

Abstract
Glutathione (GSH) exerted a profound effect on the oxidation of 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) and 7-benzyloxyquinoline (BQ) by human liver microsomes as well as by CYP3A4-containing insect cell microsomes (Baculosomes). The cooperativity in O-debenzylation of both substrates is eliminated in the presence of 1–4 mM GSH. Addition of GSH also increased the amplitude of the 1-PB induced spin shift with purified CYP3A4 and abolished the cooperativity of 1-PB or BFC binding. Changes in fluorescence of 6-bromoacetyl-2-dimethylaminonaphthalene attached to the cysteine-depleted mutant CYP3A4(C58,C64) suggest a GSH-induced conformational changes in proximity of α-helix A. Importantly, the KS value for formation of the GSH complex and the concentrations in which GSH decreases CYP3A4 cooperativity are consistent with the physiological concentrations of GSH in hepatocytes. Therefore, the allosteric effect of GSH on CYP3A4 may play an important role in regulation of microsomal monooxygenase activity in vivo.
Keywords: cytochrome P450 3A4, glutathione, regulatory effect, allostery, human liver microsomes, 7-benzyloxy-4-(trifluoromethyl)coumarin, 7-benzyloxyquinoline, testosterone, 1-pyrenebutanol, α-naphthoflavone
Introduction
Homo- and heterotropic cooperativity in cytochromes P450 has attracted considerable attention in the past decade. Initial “space-filling” models proposed that the large substrate-binding pocket of microsomal drug metabolizing cytochromes P450 sometimes requires more than one substrate molecule to assure a productive binding orientation of at least one of them [1–9]. However, this hypothesis fails to explain the entire body of observations obtained with cytochrome P450 3A4 (CYP3A4), the principal human drug-metabolizing enzyme [10]. Although the presence of at least two substrates in the same binding pocket is well established [7, 11–16]; the additive effects of different effectors and lack of competition between certain substrates possessing cooperativity [4, 7–9, 17–22] have given rise to a set of complex and mutually incompatible models suggesting the presence of three or even more substrate-selective binding sites per molecule of the enzyme [18, 23–28].
The major trend in much current research on CYP3A4 is to assess whether the “non-Michaelis-Menten” behavior represents a manifestation of true allosteric regulation based on an effector-induced conformational transition in the enzyme [29–35]. One of the first pieces of evidence for the involvement of such changes was obtained in our studies with P450eryF, the best-studied bacterial enzyme known to exhibit cooperativity [32, 33]. An evidence of a conformational transition in CYP3A4 caused by α-naphthoflavone (ANF), a prototypic heterotropic activator, was obtained in our recent work using site-directed incorporation of a fluorescent probe into a cysteine-depleted mutant bearing a fluorescent probe [34]. More recently, the use of pressure-perturbation spectroscopy allowed us to demonstrate unusual stabilization of the substrate-bound high-spin state and a prominent increase in the cooperativity of CYP3A4 at elevated pressure, suggesting that allosteric mechanisms involve decreased protein hydration [35]. Decisive support for substantial ligand-induced conformational changes has been provided by a recently resolved crystal structure of a ketoconazole complex of CYP3A4, which revealed two ligand molecules in the active site [36].
An alternative to the multiple binding sites model is the concept of persistent heterogeneity of CYP3A4 proposed by Koley and co-authors. Heterotropic cooperativity was hypothesized to reflect modulation of the partitioning of CYP3A4 between two functionally different conformers, which are otherwise static. This concept is supported by our studies on the pressure-induced P450-to-P420 transition in solution and in microsomes [37] and our investigation of the kinetics of dithionite-dependent reduction of CYP3A4 [38]. The results suggest that the functional heterogeneity of the CYP3A4 pool may be caused by diverse conformations and/or orientation of the subunits in the enzyme oligomer.
This high level of complexity may indicate that this finely tuned allosteric mechanism is designed to play an important regulatory function in the cell. Therefore, the question of a possible physiological role of CYP3A4 cooperativity is equally intriguing as the molecular mechanisms of cooperativity. One attractive possibility is that the cooperativity of xenobiotic oxidation by microsomal P450 enzymes may simply represent "incidental manifestations" of an allosteric regulatory mechanism that is triggered by some physiological effector(s). With typical turnover numbers of 5–50 min−1 the microsomal monooxygenase is fairly uncoupled and characterized by a high rate of futile cycling and production of reactive oxygen species (see [39, 40] for review). Allosteric regulation may reflect the need to minimize uncoupling and to coordinate P450 function with the oxidative status of the cell and the function of metabolically related enzymes. One of possible candidates for the role of allosteric effector of cytochromes P450 is reduced glutathione (GSH). Besides the metabolic connections between P450 and GSH based on the role of the latter in the antioxidant system and redox signaling [41–43], another obvious junction between P450 and GSH arises from irreversible consumption of GSH upon the formation by glutathione S-transferases of conjugates of the products of P450-catalysed reactions.
There are several reports that demonstrate activation of CYP3A4-dependent reactions by GSH in reconstituted systems in vitro [44–46]. Since the maximal activation is observed immediately after addition of GSH, GSH-dependent reduction of protein thiols does not appear to be involved [44, 46]. The effect of GSH, which apparently involves a strengthening of the interactions of CYP3A4 with NADPH-cytochrome P450 reductase (CPR) [46], is amplified considerably in the presence of cytochrome b5 [46] and increased at high ionic strength [45]. The fact that the activating effect of GSH is eliminated in reconstituted systems containing 0.5 mg/ml CHAPS [47] also suggests an involvement of protein-protein interactions in the mechanism.
Although the physiological regulation of cytochromes P450 by GSH appears to be quite logical, it has never been demonstrated or hypothesized to our knowledge. Quite the opposite, it has been suggested that the activation of CYP3A4 by GSH may cause a disruption of regulation of cellular GSH level due to increased production of reactive oxygen species by P450 [48]. Furthermore, the effect of GSH on CYP3A4 observed in reconstituted systems has never been reproduced in microsomes. The only known attempt to probe such an effect showed no modulation of the rate of oxidation 200 µM nifedipine by human liver microsomes in the presence of 0.5 – 5 mM GSH [44].
Here we undertook a rigorous investigation of the effect of GSH on activity and cooperativity of CYP3A4 with 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC) and 7-benzyloxyquinoline (BQ) in model microsomes (Baculosomes) and in human liver microsomes. We also probed the impact of GSH on the activation of CYP3A4-dependent O-debenzylation of BFC by α-naphthoflavone (ANF). Our results provide the first evidence of an effect of GSH on CYP3A4 in the microsomal membrane and show that in the presence of GSH the homotropic cooperativity with BFC and BQ is eliminated. In contrast, GSH amplifies considerably the activation by ANF of BFC oxidation by human liver microsomes or Baculosomes enriched with cytochromes P450 3A4 and b5. These experiments were complemented by studies of the effect of GSH on cooperativity of substrate binding to purified CYP3A4 in solution, the formation of spectral complexes of GSH with CYP3A4, and the effect of GSH on the fluorescence of a BADAN fluorophore introduced at Cys-64. Our results reveal GSH as a prominent heterotropic effector of CYP3A4 that exhibits at least two different modes of binding, one of which involves coordination of the glutathione SH-group to the heme iron.
Materials and Methods
Materials
Water-soluble testosterone (HPCD-encapsulated testosterone), glucose-6-phosphate, glucose-6-phosphate dehydrogenase, glutathione, NADPH, protocatechuic acid, and protocatechuate-3,4-dioxygenase were the products of Sigma Chemicals (St. Louis, MO). 1-Pyrenebutanol (1-PB), 7-benzyloxy-4-(trifluoromethyl)coumarin (BFC), 7-hydroxy-4-(trifluoromethyl)coumarin (HFC), 7-benzyloxyquinoline (BQ), 7- hydroxyquinoline (HQ) and 6-bromoacetyl-2-dimethylaminonaphthalene (BADAN) were from Invitrogen/Molecular Probes Inc. (Eugene, CA). All other chemicals were of ACS grade and were used without further purification. Rat liver microsomal cytochrome b5 purified from recombinant E. coli was a generous gift of Prof. Sergei Usanov (Institute of Bioorganic Chemistry, Minsk, Belarus). Pooled human liver microsomes (pool H0620) were obtained from XenoTech LLC (Lenexa, KS). CYP3A4 microsomes prepared from insect cells infected with recombinant baculovirus containing CYP3A4 and rabbit NADPH-P450 reductase (Baculosomes) were obtained from Invitrogen (Carlsbad, CA).
Expression, purification, and chemical modification of CYP3A4
Wild-type CYP3A4 and its cysteine-depleted mutant CYP3A4 (C58,C64) [34] were expressed as His-tagged proteins in E. coli TOPP3 cells and purified by chromatography on Ni-NTA resin followed by ion-exchange chromatography on Macro-Prep CM Support resin (Bio-Rad Laboratories, Hercules, CA) [34]. Modification of CYP3A4 (C58,C64) by BADAN was performed as described earlier, except for the final step where the removal of detergent and unreacted label was performed on Ni-NTA Agarose as opposed to CM Sepharose CL-6B [34].
Incorporation of purified CYP3A4 and cytochrome b5 into Baculosomes
In order to better approximate the composition of the liver microsomes we incorporated cytochrome b5 and additional CYP3A4 into baculosomal membranes by incubation of Baculosomes with the purified proteins. Stock solutions of the purified CYP3A4 (100 – 200 µM) and cytochrome b5 (300 µM) were added directly to undiluted commercial preparation of Baculosomes (9.3 mg protein/ml, 0.1 nmol P450 per mg of protein) to achieve 20 µM concentration of each protein in solution. The incubation media was supplemented with an oxygen scavenging system (5 mM protocatechuic acid and 0.3 unit/ml protocatechuate-3,4-dioxygenase) to prevent oxidative destruction of CYP3A4 during the incubation. After 3 hours of incubation at room temperature (22 °C) under continuous stirring in a tightly closed flask flushed with argon gas the sample was diluted 10 times with 0.1 M Na-Hepes buffer, pH 7.4, containing 1 mM DTT, 1 mM EDTA, and 1.15% KCl. The pellet of enriched Baculosomes obtained by 90 min of centrifugation at 35,000 rpm was resuspended in 0.1 M Na-Hepes buffer, pH 7.4, containing 1 mM DTT, 1 mM EDTA, and 10% glycerol. The concentration of Baculosomes was standardized based on the activity of CPR in a cytochrome c reduction assay [49]. This procedure resulted in preparations containing 0.7–1 nmol of each of CYP3A4 and cytochrome b5 per milligram of protein.
