: 10.1038/82782. [DOI] [PubMed] [Google Scholar]
- 26.Griffiths CE, Dearman RJ, Cumberbatch M, Kimber I. Cytokines and Langerhans cell mobilisation in mouse and man. Cytokine. 2005;32:67–70. doi: 10.1016/j.cyto.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Sozzani S, Longoni D, Bonecchi R, Luini W, Bersani L, D'Amico G, Borsatti A, Bussolino F, Allavena P, Mantovani A. Human monocyte-derived and CD34+ cell-derived dendritic cells express functional receptors for platelet activating factor. FEBS Lett. 1997;418:98–100. doi: 10.1016/s0014-5793(97)01358-6. [DOI] [PubMed] [Google Scholar]
- 28.Cumberbatch M, Kimber I. Dermal tumour necrosis factor-alpha induces dendritic cell migration to draining lymph nodes, and possibly provides one stimulus for Langerhans' cell migration. Immunology. 1992;75:257–263. [PMC free article] [PubMed] [Google Scholar]
- 29.Sullivan S, Bergstresser PR, Tigelaar RE, Streilein JW. Induction and regulation of contact hypersensitivity by resident, bone marrow-derived, dendritic epidermal cells: Langerhans cells and Thy-1+ epidermal cells. J Immunol. 1986;137:2460–2467. [PubMed] [Google Scholar]
- 30.Bacci S, Alard P, Dai R, Nakamura T, Streilein JW. High and low doses of haptens dictate whether dermal or epidermal antigen-presenting cells promote contact hypersensitivity. Eur J Immunol. 1997;27:442–448. doi: 10.1002/eji.1830270214. [DOI] [PubMed] [Google Scholar]
- 31.Barber LA, Spandau DF, Rathman SC, Murphy RC, Johnson CA, Kelley SW, Hurwitz SA, Travers JB. Expression of the platelet-activating factor receptor results in enhanced ultraviolet B radiation-induced apoptosis in a human epidermal cell line. J Biol Chem. 1998;273:18891–18897. doi: 10.1074/jbc.273.30.18891. [DOI] [PubMed] [Google Scholar]
- 32.Ishii S, Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog Lipid Res. 2000;39:41–82. doi: 10.1016/s0163-7827(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 33.Silberberg-Sinakin I, Thorbecke GJ. Contact hypersensitivity and Langerhans cells. J Invest Dermatol. 1980;75:61–67. doi: 10.1111/1523-1747.ep12521144. [DOI] [PubMed] [Google Scholar]
- 34.Olson EN, Arnold HH, Rigby PW, Wold BJ. Know your neighbors: three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell. 1996;85:1–4. doi: 10.1016/s0092-8674(00)81073-9. [DOI] [PubMed] [Google Scholar]
- 35.Ullrich SE. Mechanisms underlying UV-induced immune suppression. Mutat Res. 2005;571:185–205. doi: 10.1016/j.mrfmmm.2004.06.059. [DOI] [PubMed] [Google Scholar]
- 36.Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity. 2003;19:47–57. doi: 10.1016/s1074-7613(03)00175-4. [DOI] [PubMed] [Google Scholar]
- 37.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Whynot J, Novitskaya I, Cardinale I, Haider A, Khatcherian A, Carucci JA, Bergman R, Krueger JG. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J Allergy Clin Immunol. 2007;119:1210–1217. doi: 10.1016/j.jaci.2007.03.006. [DOI] [PubMed] [Google Scholar]