

Abstract
In mammalian muscle a postnatal switch in functional properties of neuromuscular transmission occurs when miniature end plate currents become shorter and the conductance and Ca2+ permeability of end plate channels increases. These changes are due to replacement during early neonatal development of the γ-subunit of the fetal acetylcholine receptor (AChR) by the ɛ-subunit. The long-term functional consequences of this switch for neuromuscular transmission and motor behavior of the animal remained elusive. We report that deletion of the ɛ-subunit gene caused in homozygous mutant mice the persistence of γ-subunit gene expression in juvenile and adult animals. Neuromuscular transmission in these animals is based on fetal type AChRs present in the end plate at reduced density. Impaired neuromuscular transmission, progressive muscle weakness, and atrophy caused premature death 2 to 3 months after birth. The results demonstrate that postnatal incorporation into the end plate of ɛ-subunit containing AChRs is essential for normal development of skeletal muscle.
Keywords: synapse, homologous recombination, end plate channel, muscle development
Neuromuscular transmission is mediated by the release of quantal packets of acetylcholine (ACh) from the nerve terminal (1). Each packet generates, by the transient opening of end plate channels (2–4), a miniature end plate current (mEPC) that depolarizes the muscle fiber. Muscle action potentials are initiated when multiple quantal packets are released simultaneously. In mammalian muscle the functional properties of end plate channels change during postnatal development. The length of channel-opening bursts (5) decreases and, as a consequence, the duration of mEPCs decreases (6), whereas the conductance (7) and the Ca2+ permeability (8) of end plate channels increase. The underlying molecular mechanism is a switch in the expression of ACh receptor(AChR)-subunit genes shortly after birth. The γ-subunit gene is repressed while the ɛ-subunit gene is activated selectively in the myonuclei underlying the synapse (γ/ɛ-switch; refs. 7 and 9–14; see ref. 15 for review). To investigate the significance of the γ/ɛ-subunit switch for motor behavior, we ablated in mouse embryonic stem (ES) cells the ɛ-subunit gene (AChRɛ) by homologous recombination and injected correctly engineered cells of two independently isolated clones into C57BL/6 blastocysts. Chimeric male mice derived from both clones showed germ-line transmission of the targeted allele. Homozygous mutant animals showed that after apparently normal development in early neonatal life, neuromuscular transmission was progressively impaired. The lack of ɛ-subunits caused muscle weakness, defects in motor behavior, and premature death 2 to 3 months after birth.
MATERIALS AND METHODS
Gene Targeting.
Genomic clones of mouse ɛ-subunit were isolated from a library of strain 129 ISV (generously provided by Andrew Reaume, Mount Sinai Hospital, Toronto) and were characterized by restriction mapping and partial sequencing. The targeting vector consisted of a 5.2-kb XhoI/AatII genomic DNA fragment, the PGK-neo-cassette, and a 1.4-kb NsiI/XhoI genomic DNA fragment cloned into plasmid pBluescriptII KS (Stratagene). The NotI-linearized targeting vector was introduced into E14-1 ES cells (ref. 16; kindly provided by A. King, Universität Heidelberg) by electroporation. G418 selection (330 mg ml−1) was applied 36 h after the transfection and the neo-resistant colonies were isolated 7–9 days later. The genomic DNA of these clones was digested by BstEII and analyzed by Southern blotting. Of 400 neo-resistant clones, 6 were correctly targeted. C57BL/6 blastocytes were injected (BRL, Füllinsdorf, Switzerland) with clone 382 or 399 to generate two independent mutant mice lines. Chimeric males with white-grey-agouti color were crossed with C57BL/6 or C57BL/6xDBA/2 F1 hybrid females to yield heterozygous mice, which were analyzed by standard Southern blotting.
AChR Expression.
DNA extracted from tails was analyzed by Southern blot hybridization using a standard procedure. For RNA analysis, postnatal day (P) 5 and P34 mice were killed with CO2, and the gastrocnemius muscle was excised and immediately frozen in liquid nitrogen. Total RNA was isolated according to Chomczynski and Sacchi (17). Aliquots of 50–100 ng μl−1 total RNA were transcribed using Moloney murine leukemia virus reverse transcriptase from GIBCO/BRL. The resulting cDNA (10 μl) was used for PCR amplification of γ- and ɛ-subunit-specific fragments using Taq polymerase (Mobi-Tec, Göttingen, Germany). The PCR mix was heated for 5 min at 95°C. After 30 (ɛ-subunit fragments) or 32 (γ-subunit fragments) cycles for 45 sec at 95°C, 45 sec at 60°C, and 90 sec at 72°C, the temperature was kept for 10 min at 72°C and then cooled to 4°C. Primers for the ɛ-subunit were: forward primer, 5′-GAT GGG CAG TTT GGA GTG G and reverse primer, 5′-CAG AAA TGA GCA CGC AAG G (product 417 bp); for the γ-subunit: forward primer, 5′-GAT GCG AAA CTA CGA CCC C and reverse primer, 5′-AGG AGG AGC GGA AGA TGG (product 349 bp).