Activity measurements
The measurements of the rate of BFC and BQ O-debenzylation were performed by a real-time continuous fluorometric assay. Three to six µl of the suspension of Baculosomes (1 nmol CYP3A4 per ml) was added to 300 µl of 0.1 M Na-HEPES buffer, pH 7.4, containing 1 – 8 mM of GSH (if indicated), placed into a 5 × 5 mm quartz cell under continuous stirring, and thermostated at 25 °C. The cocktail (9.6 µl) of NADPH, glucose-6-phosphate, and glucose-6-phosphate dehydrogenase was added to attain the concentration of these ingredients of 200 µM, 10 mM, and 2 units/ml, respectively. After a short pre-incubation (15–20 s) the reaction was initiated by the addition of an aliquot of a 15–20 mM stock solution of BFC or BQ in acetone to attain a desired concentration in the range of 1 – 100 µM. This order of addition of the reagents was chosen to avoid a pre-stationary (lag) phase observed in the case of initiation of the reaction of BFC debenzylation by NADPH. Our control experiment showed that the maximal stationary rate of debenzylation of both BQ and BFC was not dependent on the way of initiating the reaction. The registration of the increase in the concentration of the product (HFC) was performed with a computerized Hitachi F2000 spectrofluorometer equipped with a thermostated cell holder and a magnetic stirrer [50] by monitoring the increase in the emission at 500 nm or 516 nm (for BFC and BQ respectively) with excitation at 404 nm. The slit bandwith of both excitation and emission monochromators was set to 10 and 20 nm, respectively. In the assays with BFC the excitation light beam was passed through a long-pass 400 nm cut-off filter to decrease photobleaching of HFC. The rate of the formation of a product was estimated by determining the slope of the linear part of the kinetic curve recorded over a period of 2 – 5 min. Calibration of the assay was performed at the end of each day by measuring the intensity of fluorescence in a series of 4–5 samples of the same reaction mixture containing HFC or HQ at concentrations increasing from 0.5 to 3 µM.
In the activity measurements presented here we used two batches of Baculosomes (Lots 39195B and 283776A) characterized by an NADPH-cytochrome c reductase activity of 1.1 and 3.3 µmol/min*mg and the CYP3A4 content of 82 and 100 pmol/mg protein respectively. Assuming the turnover number of cyt. c reduction for pure rat CPR to be around 4000 min−1 [51, 52] we may estimate the content of reductase in these batches to be around 280 and 800 pmol/mg, which corresponds to the CPR:P450 ratio of 3.4:1 and 8:1 respectively. Because of the considerable molar excess of reductase over CYP3A4, the latter is anticipated to be completely complexed with the flavoprotein and the functional properties of the monooxygenase are expected to be largely insensitive to possible variations in CPR content between the batches. This conclusion is supported by the observation of Yamazaki et al. that the incorporation of exogenous CPR into CYP3A4 Baculosomes does not affect the rate of testosterone 6β-hydroxylation [53]. It should be noted also that all experiments on BFC debenzylation with intact Baculosomes were done with the first batch (39195B), while BQ experiments and the experiments with BFC and enriched baculosomes were done with the second batch.
Absorbance and fluorescence measurements
Absorbance spectra were recorded with a S2000 CCD rapid scanning spectrometer (Ocean Optics, Inc., Dunedin, FL, USA) using an L7893 UV/VIS fiber-optics light source (Hammamatsu Photonics K.K., Shizuoka, Japan). Fluorescence measurements were performed with an F900 spectrofluorometer (Edinburgh Instruments Ltd., Edinburgh, UK) equipped with a thermostated cell holder and a magnetic stirrer. In the fluorometric titration experiments the excitation of fluorescence was performed at 402 nm with a bandwidth of 5 nm. The emission spectra in the 420 nm – 650 nm wavelength region were recorded with a bandwidth of 2 nm. All experiments were performed at 25 °C in 100 mM HEPES buffer, pH 7.4. 1-PB, BQ, BFC, and ANF were added as 10–20 mM stock solutions in acetone. Testosterone was added as a 600 µM solution of water-soluble testosterone-HPCD complex in 0.1M Na-Hepes buffer, pH 7.4. The HPCD complex of testosterone was used to increase the solubility of this low-affinity substrate in water and diminish increase of light scattering at high testosterone concentrations. Control studies presented in our recent publication showed that replacement of the stock solution of testosterone in acetone with its water-soluble complex with HPCD has virtually no effect on the parameters of its interactions with CYP3A4 [35].
Data Processing
The analysis of the series of absorbance and fluorescence spectra was done with our SpectraLab program using a principal component analysis (PCA) method (also known as singular value decomposition (SVD) technique), as described earlier [12, 54, 55]. To interpret the changes in absorbance spectra in terms of the changes in the concentration of P450 species, we used a least-squares fitting of the spectra of principal components by the set of the spectral standards of pure high- and low-spin of CYP3A4 [37] complemented, when appropriate, with the extinction spectra of thiol- and thiolate complexes of P450cam taken from [56]. Fitting of the titration curves to the Hill and Michaelis-Menten equations was made by non-linear regression using a combination of Nelder-Mead and Marquardt algorithms as implemented in our SpectraLab program [54].
Results
Effect of GSH on homotropic cooperativity of O-debenzylation of BFC and BQ by CYP3A4-containing Baculosomes
As shown in Fig. 1 and Table 1, addition of 1 – 4 mM GSH results in a significant decrease in the cooperativity of CYP3A4 with BFC as a substrate. For example, the Hill coefficient (n) of 2.0 ± 0.2 in the absence of GSH decreases to 1.1 ± 0.2 at 4 mM GSH (Table 1). There is no further decrease in n at 8 mM GSH, although kcat begins to decrease at that concentration. Similar results were obtained using BQ as a substrate (Table 1). The dependencies of the Hill coefficient on the GSH concentration can be fitted to a hyperbolic (Michaelis-Menten) equation that gives the effective constant of dissociation of the GSH-CYP3A4 complex of 0.5 ± 0.2 and 0.24 ± 0.2 mM for BFC and BQ debenzylation, respectively (Fig 1b).
Figure 1.
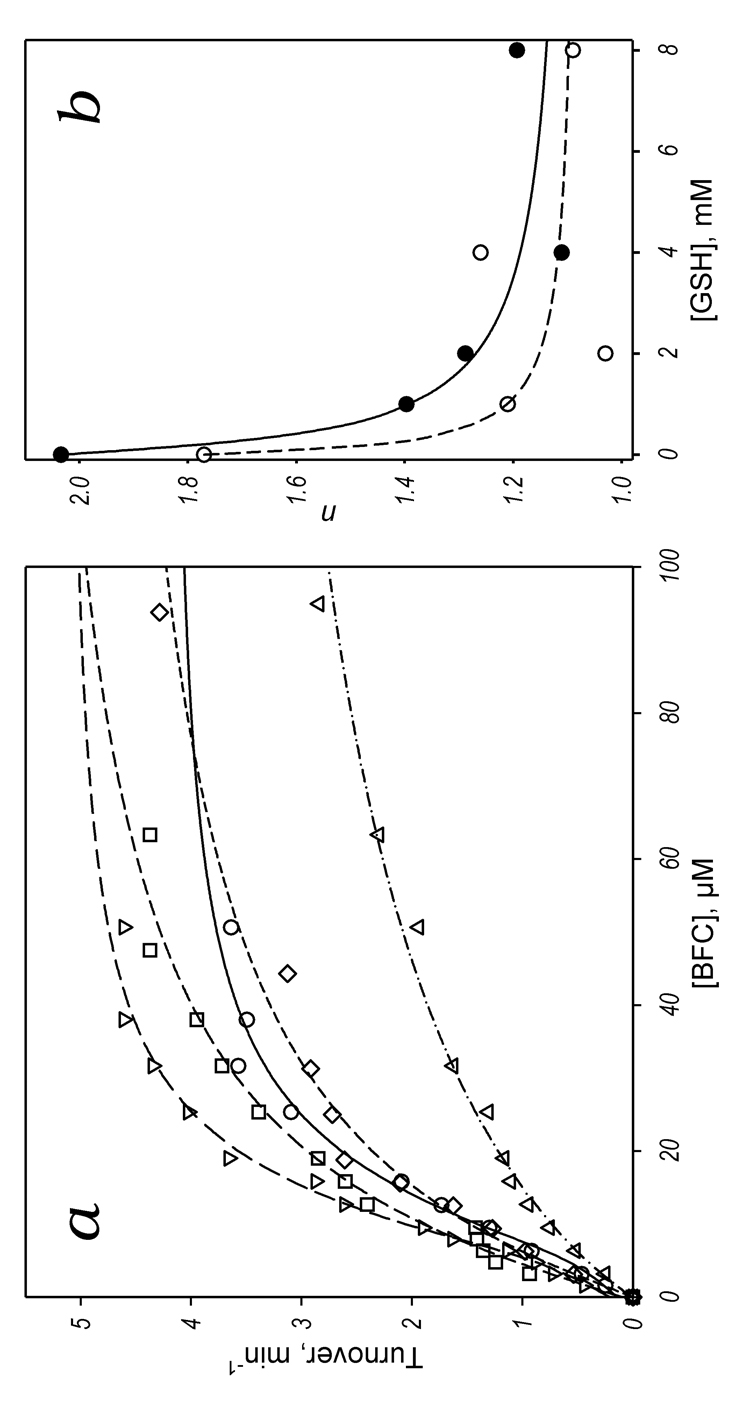
Effect of GSH on O-debenzylation of BFC and BQ by CYP3A4 in Baculosomes. Panel a shows the dependence of the rate of oxidation of BFC on the concentration of this substrate at no glutathione added (circles, solid line) and in the presence of 1 mM (inverted triangles, long dash), 2 mM (squares, medium dash), 4 mM (diamonds, short dash), and 8 mM (triangles, dash-dot) GSH. The lines represent the result of fitting of the data sets to the Hill equation. Panel b shows the dependencies of the Hill coefficient on the glutathione concentration for O-debenzylation of BFC (closed circles, solid line) and BQ (open circles dashed line). The points shown on this plot represent the averages of from two to three individual measurements. The lines on this plot show the results of the fitting of these data sets to a hyperbolic (Michaelis-Menten) equation with KDapp values of 0.5 ± 0.2 and 0.24 ± 0.2 mM, respectively.
Table 1.
