

Abstract
The lipid bilayer of the myelin membrane of the central nervous system (CNS) and the peripheral nervous system (PNS) contains the oligodendrocyte- and Schwann cell-specific glycosphingolipids galactocerebrosides (GalC) and GalC-derived sulfatides (sGalC). We have generated a UDP-galactose ceramide galactosyltransferase (CGT) null mutant mouse (cgt−/−) with CNS and PNS myelin completely depleted of GalC and derived sGalC. Oligodendrocytes and Schwann cells are unable to restore the structure and function of these galactosphingolipids to maintain the insulator function of the membrane bilayer. The velocity of nerve conduction of homozygous cgt−/− mice is reduced to that of unmyelinated axons. This indicates a severely altered ion permeability of the lipid bilayer. GalC and sGalC are essential for the unperturbed lipid bilayer of the myelin membrane of CNS and PNS. The severe dysmyelinosis leads to death of the cgt−/− mouse at the end of the myelination period.
Keywords: ceramide galactosyl transferase, cgt null allele, conduction velocity, dysmyelinosis
Axons in the central nervous system (CNS) and the peripheral nervous system (PNS) are ensheathed by oligodendrocyte and Schwann cell plasma membrane processes which assemble the multilayered myelin membrane. The lipid bilayer lends myelin the insulator function in the internodes of axons allowing saltatory conduction. It contains the myelin-specific glycosphingolipids galactocerebrosides (GalCs) and GalC-derived sulfatides (sGalCs). GalC is synthesized by UDP-galactose ceramide galactosyltransferase (CGT). We have studied the function of GalC and sGalC in a CGT-deficient mouse line completely lacking GalC and sGalC in CNS and PNS myelin lipid bilayer. The conduction velocity of myelinated axons of cgt−/− mice is reduced to that of unmyelinated axons indicating a block of saltatory conduction. The impaired insulator function of myelin due to the lack of the main glycolipids GalC and sGalC is not compensated by other lipids. The dysmyelinosis leads to body tremor, loss of locomotor activity, and to death at the end of myelination.
CGT has recently been isolated from rat brain and characterized (1, 2). The coding sequence of human cgt on chromosome 4q26 as well as mouse cgt is distributed over five exons (3, 4). Oligodendrocytes of CNS, Schwann cells in PNS, and, very weakly, kidney cells show a peak expression of the CGT gene during the myelination period between p10 and p30 (1). GalC serves as an early epitope in oligodendrocyte lineage studies (5–9).
Beyond structural functions complex sphingolipids have been proposed as precursors of second messenger systems in cell signaling and in the differentiation of oligodendrocytes (ref. 10; for a review, see ref. 11).
MATERIALS AND METHODS
cgt−/− Replacement Vector.
A 7-kb SacI fragment with exon I flanked by 3-kb upstream and 3-kb downstream sequences of the recently described mouse CGT gene (3) was subcloned into pBluescript and used for preparation of the targeting construct. The blunt end 1.2-kb neo gene (12) was inserted into the unique KpnI site of exon I at position 72 of the open reading frame in sense direction of the CGT gene fragment. The tk gene was ligated as an XhoI/BamHI fragment into the NotI site of the pBluescript multiple cloning site located at the 5′ end of the CGT gene fragment. The construct was linearized with SpeI in the multiple cloning site of pBluescript, Fig. 1A.
Figure 1.
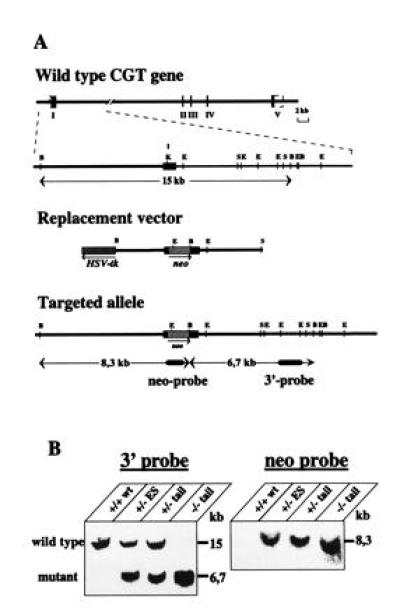
Disruption of the cgt locus in mouse ES cells and generation of CGT-deficient mice. (A) Targeting strategy: the murine cgt-coding exon I is indicated by a closed box. The neor cassette inserted into the KpnI site of exon I and the HSV-tk gene (▨) has been placed upstream of the exon I. B, BamHI; E, EcoRI; S, SacI; K, KpnI. Homologous recombination introduces an additional BamHI site through the pgk neo cassette. (B) Southern blot hybridization analysis of BamHI-digested ES cells and tail DNA from offsprings of cgt+/− intercrosses. neo probe is a 1.1-kb fragment; 3′-probe a 1.1-kb EcoRI fragment (3) coresponding to a genomic sequence downstream of the replacement sequence.