PCR products were purified by Qiagen (Chatsworth, CA) oligo purification kits and run on agarose gels. Reaction products were stained either by ethidium bromide or after transferring the PCR products on Biodyne A membranes (Pall, Portsmouth, England). DNA fragments were specifically hybridized with the fluorescein-labeled ɛ-subunit-specific probe 5′-GGA GAA TGG GCC ATA GAC TAC TGC CCA GGC ATG ATT CGC CGC TAT GAG GGA GG or the γ-subunit-specific probe 5′-GAG AGG AGG CCC TCA CAA CTA ACG TCT GGA TAG AGA TGC AAT GGT GCG A using the Enhanced Chemiluminescence detection kit from Amersham.
To detect AChRs at neuromuscular synapses, mice were killed with CO2, and their diaphragms were excised and incubated in extracellular solution (see below) containing rhodamine-labeled α-bungarotoxin at 4°C overnight. After fixation diaphragms were analyzed for rhodamine fluorescence using a Zeiss Axioskop fitted with a 40× oil immersion objective. Photographs were taken with a Zeiss MC-100 camera.
Electrophysiology.
Foot muscle (flexor digitorum brevis) was enzymatically dissociated (18). End plates were visualized with a 16× objective using differential interference contrast optics in an inverted microscope. Current records were made with patch pipettes from end plates in the cell-attached recording configuration at 20–22°C at different membrane potentials (19). Pipettes had a tip resistance of 5–10 MΩ when filled with extracellular solution consisting of 135 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM Hepes (pH 7.2). In addition, acetylcholine (10–100 nM) was added to the pipette solution. Slope conductances of single end plate channels were determined from linear regression of single channel current amplitude versus patch membrane potential. The current reversal potential was measured in each patch recording by linear interpolation between inward and outward current amplitudes or by extrapolation from inward current amplitudes. The reversal potential was assumed to correspond to 0 mV membrane potential. Slope conductances were determined between −20 mV and −100 mV membrane potential. Spontaneously occurring mEPCs were recorded in hemidiaphragms at −70 mV using a two-microelectrode voltage clamp (6). Intracellular microelectrodes were filled with 3 M KCl and had dc-resistances of 10–20 MΩ. The peak and decay time constants of mEPCs were measured on voltage-clamp currents sampled at 15 or 20 kHz. Single exponential decay time constants were fitted to mEPCs by least squares (exponentially weighted). In some neonatal muscle, mEPC frequency was increased by superfusion of the end plate region with sucrose (0.5 M) delivered from a small tipped glass pipette.
Muscle Contraction.
Isolated nerve-muscle preparations (soleus) were mounted in lucite chambers perfused with pre-aerated (95% O2/5% CO2) tyrode solution consisting of 125 mM NaCl, 25 mM NaHCO3, 5.37 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5% glucose at 25°C. Blocking tyrode solution contained in addition 0.1 μM (+)-tubocurarine. Nerves were stimulated via suction electrodes with 0.1-ms square pulses, and muscles were stimulated via bath-mounted silver wires with pulses of 0.5 ms duration at 3× the threshold intensity. The force transducer was connected to an amplifier (Hugo Sachs Elektronik, March-Hugstetten über Freiburg, Germany) and tension records were displayed on a storage oscilloscope. Nerve deficit is defined as the difference between the contraction-force time integrals measured during muscle and nerve stimulation, respectively.
Motor Behavior.
A computerized electronic pull-strain gauge (Columbus Instruments, Columbus, OH; refs. 20 and 21) was used to determine the forelimb grip strength, which can be recorded 2 to 3 weeks after birth. Animals were allowed to grasp the triangular ring and were pulled horizontally until the grip was released. Five measurements were performed per animal and the highest reading was used for statistical evaluation of mean and SD. Grip strength of wild-type and heterozygous animals showed no significant difference and were averaged together. Grip strength measurements of less than 0.1 N could not be evaluated. No significant differences in forelimb grip strength were detected between male and female mice.
RESULTS
Gene Targeting and Levels of γ- and ɛ-Subunit mRNAs.
Parts of exon 7 to intron 8 of AChRɛ were replaced by a PGK-neo-cassette by homologous recombination in ES cells (Fig. 1A). The disrupted gene region carries sequences encoding the M1 and M2 segments, which are essential for the function of AChR channels (22). Correctly engineered cell lines were identified by Southern blot analysis and cells of two independently isolated clones, 382 and 399, were injected into C57BL/6 blastocysts. Male chimeras derived from both independent clones transmitted the mutated ɛ-subunit allele to their offspring.
Figure 1.