Effect of GSH on O-debenzylation of 7-BFC and 7-BQ in Baculosomes*.
| Substrate | [GSH],mM | kcat, min−1 | S50, µM | n |
|---|---|---|---|---|
| 7-BFC | 0 | 4.2 ±0.3 | 16.7 ±2.7 | 2.03 ±0.22 |
| 1 | 4.8 ±1.1 (0.300) | 19.6 ±7.7 (0.472) | 1.40 ±0.46 (0.044) | |
| 2 | 4.7 ±1.9 (0.510) | 14.7 ±1.7 (0.413) | 1.29 ±0.30 (0.020) | |
| 4 | 5.3 ±2.0 (0.280) | 24.3 ±0.9 (0.023) | 1.11 ±0.25 (0.008) | |
| 8 | 2.5 ±0.5 (0.002) | 18.7 ±12.5 (0.250) | 1.19 ±0.30 (0.006) | |
| 7-BQ | 0 | 44.0 ±3.9 | 24.6 ±2.3 | 1.85 ±0.16 |
| 1 | 46.4 ±7.4 (0.734) | 34.7 ±13.2 (0.373) | 1.21 ±0.20 (0.018) | |
| 2 | 56.8 ±7.6 (0.140) | 34.4 ±19.2 (0.423) | 1.03 ±0.29 (0.042) | |
| 4 | 54.6 ±5.6 (0.140) | 39.2 ±11.7 (0.138) | 1.26 ±0.21 (0.048) | |
| 8 | 68.1 ±29 (0.253) | 45.3 ±27.6 (0.280) | 1.09 ±0.27 (0.043) | |
The experiments with 7-BFC were performed with Baculosomes lot 39195B, while the experiments with 7-BQ were done with the lot 283776A (see Materials and Methods for comparison of the CPR content between the lots). The values given in the Table were obtained by averaging the result of 2–4 individual measurements and the “±” values show the confidence interval calculated for p = 0.05. The values in parentheses represent the p-values of Student’s T-test for the hypothesis of equality of the respective values with that observed at no GSH added.
Effect of GSH on homotropic cooperativity of O-debenzylation of BFC by enriched Baculosomes and human liver microsomes
It is important to note that the CYP3A4 Baculosomes used in our initial experiments differ considerably from the membrane of the endoplasmic reticulum of human liver in the content of the components of the monooxygenase system. As described in Matherials and Methods, the molar ratio of CPR to CYP3A4 in the two preparations of Baculosomes used in this study was estimated to be around 3.2:1 and 8:1, while the ratio of CPR to total P450 in liver microsomes is known to be 1:10 [57] or even 1:50 [58, 59]. Another important difference is that the Baculosomes do not contain cytochrome b5. In order to probe the effect of GSH on BFC debenzylation in a system closer to the in vivo conditions we incorporated into the Baculosomal membrane cytochrome b5 and increased the concentration of CYP3A4 by incubating Baculosomes with purified recombinant proteins. As described in Materials and Methods, our treatment increased the concentration of CYP3A4 from 7 to 10 times, so that the molar ratio of CYP3A4 to CPR may be estimated to be around 2:1. The molar ratio of cytochrome b5 to CYP3A4 in these preparations, which are designated here as “Enriched Baculosomes”, was close to 1:1.
As seen from Table 2, incorporation of additional cytochrome 3A4 decreases the kcat calculated per mol of the heme protein, while the rate calculated per mole of CPR remained almost unchanged (not shown). The effect of GSH on BFC metabolism in enriched Baculosomes was qualitatively similar to that observed in the initial preparation, although the maximal effect of GSH on cooperativity was observed at considerably lower concentrations of GSH. An increase in kcat was also revealed in enriched Baculosomes at 4 mM GSH(Table 2).
Table 2.
Effects of GSH and ANF on O-debenzylation of 7-BFC in enriched Baculosomes and human liver microsomes*.
| System | [ANF], µM | [GSH], mM | kcat, min−1 | S50, µM | N |
|---|---|---|---|---|---|
| Enriched Baculosomesa | 0 | 0 | 0.65 ±0.09 | 30.6 ±1.2 | 1.47 ±0.13 |
| 0 | 0.5 | 0.64 ±0.05 (0.909) | 14.6 ±2.8 (0.013) | 0.96 ±0.06 (0.022) | |
| 0 | 1 | 0.72 ±0.23 (0.668) | 36.9 ±2.6 (0.048) | 1.06 ±0.19 (0.071) | |
| 0 | 4 | 1.32 ±0.19 (0.014) | 47.8 ±5.6 (0.011) | 1.19 ±0.42 (6.10−4) | |
| 25 | 0 | 0.68 ±0.03 (0.583) | 5.82 ±0.17 (6.10−4) | 0.98 ±0.03 (0.019) | |
| 25 | 4 | 0.91 ±0.24 (0.323) | 16.7 ±3.0 (0.030) | 1.0 ±0.08 (0.011) | |
| Human liver microsomesb | 0 | 0 | 0.42 ±0.03 | 59.4 ±17.4 | 2.00 ±0.34 |
| 0 | 0.5 | 0.61 ±0.09 (0.011) | 64.8 ±17.5 (0.680) | 1.17 ±0.20 (0.040) | |
| 0 | 1 | 0.47 ±0.07 (0.325) | 56.0 ±11.3 (0.762) | 1.24 ±0.05 (0.044) | |
| 0 | 2 | 0.46 ±0.04 (0.253) | 58.4 ±18.6 (0.951) | 0.96 ±0.09 (0.063) | |
| 0 | 4 | 0.54 ±0.10 (0.065) | 116 ±42 (0.041) | 1.00 ±0.02 (0.027) | |
| 0 | 8 | 0.40 ±0.09 (0.490) | 200 ±47 (0.002) | 1.13 ±0.34 (0.072) | |
| 25 | 0 | 0.54 ±0.18 (0.220) | 16.7 ±1.1 (0.001) | 1.34 ±0.36 (0.190) | |
| 25 | 1 | 0.69 ±0.04 (1.10−4) | 23.2 ±9.0 (0.023) | 1.23 ±0.22 (0.092) | |
The values given in the Table were obtained by averaging the result of 2–4 individual measurements and the “±” values show the confidence interval calculated for p = 0.05. The values in parentheses represent the p-values of Student’s T-test for the hypothesis of equality of the respective values with that observed at no effector (GSH and/or ANF) added.
Enriched preparations were prepared using the Baculosomes lot 283776A.
In the case of human liver microsomes kcat values are apparent and calculated per total cytochrome P450 content.
Elimination of cooperativity of BFC debenzylation in the presence of GSH was also observed with human liver microsomes (Table 2). Similar to enriched Baculosomes, the maximal effect on cooperativity and kcat in liver microsomes was observed at 0.5–2 mM GSH, which is considerably lower than the concentration of 4 mM necessary for the maximal GSH effect in unenriched Baculosomes.
Effect of GSH on heterotropic cooperativity of CYP3A4 with ANF
The studies shown above demonstrate that addition of GSH eliminates homotropic cooperativity with BFC and BQ both in human liver microsomes and in model Baculosomal membranes containing recombinant CYP3A4. To examine a possible influence of GSH on heterotropic cooperativity we studied the effect of ANF on O-debenzylation of BFC. Earlier studies have shown activation of this CYP3A4-catalyzed reaction in the presence of ANF in both a reconstituted system with DOPC [4] and in human liver microsomes [60]. Interestingly, our pilot experiments showed no measurable activating effect of 1 – 75 µM ANF on O-debenzylation of 30 µM BFC by CYP3A4-Baculosomes but rather a well-pronounced inhibition of this reaction at ANF concentrations >5 µM (data not shown).
However, in both enriched Baculosomes and in human liver microsomes Fig. 2 the addition of 25 µM ANF results in an important decrease in S50 while greatly decreasing the homotropic cooperativity with this substrate. Importantly, addition of ANF to both these systems does not affect the value of kcat for BFC O-debenzylation (Table 2), in contrast to that observed in the reconstituted micellar system [4]. Addition of GSH to human liver microsomes in the presence of ANF, while preserving these effects, results in an increase in the kcat value, similar to that observed in the absence of ANF, suggesting that ANF and GSH act independently of each other.
Figure 2.
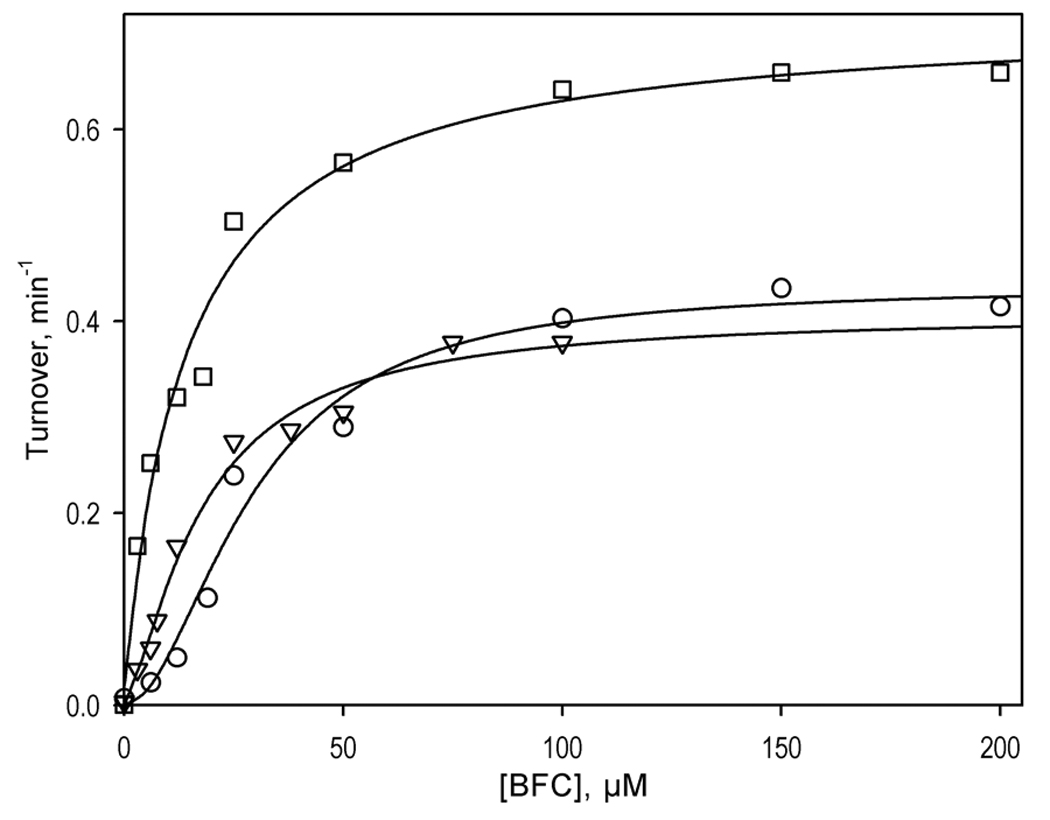
Effect of ANF on O-debenzylation of 7-BFC by human liver microsomes and effect of GSH. The titration curve obtained at no effector added is shown with circles, while the data obtained at 25 µM ANF in the absence or in the presence of 1mM GSH are represented by triangles and squares, respectively. Solid lines represent the results of the fitting of the data sets to the Hill equation.