Generation of cgt−/− Mouse Line.
A total of 107 R1 cells [(129/j × 129sv-cp)F1 background] (kindly provided by A. Nagly, Mount Sinai Hospital, Samuel Lunenfeld Research Institute, Toronto) were transfected by electroporation with 20 μg of the linearized targeting vector at 250 V and 500 μF using a Bio-Rad gene pulser. Homologous recombinant clones were selected by adding G418 (300 μg/ml) and gancyclovir (0.2 mM) to the medium. C57BL/6 blastocysts were injected with five R1-independent embryonic stem (ES) cell lines (13). Chimeric males were intercrossed with C57BL/6 mice. Heterozygous offsprings were intercrossed and homozygous cgt−/− mice were obtained in the F2 generation. ES cell clones and tail DNA were prepared according to Laird (14). C57BL/6 wild-type (wt) mice served as control animals in all comparative experiments.
Analysis of RNA Expression.
RNA was isolated by the guanidinium thiocyanate-phenol method (15). 25 μg total RNA of brain, kidney, liver, spleen and total RNA of eight sciatic nerves from 18- to 20-day-old wt, cgt+/−, and cgt−/− was fractionated by electrophoresis on formaldehyde (1.5%) agarose gels, blotted onto nitrocellulose (GeneScreen, DuPont), and hybridized with randomly 32P-labeled probes. cDNA probes included a 1180-bp EcoRI/NcoI rat CGT cDNA fragment (3), a 640-bp PstI rat fragment of myelin basic protein (MBP) cDNA encoding the 14.5 kDa isoform of MBP, a 1362-bp PstI fragment of human PLP cDNA, a 206-bp myelin-associated glycoprotein (MAG) genomic DNA fragment of mouse exon IX, a 356-bp DNA fragment of exon I of mouse oligodendrocyte myelin glycoprotein (OMgp), and a 500-bp mouse cDNA fragment of P0. Glyceraldehyde-phosphate dehydrogenase cDNA was used as internal standard. Signals were recorded with a PhosphorImager (Molecular Dynamics).
For in situ hybridization, 35S-labeled RNA probes were prepared by in vitro transcription of the 3.5-kb CGT cDNA in pGEM3Z using SP6 (BRL) or T7 RNA polymerase (Boehringer Mannheim) and [35S]UTP. CGT antisense RNA and a sense probe as control were hybridized to 6–8 mm cryosections of brains of p20-24 mice that had previously been perfused in 4% paraformaldehyde in PBS and further processed as described (16). Bright field image was ×5.6.
CGT Enzyme Assay.
CGT activity was determined as described (17) with synthetic d-2-hydroxyhexanoylsphingosine and UDP[14C]galactose (Amersham) as substrates and microsomal protein of brain, liver and kidney from p18 wt and cgt−/− mice.
Lipid Analysis.
Total lipid extracts of purified CNS myelin, sciatic nerve, liver, kidney, and spleen (1) of wt, hetero-, and homozygous CGT-deficient mice were homogenized in 10 volumes of chloroform-methanol (2:1 vol/vol) with an ultraturrax and centrifuged. The pellet was reextracted twice. The combined supernatants were taken to dryness under a stream of nitrogen. Lipids were dissolved in chloroform-methanol (2:1) to a final concentration of 20 mg/ml. Unsaponifiable sphingolipids were obtained by alkaline hydrolysis of total lipids. Aliquots were saponified in 1 ml of 0.5 N methanolic KOH at 37°C for 5 h. Water (1 ml) was added and the mixture extracted three times with chloroform. Phases were separated by centrifugation. The combined chloroform extracts were evaporated to dryness. Total and alkali-stabile lipids were separated on high-performance thin-layer chromatography plates using the solvent system chloroform/methanol/water, 65:25:4. Lipid bands were visualized by charring for phospholipids (18), glycolipids (19), sulfatides (20), and by charring for all lipid constituents with 50% H2SO4 at 120°C for 15 min. GalC and sGalC bands of wt and cgt−/− mice were separated by high-performance thin-layer chromatography, visualized by spraying with water, and scraped from plates. Individual lipids were extracted twice with chloroform/methanol (2:1), and aliquots were subjected to fast atom bombardment mass spectrometry and gas liquid chromatography.