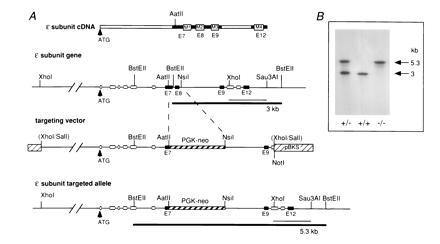
Targeted disruption of the gene encoding the ɛ-subunit of muscle AChR. (A) Schematic representation of AChR ɛ-subunit cDNA with exons E7, E8, E9, and E12 (solid bars). The regions encoding M1, M2, M3, and M4 segments (open boxes), which are essential for the assembly of functional AChR complexes, are indicated. The wild-type ɛ-subunit allele carries the putative membrane regions in exons 7, 8, 9, and 12. The targeting vector consists of 3.3 kb of 5′ untranslated sequences and 1.9 kb containing exons 1 to 7. A PGK-neo cassette (1.9 kb) was inserted to replace sequences from the AatII restriction site of exon 7 to the Nsil site located in intron 8. Deletion of this sequence region in the mutated product was expected to completely prevent the assembly of functional ɛ-AChR channels in adult muscle. The final construct contains a 5.2-kb 5′ homologous region and a 1.4-kb 3′ homologous region. The 3′ end extended from the NsiI site to the XhoI site of exon 10. Hybridization probe used for screening of ES cells and mutant mice is indicated as thin bar and represents a 1.1-kb genomic XhoI/Sau3AI DNA fragment excised from cloned genomic DNA. Thick black bars represent the different BstEII fragments expected from the wild-type and mutated allele corresponding to 3 kb and 5.3 kb, respectively. (B) Southern blot analysis performed with DNAs isolated from tails of AChRɛ+/− × AChRɛ+/− crosses. DNAs from AChRɛ+/+, AChRɛ+/−, and AChRɛ−/− mice were digested with BstEII and hybridized with XhoI/Sau3AI probe as shown in A. The wild-type allele generates a 3-kb fragment and the mutant allele a 5.3-kb fragment.
To generate heterozygous mutant AChRɛ+/− mice, chimeric males derived from clone 382 were crossed with F1 hybrids of C57BL/6xDBA/2 mice while chimeras from clone 399 were crossed with pure C57BL/6 mice. The AChRɛ+/− mutant mice grew normally and had no obvious defects. They were used to generate homozygous AChRɛ−/− mice. Among 123 offspring, the ratio of wild type/heterozygous/homozygous mutant mice was 0.9:2:1.1 as confirmed by Southern blot analysis (Fig. 1B), showing that heterozygous animals were fertile and transmission of the targeted allele occurred in a Mendelian fashion.
Analysis of RNA isolated from hindleg muscles revealed that at P5 and P34 ɛ-subunit mRNA was only present in wild-type mice but not in AChRɛ−/− mutant mice. At P5, γ-subunit mRNA levels were comparable in null mutant and wild-type mice (Fig. 2A). At P34, γ-subunit mRNA expression was strongly reduced and could not be consistently detected by Northern blot hybridization (not shown). Reverse transcription–PCR together with chemiluminescence detection demonstrated that in contrast to the faint, barely detectable signals obtained with RNA from P34 wild-type animals, RNA from null mutants yielded significant amounts of γ-subunit-specific products, indicating expression of γ-subunit transcripts (Fig. 2A). Thus, during postnatal development, two differences in γ- and ɛ-subunit mRNA levels were observed between AChRɛ−/− mutants and wild-type mice. First, the appearance of ɛ-subunit mRNA that is observed during normal development (10, 12, 14) was missing. Second, the γ-subunit mRNA did not disappear completely, but still was detectable in AChRɛ−/− animals. Consistent with the low level of γ-subunit mRNA, end plates were stained with rhodamine-α-bungarotoxin in adult animals, but the fluorescence intensity was significantly lower in AChRɛ−/− mutants than in wild-type mice (Fig. 2 B and C), suggesting a substantial reduction of AChRs in the end plates of adult AChRɛ−/− muscle.
Figure 2.
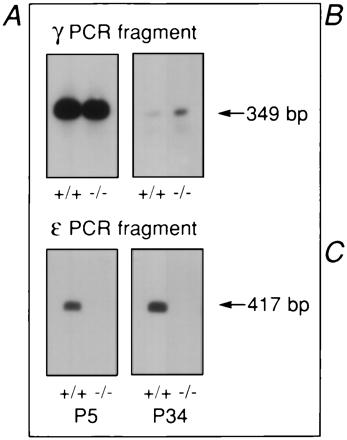
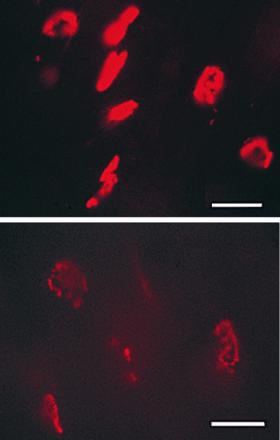
AChR γ- and ɛ-subunit transcripts and AChR expression at end plates. (A) Reverse transcription–PCR. Total RNA from hind leg muscles was extracted at P5 and P34. Transcript levels were low, and direct visualization of ethidium stained PCR products gave no clear results for γ-subunit transcripts at P34. PCR products were therefore transferred to Biodyne A membranes and were stained using an Enhanced Chemiluminescence detection kit. (B and C) AChRs visualized with rhodamine-labeled α-bungarotoxin. Diaphragm of 3-month-old wild-type (B) and AChR ɛ−/− mutant mouse (C) littermates of line 399. (Bars = 75 μm.)