Interactions of GSH with CYP3A4 monitored by changes in the heme protein absorbance in the Soret region
The above results suggest that the effect of GSH on cooperativity and kcat of BFC and BQ oxidation by CYP3A4 is attained through formation of a complex with the enzyme. Earlier studies showed that sulfur donor ligands, such as mercaptoethanol, 1-propanethiol, or p-chlorothiophenol, are capable of binding to the sixth coordination position of the heme iron [56, 61, 62]. The binding of the deprotonated SH-compounds (thiolates) results in unique hyperporphyrin UV-visible spectrum with the Soret band split into two bands located around 370–380 and 455–465 nm, respectively [56]. The complexes with SH-containing compounds in the protonated state (thiols) have absorbance spectra quite similar to those of low-spin ligand-free enzyme but with a broader and approximately 20% less extensive Soret absorbance band (λmax=418 nm) [56]. Since GSH exhibits a pK value of 8.7 [63], its SH-group is almost completely protonated in the conditions of our experiments (pH 7.4). As seen from Fig. 3a, the spectral changes observed upon addition of GSH to CYP3A4 are completely consistent with binding of a thiol to the heme iron. The titration results exhibit weakening and broadening of the Soret band, which is reflected in the appearance of a trough at 414 nm accompanied by two peaks at the left and right sides (at 378 and 460 nm, respectively) of the difference spectra shown in the inset. To interpret these changes quantitatively we used least-squares fitting of the P450 spectra by a linear combination of spectral standards of CYP3A4 low- and high-spin states [11] and the extinction spectra of thiol- and thiolate complexes of P450cam taken from [56]. The consistency of this approach is demonstrated by the fact that the correlation coefficient for the fitting of the spectra was always higher than 0.99, and the total concentration of the heme protein derived from such fitting does not exhibit any changes during the titration with GSH. The application of this approach shows that the addition of GSH results in a decrease in the concentration of the low-spin ligand-free CYP3A4 due to its conversion to the thiol-ligated state concomitant with some increase in the concentration of both high spin and thiolate-ligated states of the enzyme (Fig. 3b). As shown in Fig. 3c at saturation of CYP3A4 with GSH the fraction of the enzyme represented by thiol- and thiolate-complexes approaches 40%. The dependence of the fraction of GSH-ligated CYP3A4 on the concentration of GSH may be approximated by the Hill equation with an S50 of 8.6 mM and Hill coeffici7ent of 2.2. Positive cooperativity in GSH binding observed in these experiments indicates the presence of an additional high-affinity effector binding site, saturation of which with GSH promotes the interactions of this thiol with the heme iron.
Figure 3.
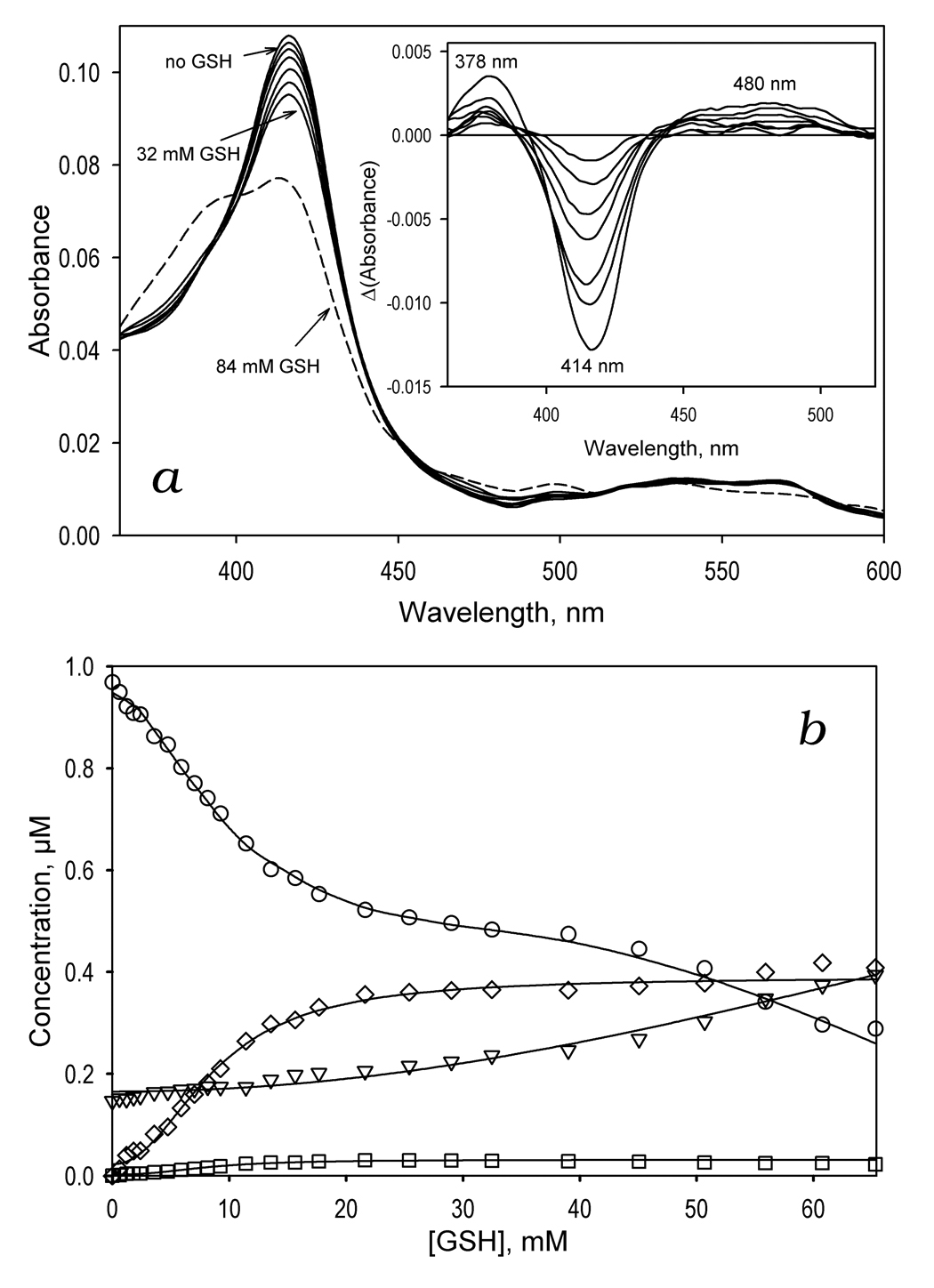
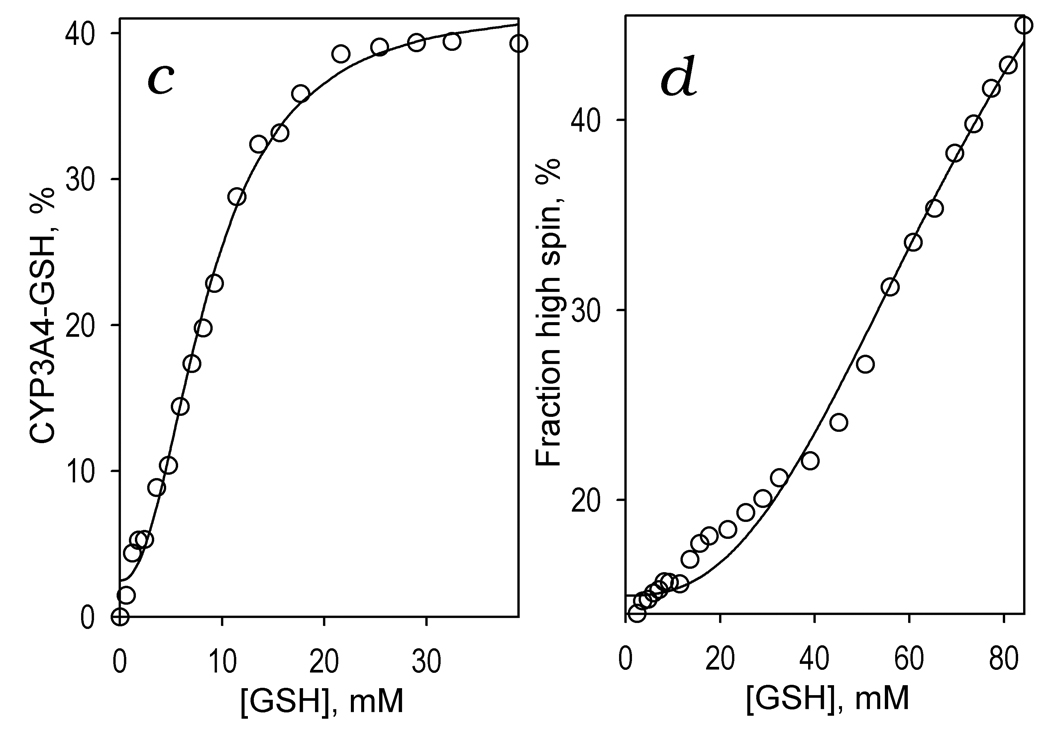
GSH-induced spectral transitions in CYP3A4 in solution. Panel a shows a series of spectra of 1 µM CYP3A4 in 0.1 M Na-Hepes buffer, pH 7.4 (25 °C) recorded at no GSH added and in the presence of GSH at the concentrations of 1.8, 4.7, 8.1, 14, 22, 32 mM (solid lines) and 84 mM (dashed line). The inset shows the difference spectra obtained by subtraction of the first spectrum of the series (no GSH added) from all subsequent spectra. Panel b illustrates the GSH-induced changes in the concentrations of the water-coordinated low spin (circles), pentacoordinated high-spin (inverted triangles), thiolate- (squares) and thiol-coordinated (diamonds) states of the heme protein. The changes in the fraction of the enzyme represented by the total of its thiolate- and thiol-coordinated states are shown in panel c. Here the solid line represent the results of the fitting of the data set by the Hill equation (S50 = 8.6 mM, n = 2.2, ΔFmax = 40 %). Changes in the high-spin fraction of the enzyme are illustrated in panel d, where the line represent the results of the fitting of the data set by the Hill equation (S50 = 76.5 mM, n = 2.5, ΔFmax = 50.5 %).