Conduction Velocity Measurement.
Nerve conduction was measured by proximal stimulation of the sciatic nerve at the knee and distal close to the hip, using a Medelec electromyograph, model MS92. Gait pattern was recorded as described (21, 22).
Electron Microscopy.
Twenty-four-day-old mice were anesthetized with Nembutal and perfused with 3% glutaraldehyde and 2% paraformaldehyde in PBS via the left cardiac ventricle. Cervical segments of the spinal cord, optic, and sciatic nerve were obtained, postfixed in 1% phosphate-buffered OsO4 in 0.1 M sucrose, and embedded in Epon 812. Ultrathin cross sections of spinal cord, optic, and sciatic nerve were contrasted with uranylacetate and lead citrate and examined as described (23).
RESULTS
We report here the generation of a CGT-deficient (cgt−/−) mouse line by gene targeting (Fig. 1). Oligodendrocyte- and Schwann cell-specific gene expression relevant for myelination of axons was studied by Northern blot hybridization analyis (Fig. 2A). Expression of cgt was completely eliminated in the homozygous mutant mouse. No enhanced expression of any mRNA of the predominant myelin proteins MBP, proteolipid protein (PLP), MAG, and OMgp of oligodendrocytes, and MBP and P0 of Schwann cells could be observed when normalized to the endogenous glyceraldehyde-phosphate dehydrogenase signal. In situ hybridization of horizontal sections of wt and cgt−/− brains with antisense CGT and MBP RNA also proved the absence of CGT mRNA in oligodendrocytes (Fig. 2B). The number of oligodendrocytes of wt and cgt−/− mice labeled with the mbp antisense RNA probe was similar, indicating a normal development of the oligodendrocyte lineage in the cgt−/− mutant mouse. Enzymatic activity of CGT was determined by the galactosyl transferase assay (17) (Fig. 2C). cgt−/− mice showed a complete loss of CGT activity in brain giving additional prove of the presence of only one CGT gene copy responsible for galactosphingolipid synthesis.
Figure 2.
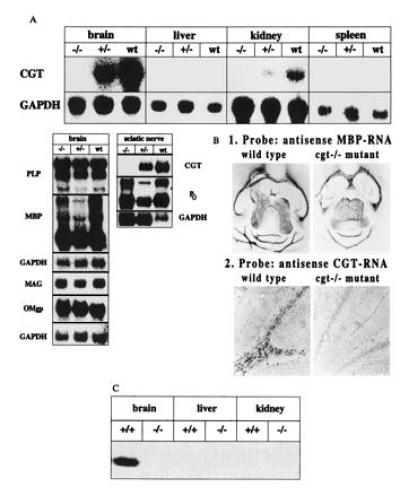
Analysis of cgt expression and relevant myelin-associated genes of oligodendrocyte by Northern blot, in situ hybridization and enzyme assay. C57BL/6 mice were used as control mice in all comparative experiments. (A) Northern blot analysis of CGT, PLP, MBP, MAG, OMgp, and P0 RNA of brain, liver, kidney, spleen, and sciatic nerve. Approximate sizes of transcripts: CGT, 3.2 kb; PLP, 1.6, 2.4 and 3.2 kb; MBP, 2.1 kb; MAG, 4 kb; OMgp, 1.8 kb; Po, 2.0 kb; glyceraldehyde-phosphate dehydrogenase, 1.2 kb. (B) Comparative in situ hybridization analysis of CGT and MBP expression in wt and homozygous cgt−/− mouse brain. (C) CGT activity of brain, liver and kidney of age matched p18-20 wt and homozygous mutant mice. Enzymatic activity was determined as described (17) with synthetic d-2-hydroxyhexanoylsphingosine and UDP[14C]galactose (Amersham) as substrates and microsomal protein of brain, liver, and kidney from p18 wt and cgt−/− mice. The reaction product, radioactive [14C]galactosyl ceramide, was detected in the lipid extract after separation by TLC as described and recorded with the phosphoimager. Only brain microsomes of wt mouse brain showed significant enzymatic activity, that of kidney was to weak to be detected by autoradiography.