Effects of ɛ-Subunit Gene Ablation on Neuromuscular Transmission.
We compared the functional properties of end plate channels and of neuromuscular transmission in wild-type and AChRɛ−/− mice between P5 to P6 and P28 to P35. At P32, body weight was 33% below normal and motor defects were obvious in the null mutants (see below). In wild-type muscles, the low conductance end plate channel (37 ± 4 pS, n = 5) of neonatal muscle (P5) was replaced at P30 by a high conductance channel (52 ± 0.4 pS, n = 3; Fig. 3). At P5 in AChRɛ−/− muscle, the conductance (36 ± 2 pS, n = 5) was not different from that in neonatal wild-type muscle (P < 0.05, t test). At P30, the conductance was 33 ± 3 pS (n = 7, Fig. 3), which was not significantly different from that of end plate channels in neonatal muscle from wild-type or null mutants mice (P < 0.05, t test). This shows that the switch from end plate channels with low-to-high conductance, which in normal mice is complete by P14–P16 (not shown), does not occur in AChRɛ−/− mice.
Figure 3.
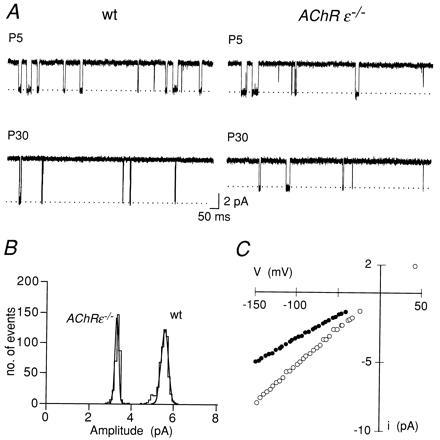
Low conductance end plate channels persist in AChRɛ−/− mice. (A) Single channel currents in wild-type (wt) (Left) and AChRɛ−/− muscle (Right) recorded from P5 (upper traces) and P30 (lower traces) end plate. Membrane potential was approximately −100 mV in each experiment. Membrane potential was estimated from current reversal potential measurement in the same patch. Temperature 20°C, inward current is downward. (B) Amplitude distribution of single channel currents recorded from the end-plate of wild-type (wt) and AChRɛ−/− muscle at P30 as shown in A. Each histogram was fitted to a single Gaussian and show that current amplitudes in both patches represent homogeneous conductance class channels. Mean amplitudes are 3.3 ± 0.1 pA (AChRɛ−/−) and 5.4 ± 0.2 pA (wt). (C) Current-voltage (I–V) relations of single end plate channels in P30 wild-type (open symbols) and AChRɛ−/− muscle (solid symbols). Voltage (V) represents shift of patch-membrane potential from extrapolated current reversal potential. Straight lines (not shown) were fitted to data points in the voltage range of −20 mV to −100 mV. Slope conductance is 32 pS (AChRɛ−/−) and 52 pS (wt), respectively. Different experiment from that shown in A and B.
In normal mice, the switch in channel properties caused a reduction of the mEPC decay time constant (6). At early neonatal stages (<P6), mEPCs from the diaphragm of wild-type and AChRɛ−/− mutant mice were comparable in size and time course (Fig. 4A), having peak amplitudes of 2–5 nA, and were characterized by decay time constants of 4–6 ms (Table 1). As expected, the mEPC decay time course in AChRɛ−/− animals at P30 was significantly (P < 0.05, t test) slower than that of wild-type littermates (Fig. 4B, Table 1). Decay time constants were not significantly different from the values measured in early neonatal end plates (P < 0.05, t test). In addition, the mEPC amplitudes, which in wild-type animals remained almost unchanged between P5 and P30, were reduced by about 40% in AChRɛ−/− muscle. The difference in mEPC amplitudes between wild-type and null mutants became more pronounced in muscle from P65 to P75 animals (Fig. 4C, Table 1).
Figure 4.