At very high concentrations of GSH (>30 mM) the spectral changes indicating the binding of GSH by coordination to the heme iron were followed by an extensive low-to-high spin shift (Fig. 3a, dashed line). This GSH-induced spin shift, which was completely reversible upon dilution of the sample, appears to signify an additional low-affinity Type-I binding event with a very strong cooperativity characterized by a Hill coefficient of 2.5 – 3 and S50 value higher than 70 mM (Fig. 3d). This additional low-affinity interaction of CYP3A4 with GSH may be therefore responsible for some decrease in kcat of BFC debenzylation observed at 8 mM GSH (Table 1).
Effect of GSH on homotropic cooperativy of substrate binding to CYP3A4 in solution
A series of spectra obtained upon titration of CYP3A4 with 1-PB in the presence of GSH are shown in Fig. 4a. Although the addition of 1-PB results in spectral changes characteristic of a substrate-induced low-to-high spin shift, there is an important difference from the prototypical Type I binding. As shown in the inset to Fig. 3a, the major trough of the difference spectra, which is located at 418 nm, has a distinct shoulder at 452 nm that is apparently indicative of the dissociation of the thiolate-ligated hyporporphyrin complex of CYP3A4 discussed above. This observation confirms the above conclusion on the binding of GSH to CYP3A4 by coordination of the SH-group of GSH to the heme iron. Similar spectral changes were also observed in the titrations of CYP3A4 with testosterone, BFC, or ANF in the presence of GSH (data not shown). The changes in the concentration of the low-spin, high-spin, and thiol(ate)-ligated forms of the enzyme shown in Fig. 4b confirm that the binding of 1-PB in the presence of GSH results in the conversion of both low-spin ligand-free and thiol(ate)-ligated states of the enzyme to the high-spin 1-PB-bound form.
Figure 4.

Titration of CYP3A4 with 1-PB in the presence of 10 mM GSH. Panel a shows a series of spectra of 1.7 µM CYP3A4 in 0.1 M Na-Hepes buffer, pH 7.4, (25 °C) containing 10 mM GSH at no substrate added and in the presence of 0.8, 2.2, 4.5, 10, 21 and 43 µM 1-PB. The inset shows the difference spectra obtained by subtraction of the first spectrum of the series (no 1-PB added) from all subsequent spectra. Panel b illustrates the 1-PB-induced changes in the concentrations of water-coordinated low spin (circles), high-spin (inverted triangles), and the thiol(ate)-corrdinate state (squares) of the heme protein. The diamonds represent the total concentration of the enzyme.
Concentration dependencies of the high spin fraction of CYP3A4 obtained in the titrations of the enzyme with 1-PB at different concentrations of GSH are shown in Fig 5a. Addition of GSH amplifies the 1-PB-induced spin shift considerably. In addition, GSH decreases the S50 from 12.4 ± 4.2 to 8.8 ± 5.0 µM (Fig. 5b). The most important observation is that GSH eliminates the homotropic cooperativity in the binding of 1-PB (Fig 5b) as evidenced by a decrease in the Hill coefficient from 1.5 ± 0.1 to 1.05 ± 0.2 upon addition of 8 mM GSH. Results of these experiments as well as those of the similar studies with three other substrates (ANF, BFC, and testosterone) are summarized in Table 3. Similar to the results with 1-PB, addition of 10 mM GSH completely eliminates the cooperativity of BFC binding. However, there is no effect on the S50 value. Most strikingly, addition of GSH has no significant effect on the interactions of CYP3A4 with ANF or testosterone.
Figure 5.
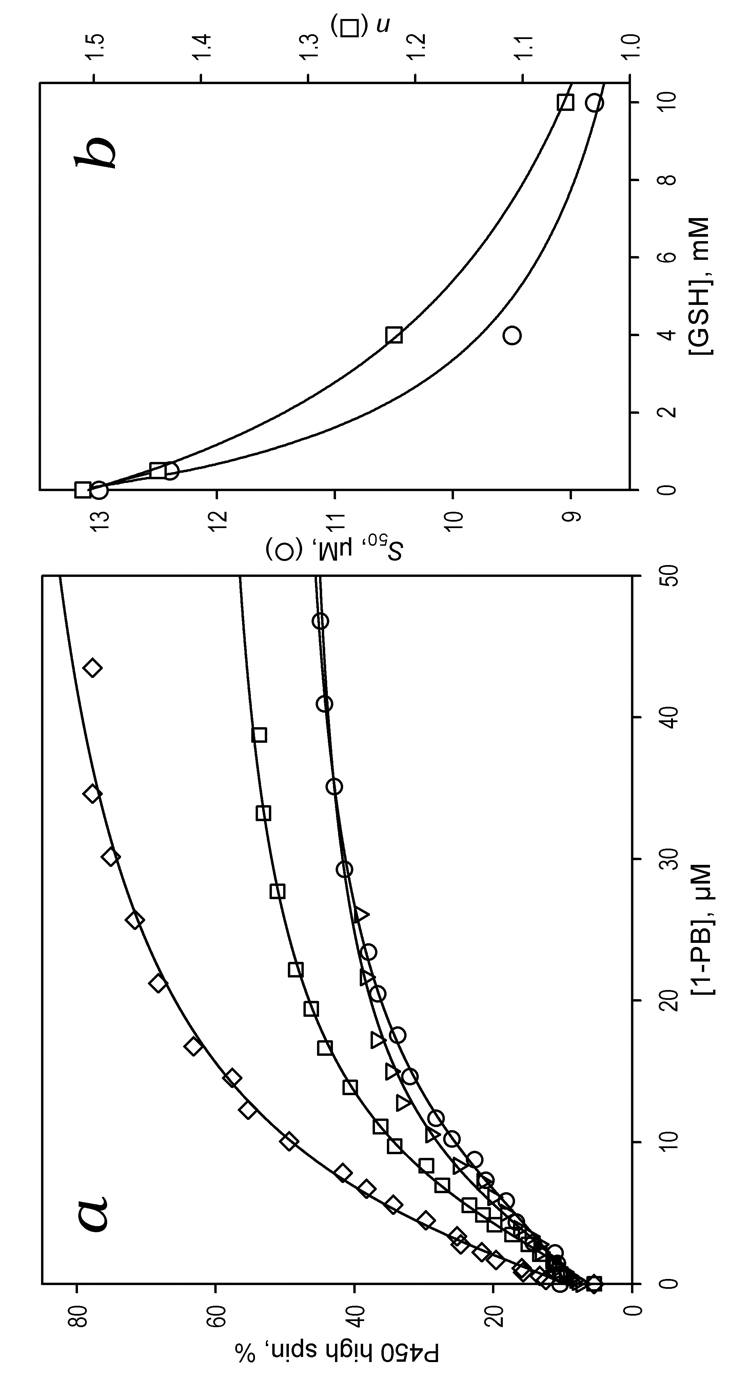
Effect of GSH on 1-PB-induced spin shift in CYP3A4. Panel a shows the changes in the high spin fraction of CYP3A4 upon addition of 1-PB at no glutathione added (circles) and in the presence of 0.5 mM (inverted triangles), 4 mM (squares), and 10 mM (diamonds) GSH. The lines represent the result of fitting of the data sets to the Hill equation. Panel b shows the dependence of the S50 value (circles) and the Hill coefficient (squares) on the glutathione concentration. The points on this plot represent the averages of two to three individual measurements. The solid lines show the fitting of these data sets with a hyperbolic (Michaelis-Menten) equation with KD app values of 1.4 ± 0.9 and 1.6 ± 1.4 mM, respectively.
Table 3.
Effect of GSH on substrate binding in CYP3A4*
| Substrate | [GSH], mM | S50. µM | n | ΔFh, %a |
|---|---|---|---|---|
| 1-PB | 0 | 13.0 ±2.6 | 1.51 ±0.09 | 49 ±6 |
| 0.5 | 12.4 ±4.2 (0.839) | 1.44 ±0.14 (0.791) | 50 ±18 (0.872) | |
| 4 | 9.5 ±2.5 (0.204) | 1.22 ±0.20 (0.133) | 53 ±8 (0.865) | |
| 10 | 8.8 ±5.7 (0.183) | 1.06 ±0.19 (0.017) | 75 ±20 (0.137) | |
| ANF | 0 | 3.87 ±0.4 | 2.0 ±0.34 | 39 ±5 |
| 0.5 | 4.54 ±0.1 (0.085) | 1.67 ±0.16 (0.254) | 43 ±1 (0.230) | |
| 4 | 4.51 ±0.9 (0.228) | 1.75 ±0.04 (0.349) | 42 ±2 (0.385) | |
| 10 | 3.59 ±1.9 (0.733) | 1.72 ±0.22 (0.327) | 40 ±3 (0.646) | |
| 7-BFC | 0 | 22.8 ±1.9 | 1.48 ±0.06 | 16 ±3 |
| 0.5 | 14.8 ±6.3 (0.137) | 1.62 ±0.01 (0.041) | 14 ±5 (0.304) | |
| 4 | 27.1 ±4.7 (0.223) | 1.29 ±0.08 (0.063) | 21 ±1 (0.097) | |
| 10 | 19.3 ±7.0 (0.430) | 1.03 ±0.05 (0.007) | 15 ±5 (0.709) | |
| TST-HPCDa | 0 | 108 ±16 | 1.19 ±0.09 | 41 ±9 |
| 0.5 | 125 ±25 (0.216) | 1.14 ±0.10 (0.540) | 33 ±5 (0.300) | |
| 4 | 136 ±14 (0.092) | 1.23 ±0.01 (0.611) | 56 ±11 (0.098) | |
| 10 | 104 ±26 (0.776) | 1.34 ±0.13 (0.114) | 47 ±11 (0.479) | |
The values given in the Table were obtained by averaging the result of 2–4 individual measurements and the “±” values show the confidence interval calculated for p = 0.05. The values in parentheses represent the p-values of Student’s T-test for the hypothesis of equality of the respective values with that observed at no GSH added.
Maximal amplitude of the substrate-induced spin shift, in percent of the total enzyme content.
Water-soluble complex of testosterone with HPCD.