Oligodendrocytes and Schwann cells of the cgt−/− null mutant mouse produce a myelin membrane, the lipid bilayer of which is completely deficient in GalC and sGalC (Fig. 3). Total lipid extract and unsaponifiable lipids were separated and visualized with specific reagents. Two new glycolipid bands with Rf values intermediate between and below normal fatty acyl (nGalC) and hydroxy fatty acyl (hGal) and nsGalC, and hsGalC, respectively (see Fig. 3, arrows 1 and 2), occurred in the myelin lipid pattern of cgt−/− CNS and PNS. Mass spectroscopy identified the unknown lipids as hexosyl-substituted with n-saturated and α-hydroxylated very long chain fatty acid (18–26 C-atoms). Their sugar moiety was identified as a glucosyl derivative after acid hydrolysis, reduction and peracetylation as glucitol hexaacetate by GLC (not shown). Fatty acid analysis of these glucocerebrosides of cgt−/− mice proved that the ceramide intermediate, which is normally the precursor of the GalC synthesis in wt mouse, was utilized by a glucosyl transferase. These ceramide species were also utilized for sphingomyelin synthesis. The sphingomyelin band with lower Rf-value corresponds to long chain α-hydroxy fatty acid containing sphingomyelin (Fig. 3, arrow 3).
Figure 3.
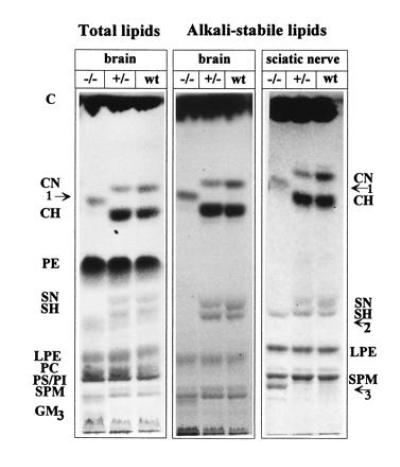
Analysis of myelin lipids of brain and sciatic nerve of wt, hetero-, and homozygous cgt−/− mice (24-day-old). Brain, total and alkali-stable lipids; sciatic nerve, alkali-stable lipids; C, cholesterol; CN, normal fatty acid substituted GalC; CH, α-hydroxy fatty acid substituted GalC; PE, phosphatidylethanolamine; SN, normal fatty acid substituted sGalC; SH, α-hydroxy fatty acid substituted sGalC; LPE, lysophosphatidylethanolamine (lysoplasmalogens); PC, phosphatidylcholine; GM3, ganglioside GM3. Arrows: 1, glucocerebroside; 2, sulfated glucocerebroside; and 3, sphingomyelin-containing long chain α-hydroxy fatty acids.
Schwann cells also transfer a sulfate group to the newly formed glucocerebroside. These glucocerebrosides and their sulfate esters are unable to compensate the loss of galactosphingolipids. Although the ceramide component specific for GalC is utilized for the reduced synthesis of glucocerebrosides, the hydrophobic core of the bilayer and particularly the carbohydrate polar head group and charge pattern of the myelin membrane surfaces of the cgt−/− mouse remain seriously altered.
The complete loss of CGT activity in brain of myelinating homozygous cgt−/− mice gives additional prove of the presence of only one single ceramide galactosyl transferase gene copy responsible for oligodendrocyte-, Schwann cell-, and kidney-specific galactosphingolipid synthesis. The in vivo experiments indicate furthermore that cerebrosides and sulfatides are not primarily responsible and essential for the differentiation of the oligodendrocyte precursor cell.
The growth of the homozygous cgt−/− mice is retarded as shown in the comparison of wt and homozygous sibling (Fig. 4). Most of the homozygous mice have died by the end of the myelination period ≈p25-p30.
Figure 4.
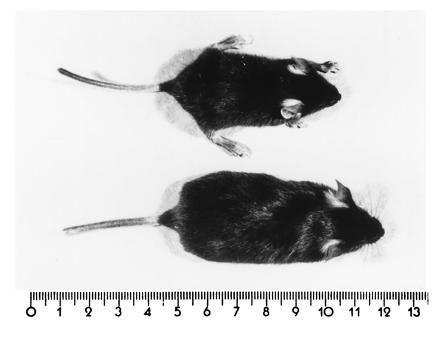
Age-matched (p24) wt (Lower) and cgt−/− (Upper) mice showing the growth retardation of cgt−/− upper mouse. (Scale in cm.)