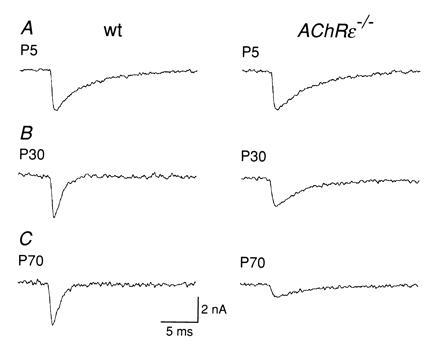
Amplitude and decay time course of miniature end plate currents. Records of representative mEPCs from wild-type (Left) and AChRɛ−/− mutant mouse muscle (Right). All records at −70 mV, 20–21°C. Inward current is downward. Sampling at 15 kHz. Scale bars in C refer to all traces. (A) P5 muscles. Peak mEPC amplitude is 3.2 nA and decay time constant is 4.6 ms for wild-type and 3.2 nA and 4.7 ms for AChRɛ−/− end plate. (B) P30 muscles: 3.3 nA and 1.0 ms for wild-type and 2.3 nA and 4.2 ms for AChRɛ−/− end plate. (C) P70 muscles: 3.3 nA and 1.0 ms for wild-type and 1.0 nA and 4.2 ms for AChRɛ−/− end plate.
Table 1.
Peak amplitude and decay time constant of mEPCs
| Age | wt
|
AChRɛ−/−
|
||
|---|---|---|---|---|
| A, nA | τ, ms | A, nA | τ, ms | |
| P4–P5 | 3.3 ± 0.5 | 4.2 ± 0.3 | 3.4 ± 0.4 | 5.1 ± 0.6 |
| P30–P32 | 3.3 ± 0.5 | 1.3 ± 0.1 | 2.1 ± 0.3 | 4.7 ± 0.6 |
| P65–P75 | 3.1 ± 0.4 | 1.1 ± 0.1 | 0.9 ± 0.2 | 4.4 ± 0.7 |
The mean peak amplitude (A) and decay time constants (τ) of mEPCs recorded from hemidiaphragms of wild-type (wt) or AChRɛ−/− mice were determined for each end plate from between 45 and 210 mEPCs measured individually. All mEPCs had fast rise times (<0.5 ms for 20–80% peak amplitude rise time). Means ± 1 SD are given, and the number of end plates was between 7 and 9 for each mean. All experiments at −70 mV, temperature 20–21°C.
The longer decay time course and smaller peak amplitude of mEPCs in null mutants is consistent with the AChR channels in the end plate lacking an ɛ-subunit. Assuming that the amount of ACh packaged in a vesicle is unchanged in null mutants, the smaller mEPC amplitudes indicate also a substantial reduction in the packing density of channels in the end plate.
Effects of ɛ-Subunit Gene Ablation on Muscle Contraction and Motor Performance.
The most conspicuous phenotypic difference between wild-type and AChRɛ−/− mice was the reduction in body weight and motor performance with age. Mutant mice lost the ability to climb or to bend their body upward when suspended by their tail. To elucidate whether the cause of these motor deficits was impaired neuromuscular transmission, we measured the isometric twitch and tetanic contraction forces following direct and indirect stimulation of the muscle. The strength of muscle twitch was significantly (P < 0.05, t test) smaller in the AChRɛ−/− mice (twitch force 24 ± 6.7 mN, n = 6) than in heterozygous AChRɛ+/− or wild-type mice (35 ± 6.2 mN, n = 3; Fig. 5). Fig. 5 also compares contraction forces at P32 elicited by nerve and muscle stimulation, respectively, in a heterozygous AChRɛ+/− and a homozygous AChRɛ−/− mutant mouse muscle. The muscle of heterozygous animals showed no deficits in nerve-evoked forces upon single or tetanic stimulation (Fig. 5A) either in normal Tyrode solution or in a blocking solution that contained, in addition, 0.1 μM (+)-tubocurarine (dTC) (Table 2), indicating a normal safety factor of transmission. Marked impairment of transmission, however, was observed in muscles of AChRɛ−/− mice. While single twitches evoked by nerve or by muscle stimulation were similar, differences became obvious with tetanic stimulation (Fig. 5B, Table 2). In three soleus muscles from either heterozygous or null mutant animals, the mean nerve deficit, calculated as the difference in tension time integrals at 100 Hz during muscle or nerve stimulation, was 0% versus 16% (range 1 to 38%) in normal Tyrode (Fig. 5B) and 1 ± 1.7% versus 54 ± 30% in the dTC blocking solution, respectively. These differences were significant (P < 0.05, t test) and increased with higher frequency of stimulation and with the number of stimuli during tetanic stimulation (Table 2). It indicated impaired neuromuscular transmission (23) with a reduced safety factor in AChRɛ−/− mice.
Figure 5.