Effect of GSH on the fluorescence of CYP3A4(C58,C64) labeled with BADAN
In order to probe whether the interactions of CYP3A4 with GSH result in a conformational transition similar to that described for the binding of ANF [34], we studied the effect of GSH on the fluorescence of BADAN attached to Cys-64 in our CYP3A4(C58,C64) construct. As shown in Fig. 6a addition of GSH results in a decrease in the intensity of fluorescence similar to that observed earlier upon addition of ANF [34]. The dependence of the relative intensity of fluorescence on the concentration of GSH (Fig. 6b) may be adequately approximated by a hyperbolic (Michaelis-Menten) equation. The values of the maximal amplitude of the fluorescence decrease and the apparent KD of the CYP3A4-GSH complex found by averaging the results of three individual experiments are 20 ± 14% and 1.2 ± 0.5 mM, respectively. The latter value is reasonably close to the estimates of 0.5 and 1.3 mM obtained for KD app for the effect of GSH on the Hill coefficient for O-debenzylation of BFC and BQ by CYP3A4-containing Baculosomes. We may conclude therefore that the CYP3A4 transition revealed in the effect of GSH on the fluorescence of CYP3A4(C58,C64)-BADAN is similar or directly related to the GSH-dependent transition that abolishes cooperativity of BFC and BQ oxidation.
Figure 6.
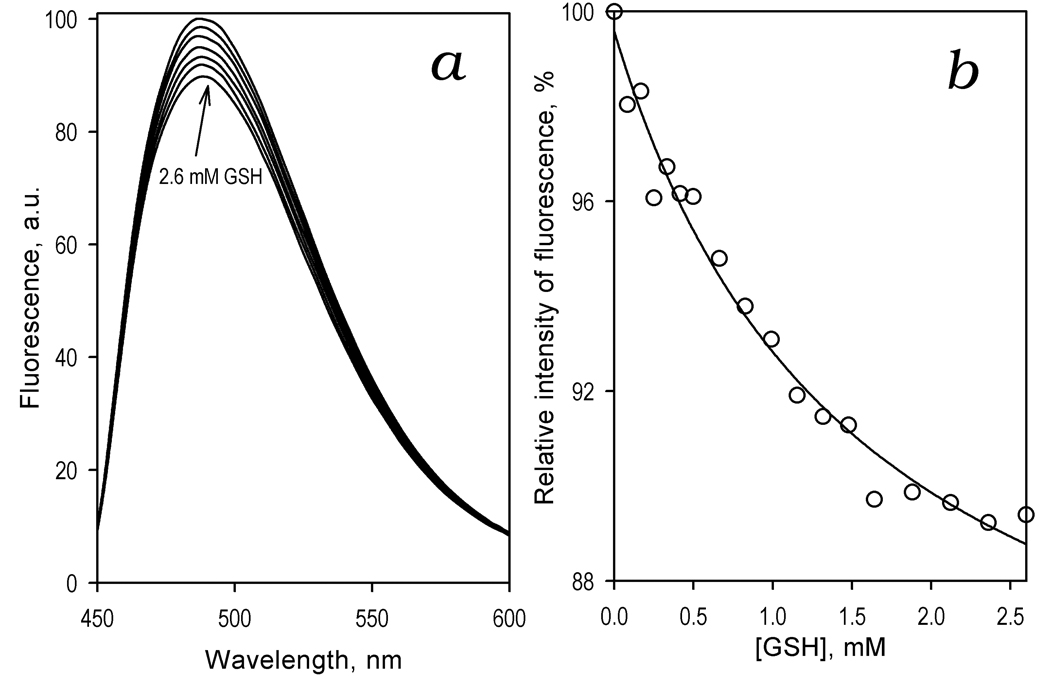
Interaction of CYP3A4(C58,C64)-BADAN with GSH monitored by the changes in the fluorescence of the probe. (a) A series of emission spectra of 1.5 µM enzyme recorded at 0, 0.17, 0.33, 0.66, 1.0, 1.5, and 2.6 mM GSH. (b) Relative fluorescence intensity versus GSH concentration. The line shows the fitting of the data sets to a hyperbolic (Michaelis-Menten) equation with KDapp = 1.6 mM and the maximal amplitude of the fluorescence decrease of 17%.
In order to probe the relationship between ANF and GSH binding we studied the effect of ANF on the fluorescence of the labeled protein in the presence of 3 mM GSH (Fig. 7). Similar to previous results in the absence of GSH [34], addition of ANF to CYP3A4(C58,C64)-BADAN in the presence of GSH results in a decrease in the fluorescence of the probe (Fig. 7a). The dependence of the relative intensity of fluorescence on the concentration of ANF (Fig. 7b) obeys the Hill equation. The values of S50, n, and the maximal amplitude of the fluorescence decrease obtained by averaging the results of three individual experiments are equal to 14.2 ± 4.6 µM, 1.8 ± 0.3, and 51.5 ± 3.1% respectively. Since these estimates are similar to the values obtained earlier in the absence of GSH (18.2 ± 0.7 µM, 1.7 ± 0.1 and 49 ± 8% respectively), we may conclude that the effects of ANF and GSH on CYP3A4 are additive, and the binding site for GSH appears to be distinct from the low-affinity ANF-binding site. However, the similarity of GSH- and ANF-induced changes in the fluorescence of the BADAN probe suggests that the mechanisms of action of these effectors are closely interrelated.
Figure 7.
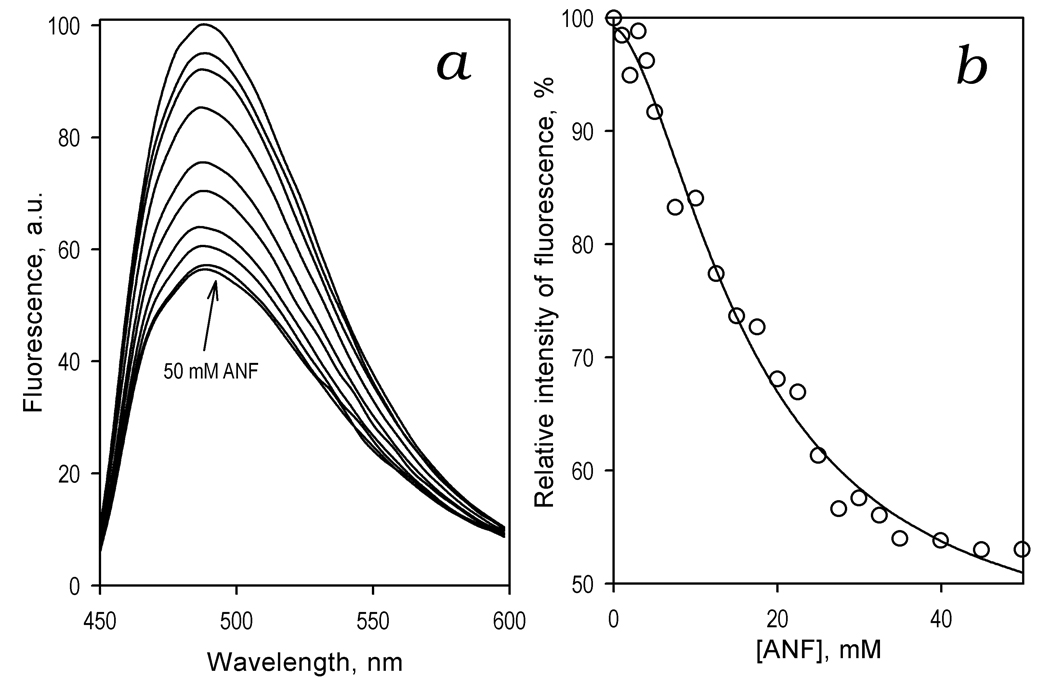
Effect of ANF on fluorescence of CYP3A4(C58,C64)BADAN in the presence of 3 mM GSH. (a) A series of emission spectra of 1.5 µM protein recorded at 0, 2, 5, 10, 15, 20, 25, 30, 35, and 50 µM ANF. (b) Relative fluorescence intensity versus substrate concentration. The line shows the fitting of the data sets to the Hill equation with S50 = 16.6 µM, n=1.7, and the maximal amplitude of the fluorescence decrease of 56%.
Discussion
Although the activating effect of GSH on CYP3A4 catalyzed reactions in model systems has been documented previously, the present study provides the first evidence of a role of glutathione as an allosteric effector of CYP3A4 in microsomal membranes. We demonstrated here that the homotropic cooperativity of human liver microsomes with the CYP3A4 marker substrate BFC is eliminated at low milimolar concentrations of GSH. As this effect is well reproduced in CYP3A4 Baculosomes with BFC as well as with another CYP3A4 marker substrate, BQ, we may conclude that the loss of cooperativity caused by GSH in liver microsomes is determined primarily by the interactions with CYP3A4. The effect of GSH is not limited to suppression of CYP3A4 cooperativity but also involves some increase in kcat. for BQ oxidation in Baculosomes and BFC metabolism in enriched Baculosomes and human liver microsomes.
The loss of cooperativity of CYP3A4 in the presence of GSH was also revealed in our studies of interactions with BFC monitored by a substrate-induced spin shift in the purified enzyme in solution. This effect was found to be even more pronounced with 1-PB, another substrate possessing homotropic cooperativity. In the case of 1-PB there was also a remarkable increase of the amplitude of the 1-PB-induced spin shift in the presence of GSH, which may indicate an effect of the latter on the degree of hydration and the openness of the heme pocket of the 1-PB-bound enzyme. It seems therefore likely that the binding of GSH displaces the equilibrium between the recently postulated “P” and “R” conformations of the enzyme [35] towards the “P” state, where water flux into the heme pocket is hampered and the high-spin state of the substrate-bound CYP3A4 is stabilized.
In contrast to 1-PB and BFC, interactions of CYP3A4 with GSH have virtually no effect on the binding of testosterone or ANF, suggesting differences in the mechanisms of binding of the substrates. This conclusion agrees with the fact that in our recent study of CYP3A4(C58,C64) labeled with fluorescent probes, only ANF but not 1-PB or testosterone had a pronounced effect on the fluorescence, and only testosterone but not 1-PB affected the fluorescence in the presence of ANF.
Titration of CYP3A4 showed that GSH is capable of binding to the sixth coordination position of the heme iron. The positive cooperativity in GSH binding observed in these experiments indicates the presence of an additional high-affinity effector binding site, saturation of which by GSH promotes the interactions of this thiol with the heme iron. This is also supported by the results of our experiments with CYP3A4(C58,C64)-BADAN, which revealed a GSH binding site with KDapp of 1.2 ± 0.5 mM, which is consistent with its role as an effector promoting the coordination of the second molecule of GSH to the heme iron characterized by an S50 of 8.6 mM and Hill coefficient of 2.2. The above KD is also consistent with the estimates of 0.8 – 1.2 mM for the apparent dissociation constant obtained from the dependencies of the Hill coefficient in BFC and BQ debenzylation on GSH concentration. Therefore, we may postulate a presence of a high-affinity effector binding site for GSH characterized with a low millimolar KD. The interaction of GSH with this site is likely to be responsible for the effect on CYP3A4 cooperativity with BFC, BQ and 1-PB and also promote the binding of GSH to the heme iron as a thiol ligand.