Electromyographic measurement of the conduction velocity of the sciatic nerve of age-matched wt and cgt−/− mice indicated that the velocity observed had decreased from 22–25 m/s in wt C57BL/6 to 4–8 m/s in cgt−/− mice (n = 6, 18–20 days old) which is that of unmyelinated axons (24) (Fig. 5A).
Figure 5.
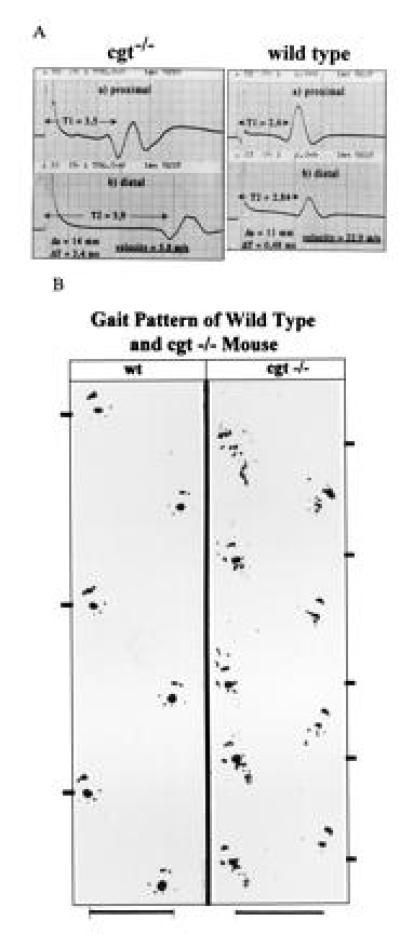
(A) Nerve conduction velocity of cgt−/− is decreased from 22–25 m/s to 4–8 m/s when p18-20 mice were compared. (B) Gait pattern of wt and cgt−/− mouse.
A whole body tremor of cgt−/− mice is observed after day 10–12 accompanied by increasing loss of locomotor activity and a very conspicuous gait pattern, as documented by the hind foot pattern (25), Fig. 5B. In the open field test the homozygous cgt−/− mutant mouse showed only minute activity. Furthermore, homozygotes were unable to perform the horizontal wire test (22) due to an increasing weakness of their front and hind legs, which were severely paralyzed around day 21. Heterozygous mice showed no neurological symptoms.
In electron microscopy the myelin sheath of CNS (optic nerve) and PNS (sciatic nerve) axons showed a regular periodicity of the main and intermediate dense lines in wt and cgt−/− mice (Fig. 6 A–F). This is surprising in view of the striking phenotype of the cgt−/− mutant comparable with the dysmyelinosis with severe hypomyelination seen in shiverer, jimpy mouse, or myelin-deficient rat.
Figure 6.
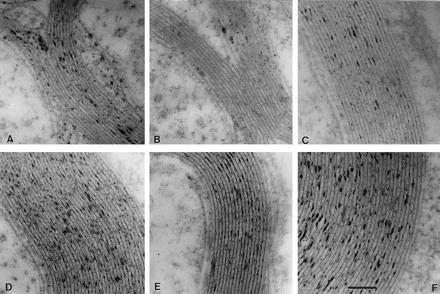
Electron microscopy of cross sections of the optic and sciatic nerve of p24 wt, hetero- and homozygous mutant mice. (A–C) Optic nerve. Cross section of optic nerve of A, cgt−/−; B, cgt+/−; C, wt. (D–F) Sciatic nerve. Cross section of sciatic nerve of D, cgt−/−; E, cgt+/−; F, wt. (Bar = 1.0 mm.)
These results indicate that although ultrastructurally the myelin membrane system of CNS and PNS is inconspicuous, the lipid bilayer structure on the molecular level must be severely altered by the lack of the membrane-stabilizing galactocerebrosides and sulfatides.