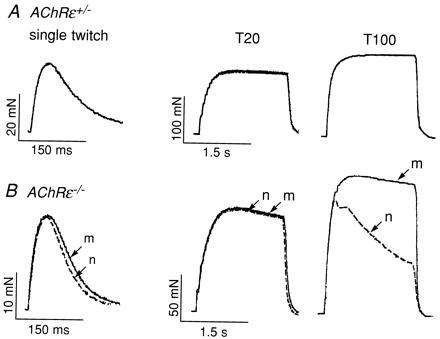
Reduction of isometric tetanic muscle contraction force. (A) Isometric tension measurements on soleus muscle in vitro from a heterozygous AChRɛ+/− animal at P32. Single twitches and tetanic contractions were evoked by direct muscle stimulation or by nerve stimulation in normal Tyrode solution. The records of contractions evoked by nerve or muscle stimulation superimposed perfectly indicating no difference in contraction force upon nerve or muscle stimulation. (B) Isometric tension measurements on soleus muscle from an AChRɛ−/− animal at P32. Single twitches and tetanic contractions evoked by direct muscle stimulation (m; solid lines) and nerve stimulation (n; dashed lines) are superimposed for direct comparison. Note different scales in A and B. Tetanic stimulation frequencies (T) were 20 or 100 Hz as indicated. Vertical scale bars indicate muscle contraction force in mN; horizontal ones indicate duration of stimulation in seconds (s). Body weight of the three animals studied in each group was on average 22 ± 4.1 g for the AChRɛ+/− mice and 11 ± 1.1 g for AChRɛ−/− mice; wet weight of soleus muscles was 11.5 mg ± 2.2 and 7.5 mg ± 1.1, respectively (n = 6 each) (P < 0.005 for each comparison, t test).
Table 2.
Maximal force of contraction of soleus muscle during electrical stimulation of muscle or nerve
| Stimulation | force (mN)
|
|||
|---|---|---|---|---|
|
AChRɛ+/−
|
AChRɛ−/−
|
|||
| Muscle | Nerve | Muscle | Nerve | |
| Twitch | 28.7 | 28.7 | 20.4 | 20.4 |
| 20 Hz | 125 | 125 | 119 | 112 |
| 100 Hz | 168 | 167 | 154 | 126 |
| Twitch/dTC | 27.5 | 27.5 | 14.4 | 13.7 |
| 20 Hz/dTC | 130 | 130 | 83.7 | 43.1 |
| 100 Hz/dTC | 172 | 168 | 116 | 62 |
Contraction force measurements on three soleus muscles (P32) of heterozygous (AChRɛ+/−) or null mutant mice (AChRɛ−/−) in normal Tyrode solution and in blocking solution [Tyrode solution containing in addition 0.1 μM (+)-tubocurarine, (dTC)] at 25°C. Force refers to mean maximal amplitude of contraction force measured during direct electrical stimulation of the muscles or during electrical stimulation of the ischiadic nerve.
The compromised motor performance of AChRɛ−/− mutant mice was also quantified by the grip strength test (20, 21). Fig. 6A compares the development of forelimb “grip strength” in normal and heterozygous animals and in AChRɛ−/− mutant mice. At P20, grip strength appeared similar in the two groups. Later, however, it decreased progressively in AChRɛ−/− mice, whereas in wild-type and heterozygous animals, it increased with age. Around P40, grip strength of AChRɛ−/− mice decreased to 10–20% in wild-type or heterozygous mice, and later on, it became too small to be quantified.
Figure 6.
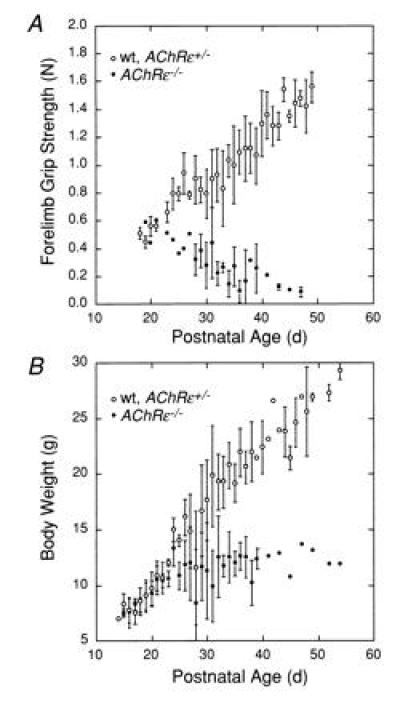
Differential changes in muscle strength and body weight during development. (A) Forelimb grip strength (measured in N) of male wild-type or heterozygous AChRɛ+/− mice (○), and homozygous AChRɛ−/− mutant mice (•). (B) Body weight (measured in g) of male wild-type or heterozygous AChRɛ+/− mice (○) and of male AChRɛ−/− mice (•) during postnatal development. Mean values ± 1 SD (three to five animals) are shown in A and B. Symbols without error bars represent individual animals.
At P28, the body weight of AChRɛ−/− male animals was at least 40% smaller than that of wild-type mice (Fig. 6B). Reduced weight was accompanied by muscle atrophy since at P28-P35 the net weight of slow and fast twitch leg muscles of mutant mice was less than half the weight of muscles from wild-type animals. Homozygous mutant animals deceased prematurely 2 to 3 months after birth (n = 7).