Importantly, the effect of ANF, a prototype heterotropic activator of CYP3A4, was found to be additive with the effect of GSH. The effects of ANF and GSH on the fluorescence of BADAN attached to Cys-64 were also additive. Therefore, the high affinity binding site for GSH appears to be distinct from the effector binding site for ANF described in our recent study with CYP3A4(C58,C64) construct [34].
Coordination of GSH to the CYP3A4 heme iron is expected to compete with the interactions of CYP3A4 with substrates. However, the addition of 1 – 10 mM GSH results in an increased affinity of CYP3A4 for 1-PB and does not affect the S50 values for the binding of BFC, testosterone, or ANF, suggesting that there is no competition between coordination of GSH to the heme iron and the interactions of CYP3A4 with these substrates. A possible explanation for this observation may be provided by the concept of persistent heterogeneity of CYP3A4 [37, 38, 64–66] caused by dissimilarity of the orientation and/or conformation of the subunits in the oligomers (aggregates) in solution [38, 66]. Lack of competition between GSH ligation to the heme and substrate binding may be caused by heterogeneity of oligomeric P450 in solution, similar to that revealed in barotropic behavior of CYP3A4 [37], kinetics of the enzyme reduction by dithionite [38], and CO-binding kinetics [64]. A suggestion that only some of the P450 molecules constituting the oligomer in solution have an orientation permitting them to bind GSH by ligation to the heme iron is consistent with our observation that, upon saturation of CYP3A4 with GSH the fraction of the enzyme represented by its thiol- and thiolate-ligated heme complexes does not exceed 40%. This is in contrast to soluble monomeric P450cam, where the binding of thiols and thiolates was shown to be competitive with substrate binding, and the complete conversion of P450cam to the thiol(ate)-bound forms was observed upon saturation of this enzyme with thiols and thiolates [56]. Since the binding of GSH binding by coordination to the heme iron reveals no competition with substrate binding in CYP3A4 in oligomers in solution, we may infer that the CYP3A4 molecules interacting with GSH do not participate in the interactions with BFC, 1-PB, ANF, or testosterone and/or do not respond to substrate binding by the low-to-high spin shift.
A similar mechanism may also take place in the membrane, where microsomal cytochromes P450 are also known to exist in equilibrium between monomers and oligomers [67–72]. However, the reversibility of oligomerization in the membrane, as opposed to virtual irreversibility in solution, would blunt the manifestations of the proposed conformational heterogeneity in the membrane-bound enzyme. Consistent with this inference, increasing concentrations of GSH in Baculosomes and human liver microsomes caused an increase in S50 with BQ and BFC, respectively, possibly resulting from competition of GSH ligation to the heme with substrate binding (Table 1).
Regardless of the mechanisms of GSH action, our study provides convincing evidence for the role of GSH as a heteroropic activator and a modulator of homotropic cooperativity in CYP3A4. This finding may indicate that the abundant evidence of CYP3A4 homo- and heterotropic cooperativity represent a manifestation of an allosteric mechanism designed to be triggered by physiological effectors, such as GSH, the concentration of which in tissues is consistent with this role. For instance, the average concentration of GSH in rat liver was shown to be in the range of 4 – 8 mM [73]. Moreover, the GSH level in primary cultures of rat hepatocytes is reported to be 40 – 60 nmol/106 cells [74, 75], which is equivalent to a concentration of 8 – 12 mM (calculated assuming the volume of a single hepatocyte cell equal to 4940 µm3 [76]). This concentration is subject to substantial changes in response to various stimuli [41, 48, 73, 77, 78]. In particular, exposure of human hepatocarcinoma Hep2 cells to organochlorine insecticides, such as DDT, decreases the GSH concentration from 4 – 5 down to 1.2 – 1.5 mM (calculated from [48] assuming the protein content in hepatocytes to be of 220 mg/g [79]). Therefore, the estimate of 0.5 – 1.3 mM for the concentration of GSH exhibiting a half-maximal effect on the Hill coefficient for O-debenzylation of BFC and BQ is certainly consistent with the physiological relevance of this newly-discovered regulatory interaction. Moreover, an inhibition of CYP3A4 at high GSH concentrations observed in our experiments may also be physiologically significant under certain conditions. The fact that the GSH concentration necessary for its maximal effect on cooperativity is decreased upon incorporation of cytochrome b5 and increase in CYP3A4 concentration in Baculosomes shows that the mechanism of action of GSH may also involve an effect on protein-protein interactions, such as CYP3A4 interactions with cytochrome b5, CPR and/or CYP3A4 oligomerization. Therefore, the response of the microsomal monooxygenase to GSH appears to be sensitive to the composition of the microsomal electron transfer chain, and a regulatory action of GSH in vivo may change upon variation of the content of cytochromes b5 and P450 in the membrane.
In view of possible physiological role of observed effect, further detailed study of the mechanism of binding of GSH to CYP3A4 and its effect on the structure and functional properties of the enzyme appear to be important. Of particular interest is the nature of the high-affinity GSH binding site and its possible overlap with the effector binding site for substrates exhibiting homotropic cooperativity. Another important question is whether the interactions of CYP3A4 with GSH affect the stoichiometry of monooxygenation. The functional role of CYP3A4 modulation by glutathione may be especially important if in addition to the effect on cooperativity and turnover rate interactions with GSH modulate the coupling of NADPH oxidation to substrate monooxygenation. A possible effect of glutathione on coupling is supported by the observation that the addition of GSH results in a remarkable activation of testosterone hydroxylation by cytochrome b5, [46], which is known to increase the coupling of microsomal P450 enzymes (see [39, 40] for review). Furthermore, an important practical inference that may be derived from our study is that the effects of GSH on CYP3A4 cooperativity and the rate of monooxygenation appear to be important factors that, which must be taken into account when extrapolating the results of in vitro studies of drug metabolism and drug-drug interactions to the situation in vivo.
Aknowledgements
The authors are grateful to Dr. Santosh Kumar (Department of Pharmacology and Toxicology, UTMB) for his help with the experiments on the effect of GSH on BQ metabolism by Baculosomes. This research was supported by NIH grant GM54995, Center grant ES06676, and Robert A. Welch Foundation grant H1458
Abbreviations used
- CYP3A4
cytochrome P450 3A4
- Hepes
N-[2-hydroxyethylpiperazine-N"-[2-ethanesulfonic acid]
- 1-PB
1-pyrenebutanol
- BFC
7-benzyloxy-4-(trifluoromethyl)coumarin
- HFC
7-hydroxy-4-(trifluoromethyl)coumarin
- BQ
7-benzyloxyquinoline
- HPCD
2-hydroxypropyl-β-cyclodextrin
- BADAN
6-bromoacetyl-2-dimethylaminonaphthalene
- GSH
reduced glutathione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shou M, Grogan J, Mancewicz JA, Krausz KW, Gonzalez FJ, Gelboin HV, Korzekwa KR. Biochemistry. 1994;33:6450–6455. doi: 10.1021/bi00187a009. [DOI] [PubMed] [Google Scholar]
- 2.Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise RA, Rettie AE, Gonzalez FJ, Tracy TS. Biochemistry. 1998;37:4137–4147. doi: 10.1021/bi9715627. [DOI] [PubMed] [Google Scholar]
- 3.Ueng YF, Kuwabara T, Chun YJ, Guengerich FP. Biochemistry. 1997;36:370–381. doi: 10.1021/bi962359z. [DOI] [PubMed] [Google Scholar]
- 4.Domanski TL, He YA, Khan KK, Roussel F, Wang Q, Halpert JR. Biochemistry. 2001;40:10150–10160. doi: 10.1021/bi010758a. [DOI] [PubMed] [Google Scholar]
- 5.Ekins S, Stresser DM, Williams JA. Trends Pharm. Sci. 2003;24:161–166. doi: 10.1016/s0165-6147(03)00049-x. [DOI] [PubMed] [Google Scholar]
- 6.Yoon MY, Campbell AP, Atkins WM. Drug Metab. Rev. 2004;36:219–230. doi: 10.1081/dmr-120033998. [DOI] [PubMed] [Google Scholar]
- 7.He YA, Roussel F, Halpert JR. Arch. Biochem. Biophys. 2003;409:92–101. doi: 10.1016/s0003-9861(02)00484-8. [DOI] [PubMed] [Google Scholar]
- 8.Hosea NA, Miller GP, Guengerich FP. Biochemistry. 2000;39:5929–5939. doi: 10.1021/bi992765t. [DOI] [PubMed] [Google Scholar]
- 9.Ngui JS, Chen Q, Shou MG, Wang RW, Stearns RA, Baillie TA, Tang W. Drug Metab. Disp. 2001;29:877–886. [PubMed] [Google Scholar]