DISCUSSION
Pivotal for the insulator function of the myelin membrane structure of CNS and PNS and saltatory conduction of axons is the unperturbed lipid bilayer structure. Galactolipids GalC and sGalC contribute to the structure and stability of the myelin lipid bilayer on three levels. (i) The all-trans configuration of the long chain (C18–C26), mostly saturated and α-hydroxylated fatty acyl chains together with the alkane chain of the long chain sphingosine base contribute to the tight packing of the hydrophobic core domain. A high concentration of cholesterol and highly unsaturated ethanolamine plasmalogen adjusts its fluidity. (ii) The polar head groups of GalC and sGalC contribute significantly to the inner and outer membrane surface. sGalC together with the polar head groups of phosphatidylserine and -inositol (phosphates) provide a polyanionic surface array. Strong interactions with both the positively charged MBP at the cytosolic and hydrophobic domains of PLP at the extracytoplasmic surface might contribute to the tight compaction of the multilayer membrane system. (iii) At the interphase of the membrane the abundant galactosphingolipids contribute to a dense network of H-bonding between three β-hydroxy groups of cholesterol, the 3R hydroxy group of the sphingosine moiety of sphingolipid, the α-D-hydroxy groups of the acyl chains and the amide bond of the sphingolipids. It is likely though not proven that these interactions are concentrated at the extracytoplasmic surface of the myelin lipid bilayer. The cooperativity of these structures renders the lipid bilayer of myelin impermeable for ions, a prerequisite of its insulation properties.
In the null mutant cgt−/− mouse lack of GalC and sGalC in the lipid bilayer of myelin leaves cholesterol and highly unsaturated phospholipids as main constituents for the assembly of the lipid bilayer. Consequently the phase transition temperature of the lipid bilayer should be considerably lowered and membrane fluidity and permeability increased. The dramatically altered electrophysiology due to the structural changes in the lipid bilayer of CNS and PNS myelin membrane is apparent in the loss of insulating properties of the internodes and blocked saltatory conduction. The velocity of nerve conduction of the sciatic nerve of 24-day-old wt mice (n = 6) of 25–28 m/s was reduced in age matched homozygous cgt−/− mice to 4–8 m/s, comparable with that of unmyelinated axons (24) For nonmyelinated axons (1.1 μm diameter) originating from dorsal root ganglia a conduction velocity of 2.3 m/s has been measured. The saltatory conduction of homozygous cgt−/− mice has turned into the propagation of the action potential by continuous depolarization of the axon, due to the complete loss of the insulating properties of the multilayered internodal membrane system. Ectopic discharges along the axons of CNS and PNS block saltatory conduction and cause whole body tremor of the cgt−/− mouse similar to the homozygous shiverer mouse in which the myelination is fully down regulated. Rapidly developing paresis of the limbs and facial nerves monitors a demyelinating phenotype comparable with that observed in chronic peripheral neuropathies.
The structure-function relation of the main myelin proteins—the peripheral MBP, the integral PLP and MAG, synthesized by oligodendrocytes and P0 by Schwann cells—is rapidly emerging now particularly from the natural MBP mutant shiverer 25, the null-allelic mouse mutants of PLP (plp−/−) (26), (plp−/−, mbp−/−), MAG (mag−/−) (27), and P0 (28) generated by gene targeting with homologous recombination.
The in vivo analysis of the function of the GalC and sGalC constituents of the lipid bilayer of the myelin membrane of mammalian CNS and PNS is, to our knowledge, the first example which demonstrates the great potential of gene targeting by homologous recombination in studies addressing functional aspects of lipid structures in biological membranes.
Acknowledgments
The support by Dr. A. Walsh, Laboratory of Animal Research Center, Rockefeller University, New York, during the animal experiments is gratefully acknowledged. We thank Prof. Dr. Haupt, Neurologische Universitätsklinik Köln, for the electromyographic measurements, and Dr. G. Pohlentz, Physiologisch-Chemisches Institut, Universität Bonn, for mass spectroscopic analyses. This work was generously supported by the Deutsche Forschungsgemeinschaft, Grant SFB 243, project A4, (W.S.), and by the Alexander von Humboldt Foundation through the Max Planck Research Prize (W.S.).
Footnotes
Abbreviations: CNS, central nervous system; PNS, peripheral nervous system; GalC, galactocerebroside; sGalC, GalC-derived sulfatides; CGT, ceramide galactosyltransferase; MBP, myelin basic protein; wt, wild type; MAG, myelin-associated glycoprotein; ES cell, embryonic stem cell; OMgp, oligodendrocyte myelin glycoprotein; PLP, proteolipid protein.