DISCUSSION
Effects of ɛ-Gene Ablation on Adult End Plate Channels.
In wild-type mice, a switch in end plate channels from fetal to adult subtypes occurs normally between P7 and P14 (not shown). In AChRɛ−/− mutant mice, this switch was absent. At P30–P32 in null mutant mice, end plate channels were functionally indistinguishable from those found in fetal muscle. Furthermore, the mEPCs at P30–P32, which normally decay 3- to 5-fold faster in wild-type mice, decayed as slowly in null mutant mice as in P5 animals. Channels without a γ-subunit which can be generated as recombinant αβδ-AChRs in Xenopus oocytes (24), have a smaller inward conductance than fetal γ-AChRs. The predominant end plate channel in juvenile null mutant muscle thus is presumably of the fetal subtype. Nevertheless, end plate channels in null mutant mice are expressed at a lower density than in wild-type mice as indicated by the substantial decrease in fluorescence intensity of rhodamine-α-bungarotoxin-labeled end plates (Fig. 2 B and C) and by the 3- to 4-fold reduction in the peak amplitudes of mEPCs at P65-P75. The latter result suggests that the density of end plate channels is less than 10% of that in a normal end plate (25).
Functionality of the Neuromuscular Synapse in AChRɛ−/− Animals.
The relatively long survival of AChRɛ−/− animals (>P60) is based on the continued presence of fetal type receptors in adult muscle, a situation that does not normally occur. These end plate channels could reflect remaining fetal channels from the early neonatal developmental stage (<P5) or the incorporation of new AChRs. The degradation half-life (t1/2) of mouse end plate AChRs is ≈8 days (26), and the replacement of γ-AChRs by ɛ-AChRs in normal mice occurred between P8 and P14. Hence at most, about 10% of the original γ-AChRs from the early neonatal stages would be present at P32 and less than 1% would be present at P64. An estimate of the effective AChR density, derived from mEPC peak amplitudes (25), suggests that the receptor density in AChRɛ−/− animals at P30–P32 is about 30% and at P65–P75, it is about 10% of the density in normal end plates. Therefore, newly synthesized AChRs must have been incorporated after P8 assuming that t1/2 is not prolonged substantially in AChRɛ−/− mice. Consistent with that, the γ-subunit gene expression in AChRɛ−/− mice was not shut off postnatally as happens during normal development (10, 14, 27). Persistent γ-subunit expression in AChRɛ−/− adult muscle could reflect a decrease in its electrical activity (i.e., action potentials) and/or the lack of an altered influx of Ca2+ at the end plate associated with the ɛ-AChR (8). The latter alternative seems likely since the expression of the γ-subunit gene at the adult end plate is controlled by a “neural” factor acting via AChRs rather than only by electrical activity (13, 28).
In any case, the persistent expression of γ-subunit transcripts in AChRɛ−/− animals shows that the deletion of one AChR subunit alters not only the functional properties of the end plate channel in adult muscle, but also disrupts a regulatory mechanism controlling the subunit expression pattern.
Effect of ɛ-Subunit Gene Ablation on Motor Behavior.
The functional consequences of ablating the ɛ-subunit gene became apparent during the third postnatal week as a stagnation in body and muscle weights and a progressive reduction in grip strength. The atrophy presumably reflects a lack of muscle exercise due to the impairment of neuromuscular transmission until premature death.
The motor phenotype of juvenile and adult null mutants appears to be the consequence of the reduced density of end plate channels, rather than being caused by their different functional properties. In normal soleus muscle, even with an ≈3-fold reduction of functional AChRs in the presence of 0.1 μM (+)-tubocurarine (25), muscle contraction upon nerve stimulation was normal (Table 2). In the null mutants, however, a 3-fold reduction in receptor density caused deficits in nerve-evoked contraction (Table 2). Therefore, additional presynaptic alterations such as a reduction in quantal content of end plate potentials could contribute to the impairment of neuromuscular transmission in AChRɛ−/− mice. It has been demonstrated that nerve terminals retract as a consequence of long-term blockade of AChRs (29) and loss of terminals could also happen at end plates of AChRɛ−/− mice.
To delineate more precisely the functional consequence of the γ/ɛ-subunit-switch for muscle development and motor behavior, it will be necessary to maintain a normal density and number of fetal AChRs in the end plate of adult muscle without expression of the ɛ-subunit. This could be achieved by replacing the ɛ-subunit gene by the γ-subunit (gene “knock in”) rather than by simple ablation of the ɛ-subunit gene.
Acknowledgments
We thank Dr. A. King (Universität Heidelberg, Department of Neurobiology, Germany) for ES cells, A. Härri (BRL, Füllinsdorf, Switzerland) for blastocyst transfer to generate chimeric mice, Dr. A. Irintchev and J. Krischler (Universität Bonn, Germany) for help on muscle contraction measurements, and U. Warncke and B. Mandery for excellent technical assistance. H.R.B. was supported by a grant from the Swiss National Science Foundation. This work was supported by the Deutsche Forschungsgemeinschaft (Wi 487/8-1).
Footnotes
Abbreviations: AChR, acetylcholine receptor; AChRɛ, acetylcholine receptor ɛ-subunit gene; mEPC, miniature end plate current; P, postnatal day; ES, embryonic stem.
References
- 1.Katz B. The Release of Neural Transmitter Substances. Springfield, IL: Thomas; 1969. [Google Scholar]
- 2.Katz B, Miledi R. J Physiol (London) 1972;224:665–699. doi: 10.1113/jphysiol.1972.sp009918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson C R, Stevens C F. J Physiol (London) 1973;235:655–691. doi: 10.1113/jphysiol.1973.sp010410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neher E, Sakmann B. Nature (London) 1976;260:799–802. doi: 10.1038/260799a0. [DOI] [PubMed] [Google Scholar]
- 5.Colquhoun D, Sakmann B. J Physiol (London) 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakmann B, Brenner H R. Nature (London) 1978;276:401–402. doi: 10.1038/276401a0. [DOI] [PubMed] [Google Scholar]
- 7.Mishina M, Takai T, Imoto K, Noda M, Takahashi T, Numa S, Methfessel C, Sakmann B. Nature (London) 1986;321:406–411. doi: 10.1038/321406a0. [DOI] [PubMed] [Google Scholar]
- 8.Villarroel A, Sakmann B. J Physiol (London) 1996;496:331–338. doi: 10.1113/jphysiol.1996.sp021688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Witzemann V, Barg B, Nishikawa Y, Sakmann B, Numa S. FEBS Lett. 1987;223:104–112. doi: 10.1016/0014-5793(87)80518-5. [DOI] [PubMed] [Google Scholar]
- 10.Witzemann V, Barg B, Criado M, Stein E, Sakmann B. FEBS Lett. 1989;242:419–424. doi: 10.1016/0014-5793(89)80514-9. [DOI] [PubMed] [Google Scholar]
- 11.Witzemann V, Stein E, Barg B, Konno T, Koenen M, Kues W, Criado M, Hofmann M, Sakmann B. Eur J Biochem. 1990;194:437–448. doi: 10.1111/j.1432-1033.1990.tb15637.x. [DOI] [PubMed] [Google Scholar]
- 12.Brenner H R, Witzemann V, Sakmann B. Nature (London) 1990;344:544–547. doi: 10.1038/344544a0. [DOI] [PubMed] [Google Scholar]
- 13.Witzemann V, Brenner H R, Sakmann B. J Cell Biol. 1991;114:125–141. doi: 10.1083/jcb.114.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinou J-C, Merlie J P. J Neurosci. 1991;11:1291–1299. doi: 10.1523/JNEUROSCI.11-05-01291.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hall, Z. W. & Sanes, J. R. (1993) Cell 72/Neuron 10, 99–121. [DOI] [PubMed]
- 16.Kühn R, Rajewsky K, Müller W. Science. 1991;254:707–710. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- 17.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 18.Bekoff A, Betz W J. J Physiol (London) 1977;271:25–40. doi: 10.1113/jphysiol.1977.sp011988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 20.Meyer O A, Tilson H A, Byrd W C, Riley M T. Neurobehav Toxicol. 1979;1:233–236. [PubMed] [Google Scholar]
- 21.Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H. Nature (London) 1993;365:27–32. doi: 10.1038/365027a0. [DOI] [PubMed] [Google Scholar]
- 22.Herlitze S, Villarroel A, Witzemann V, Koenen M, Sakmann B. J Physiol (London) 1996;492:775–787. doi: 10.1113/jphysiol.1996.sp021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badke A, Irintchev A, Wernig A. Muscle and Nerve. 1989;12:580–586. doi: 10.1002/mus.880120709. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Brehm P. J Physiol (London) 1993;470:349–363. doi: 10.1113/jphysiol.1993.sp019862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pennefather P, Quastel D M J. J Gen Physiol. 1981;78:313–344. doi: 10.1085/jgp.78.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shyng S-L, Salpeter M M. J Neurosci. 1990;10:3905–3915. doi: 10.1523/JNEUROSCI.10-12-03905.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kues W A, Sakmann B, Witzemann V. Eur J Neurosci. 1995;7:1376–1385. doi: 10.1111/j.1460-9568.1995.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 28.Kues W A, Brenner H R, Sakmann B, Witzemann V. J Cell Biol. 1995;130:949–957. doi: 10.1083/jcb.130.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balice-Gordon R J, Lichtman J W. Nature (London) 1994;372:519–524. doi: 10.1038/372519a0. [DOI] [PubMed] [Google Scholar]