References
- 1.Schulte S, Stoffel W. Proc Natl Acad Sci USA. 1993;90:10265–10269. doi: 10.1073/pnas.90.21.10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulte S, Stoffel W. Eur J Biochem. 1995;233:947–953. doi: 10.1111/j.1432-1033.1995.947_3.x. [DOI] [PubMed] [Google Scholar]
- 3.Bosio A, Binczek E, Stoffel W. Genomics. 1996;35:223–226. doi: 10.1006/geno.1996.0342. [DOI] [PubMed] [Google Scholar]
- 4.Bosio A, Binczek E, Le Beau M M, Fernald A A, Stoffel W. Genomics. 1996;34:69–75. doi: 10.1006/geno.1996.0242. [DOI] [PubMed] [Google Scholar]
- 5.Gard A L, Pfeiffer S E. Neuron. 1990;5:615–625. doi: 10.1016/0896-6273(90)90216-3. [DOI] [PubMed] [Google Scholar]
- 6.Raff M, Mirsky R, Fields K, Lisak R, Dorfman S, Silberberg D, Gregson N, Leibowitz S, Kennedy M. Nature (London) 1978;274:813–816. [PubMed] [Google Scholar]
- 7.Raff M, Fields K, Hakomori S, Mirsky R, Pruss R, Winter J. Brain Res. 1979;174:383–308. doi: 10.1016/0006-8993(79)90851-5. [DOI] [PubMed] [Google Scholar]
- 8.Raff M, Abney E, Miller R. Dev Biol. 1984;106:53–60. doi: 10.1016/0012-1606(84)90060-5. [DOI] [PubMed] [Google Scholar]
- 9.Raff M, Abney E, Fok-Seang J. Cell. 1985;42:61–69. doi: 10.1016/s0092-8674(85)80101-x. [DOI] [PubMed] [Google Scholar]
- 10.Bansal R, Pfeiffer S E M. Proc Natl Acad Sci USA. 1989;86:6181–6185. doi: 10.1073/pnas.86.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyer C A. Mol Neurobiol. 1993;7:1–22. doi: 10.1007/BF02780606. [DOI] [PubMed] [Google Scholar]
- 12.Mansour S, Thomas K, Capecchi M. Nature (London) 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 13.Kuehn M R, Bradley A, Robertson E J, Evans M J. Nature (London) 1987;326:295–298. doi: 10.1038/326295a0. [DOI] [PubMed] [Google Scholar]
- 14.Laird P W, Zijderveld A, Linders K, Rudnicki M A, Jaenisch R, Berns A. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 16.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1987. [Google Scholar]
- 17.Neskovic N, Roussel G, Nussbaum J. J Neurochem. 1986;47:1412–1418. doi: 10.1111/j.1471-4159.1986.tb00773.x. [DOI] [PubMed] [Google Scholar]
- 18.Dittmer J, Lester R. J Lipid Res. 1964;5:126–127. [PubMed] [Google Scholar]
- 19.Ledeen R W, Yu R K. Methods Enzymol. 1982;83:139–191. doi: 10.1016/0076-6879(82)83012-7. [DOI] [PubMed] [Google Scholar]
- 20.Schnaar R L, Needham L K. Methods Enzymol. 1994;230:371–389. doi: 10.1016/0076-6879(94)30025-9. [DOI] [PubMed] [Google Scholar]
- 21.Xu M, Moratalla R, Gold L H, Hiroi N, Koob G F, Graybiel A M, Tonegawa S. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 22.Wallace E J, Krauter E E, Campbell B A. J Gerontol. 1980;35:364–370. doi: 10.1093/geronj/35.3.364. [DOI] [PubMed] [Google Scholar]
- 23.Büssow H. J Neurocytol. 1978;7:207–214. doi: 10.1007/BF01217919. [DOI] [PubMed] [Google Scholar]
- 24.Gasser H S. J Gen Physiol. 1950;33:651–690. doi: 10.1085/jgp.33.6.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aiba A, Kano M, Chen C, Stanton M E, Fox G D, Herrup K, Zwingman T A, Tonegawa S. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 26.Boison D, Stoffel W. Proc Natl Acad Sci USA. 1994;91:11709–11713. doi: 10.1073/pnas.91.24.11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Tropak M B, Gerlai R, Clapoff S, Abramow-Newerly W, Trapp B, Peterson A, Roder J. Nature (London) 1994;369:747–750. doi: 10.1038/369747a0. [DOI] [PubMed] [Google Scholar]
- 28.Giese K P, Martini R, Lemke G, Soriano P, Schachner M. Cell. 1992;71:565–576. doi: 10.1016/0092-8674(92)90591-y. [DOI] [PubMed] [Google Scholar]