- 10.Guengerich FP. Annu. Rev. Pharmacol. Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- 11.Fernando H, Halpert JR, Davydov DR. Biochemistry. 2006;45:4199–4209. doi: 10.1021/bi052491b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davydov DR, Femando H, Halpert JR. Biophys. Chem. 2006;123:95–101. doi: 10.1016/j.bpc.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Cameron MD, Wen B, Allen KE, Roberts AG, Schuman JT, Campbell AP, Kunze KL, Nelson SD. Biochemistry. 2005;44:14143–14151. doi: 10.1021/bi051689t. [DOI] [PubMed] [Google Scholar]
- 14.Roberts AG, Campbell AP, Atkins WM. Biochemistry. 2005;44:1353–1366. doi: 10.1021/bi0481390. [DOI] [PubMed] [Google Scholar]
- 15.Baas BJ, Denisov IG, Sligar SG. Arch. Biochem. Biophys. 2004;430:218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 16.Denisov IG, Baas BJ, Grinkova YV, Sligar SG. J. Biol. Chem. 2007;282:7066–7076. doi: 10.1074/jbc.M609589200. [DOI] [PubMed] [Google Scholar]
- 17.Wang RW, Newton DJ, Liu N, Atkins WM, Lu AYH. Drug Metab. Disp. 2000;28:360–366. [PubMed] [Google Scholar]
- 18.Galetin A, Clarke SE, Houston JB. Drug Metab. Disp. 2002;30:1512–1522. doi: 10.1124/dmd.30.12.1512. [DOI] [PubMed] [Google Scholar]
- 19.Galetin A, Clarke SE, Houston JB. Drug Metab. Disp. 2003;31:1108–1116. doi: 10.1124/dmd.31.9.1108. [DOI] [PubMed] [Google Scholar]
- 20.Funahashi T, Tanaka Y, Yamaori S, Kimura T, Matsunaga T, Ohmori S, Kageyama T, Yamamoto I, Watanabe K. Drug Metab. Pharmacokinetics. 2005;20:358–367. doi: 10.2133/dmpk.20.358. [DOI] [PubMed] [Google Scholar]
- 21.Lu P, Lin Y, Rodrigues AD, Rushmore TH, Baillie TA, Shou MG. Drug Metab. Disp. 2001;29:1473–1479. [PubMed] [Google Scholar]
- 22.Ngui JS, Tang W, Stearns R, A, Shou MG, Miller RR, Zhang Y, Lin JH, Baillie T. Drug Metab. Disp. 2000;28:1043–1050. [PubMed] [Google Scholar]
- 22.Houston B, Galetin A. Drug Metab. Rev. 2003;35:393–415. doi: 10.1081/dmr-120026870. [DOI] [PubMed] [Google Scholar]
- 24.Houston JB, Galetin A. Arch. Biochem. Biophys. 2005;433:351–360. doi: 10.1016/j.abb.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Schrag ML, Wienkers LC. Arch. Biochem. Biophys. 2001;391:49–55. doi: 10.1006/abbi.2001.2401. [DOI] [PubMed] [Google Scholar]
- 26.Egnell AC, Houston JB, Boyer CS. J. Pharm. Exp. Ther. 2005;312:926–937. doi: 10.1124/jpet.104.078519. [DOI] [PubMed] [Google Scholar]
- 27.Kenworthy KE, Clarke SE, Andrews J, Houston JB. Drug Metab. Disp. 2001;29:1644–1651. [PubMed] [Google Scholar]
- 28.Kenworthy KE, Bloomer JC, Clarke SE, Houston JB. Brit. J. Clin. Pharm. 1999;48:716–727. doi: 10.1046/j.1365-2125.1999.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atkins WM, Wang RW, Lu AYH. Chem. Res. Toxicol. 2001;14:338–347. doi: 10.1021/tx0002132. [DOI] [PubMed] [Google Scholar]
- 30.Atkins WM. Expert Opinion Drug Metab. Toxicol. 2006;2:573–579. doi: 10.1517/17425255.2.4.573. [DOI] [PubMed] [Google Scholar]
- 31.Lampe JN, Atkins WM. Biochemistry. 2006;45:12204–12215. doi: 10.1021/bi060083h. [DOI] [PubMed] [Google Scholar]
- 32.Davydov DR, Botchkareva AE, Kumar S, He YQ, Halpert JR. Biochemistry. 2004;43:6475–6485. doi: 10.1021/bi036260l. [DOI] [PubMed] [Google Scholar]
- 33.Davydov DR, Botchkareva AE, Davydova NE, Halpert JR. Biophys. J. 2005;89:418–432. doi: 10.1529/biophysj.104.058479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsalkova TN, Davydova NE, Halpert JR, Davydov DR. Biochemistry. 2007;46:106–119. doi: 10.1021/bi061944p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davydov DR, Baas BJ, Sligar SG, Halpert JR. Biochemistry. 2007;46:7852–7864. doi: 10.1021/bi602400y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekroos M, Sjögren T. Proc. Natl. Acad. Sci. USA. 2006;103:13684–13687. doi: 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davydov DR, Halpert JR, Renaud JP, Hui Bon Hoa G. Biochem. Biophys. Res. Commun. 2003;312:121–130. doi: 10.1016/j.bbrc.2003.09.247. [DOI] [PubMed] [Google Scholar]
- 38.Davydov DR, Fernando H, Baas BJ, Sligar SG, Halpert JR. Biochemistry. 2005;44:13902–13913. doi: 10.1021/bi0509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davydov DR. Trends Biochem. Sci. 2001;26:155–160. doi: 10.1016/s0968-0004(00)01749-7. [DOI] [PubMed] [Google Scholar]
- 40.Zangar RC, Davydov DR, Verma S. Toxicol. Appl. Pharm. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 41.Lu SC. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 42.Sies H. Free Radical Biol. Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- 43.Meyer AJ, Hell R. Photosynth. Res. 2005;86:435–457. doi: 10.1007/s11120-005-8425-1. [DOI] [PubMed] [Google Scholar]
- 44.Gillam EM, Baba T, Kim BR, Ohmori S, Guengerich FP. Arch. Biochem. Biophys. 1993;305:123–131. doi: 10.1006/abbi.1993.1401. [DOI] [PubMed] [Google Scholar]
- 45.Ingelman-Sundberg M, Hagbjork A, L, Ueng Y, Yamazaki FH, Guengerich FP. Biochem. Biophys. Res. Commun. 1996;221:318–322. doi: 10.1006/bbrc.1996.0593. [DOI] [PubMed] [Google Scholar]
- 46.Kim BR, Kim DH. Biochem. Biophys. Res. Commun. 1998;242:209–212. doi: 10.1006/bbrc.1997.7861. [DOI] [PubMed] [Google Scholar]
- 47.Shaw PM, Hosea NA, Thompson DV, Lenius JM, Guengerich FP. Arch. Biochem. Biophys. 1997;348:107–115. doi: 10.1006/abbi.1997.0378. [DOI] [PubMed] [Google Scholar]
- 48.Dehn PF, Allen-Mocherie S, Karek J, Thenappan A. Toxicol. in vitro. 2005;19:261–273. doi: 10.1016/j.tiv.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 49.French JS, Coon MJ. Arch. Biochem. Biophys. 1979;195:565–577. doi: 10.1016/0003-9861(79)90383-7. [DOI] [PubMed] [Google Scholar]
- 50.Davydov DR, Kumar S, Halpert JR. Biochem. Biophys. Res. Commun. 2002;294:806–812. doi: 10.1016/S0006-291X(02)00565-X. [DOI] [PubMed] [Google Scholar]
- 51.Masters BS, Okita RT. Pharm. Ther. 1980;9:227–244. doi: 10.1016/s0163-7258(80)80020-9. [DOI] [PubMed] [Google Scholar]
- 52.Yim SK, Yun SJ, CH Y. J. Biochem. Mol. Biol. 2004;37:629–633. doi: 10.5483/bmbrep.2004.37.5.629. [DOI] [PubMed] [Google Scholar]
- 53.Yamazaki H, Nakajima M, Nakamura M, Asahi S, Shimada N, Gillam EMJ, Guengerich FP, Shimada T, Yokoi T. Drug Metab. Disp. 1999;27:999–1004. [PubMed] [Google Scholar]
- 54.Davydov DR, Deprez E, Hui Bon Hoa G, Knyushko TV, Kuznetsova GP, Koen YM, Archakov AI. Arch. Biochem. Biophys. 1995;320:330–344. doi: 10.1016/0003-9861(95)90017-9. [DOI] [PubMed] [Google Scholar]
- 55.Renaud JP, Davydov DR, Heirwegh KPM, Mansuy D, Hui Bon Hoa G. Biochem. J. 1996;319:675–681. doi: 10.1042/bj3190675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sono M, Andersson LA, Dawson JH. J. Biol. Chem. 1982;257:8308–8320. [PubMed] [Google Scholar]
- 57.Estabrook RW, Franklin MR, Cohen B, Shigamatzu A, Hildebrandt A. Metabolism. 1971;20:187–199. doi: 10.1016/0026-0495(71)90091-6. [DOI] [PubMed] [Google Scholar]
- 58.Karyakin AV, Davydov DR. Vestnik Akad. Med. Nauk SSSR. 1988;1988(1):53–62. [PubMed] [Google Scholar]
- 59.Watanabe J, Asaka Y, Fujimoto S, Kanamura S. J. Histochem. Cytochem. 1993;41:43–49. doi: 10.1177/41.1.8417111. [DOI] [PubMed] [Google Scholar]
- 60.Stresser DM, Turner SD, Blanchard AP, Miller VP, Crespi CL. Drug Metab. Disp. 2002;30:845–852. doi: 10.1124/dmd.30.7.845. [DOI] [PubMed] [Google Scholar]
- 61.Ullrich V, Nastainczyk W, Ruf HH. Biochemi. Soc. Trans. 1975;3:803–807. [Google Scholar]
- 62.Nastainzcyk W, Ruf H, Ullrich V. Eur. J. Biochem. 1975;60:615–620. doi: 10.1111/j.1432-1033.1975.tb21039.x. [DOI] [PubMed] [Google Scholar]
- 63.Gough JD, Williams RH, Donofrio AE, Lees WJ. J. Am. Chem. Soc. 2002;124:3885–3892. doi: 10.1021/ja016938p. [DOI] [PubMed] [Google Scholar]
- 64.Koley AP, Buters JT, Robinson RC, Markowitz A, Friedman FK. J. Biol, Chem. 1995;270:5014–5018. doi: 10.1074/jbc.270.10.5014. [DOI] [PubMed] [Google Scholar]
- 65.Koley AP, Buters JTM, Robinson RC, Markowitz A, Friedman FK. J. Biol. Chem. 1997;272:3149–3152. doi: 10.1074/jbc.272.6.3149. [DOI] [PubMed] [Google Scholar]
- 66.Fernando H, Davydov DR, Chin CC, Halpert JR. Arch. Biochem. Biophys. 2007;460:129–140. doi: 10.1016/j.abb.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Greinert R, Finch SA, Stier A. Bioscience Reports. 1982;2:991–994. doi: 10.1007/BF01122166. [DOI] [PubMed] [Google Scholar]
- 68.Kawato S, Gut J, Cherry RJ, Winterhalter KH, Richter C. J. Biol. Chem. 1982;257:7023–7029. [PubMed] [Google Scholar]
- 69.Schwarz D, Pirrwitz J, Meyer HW, Coon MJ, Ruckpaul K. Biochem. Biophys. Res. Commun. 1990;171:175–181. doi: 10.1016/0006-291x(90)91373-z. [DOI] [PubMed] [Google Scholar]
- 70.Alston K, Robinson RC, Park SS, Gelboin HV, Friedman FK. J. Biol. Chem. 1991;266:735–739. [PubMed] [Google Scholar]
- 71.Szczesna-Skorupa E, Mallah B, Kemper B. J. Biol. Chem. 2003;278:31269–31276. doi: 10.1074/jbc.M301489200. [DOI] [PubMed] [Google Scholar]
- 72.Schwarz D, Chernogolov L, Kisselev P. Biochemistry. 1999;38:9456–9464. doi: 10.1021/bi990584l. [DOI] [PubMed] [Google Scholar]
- 73.Huang ZZ, Li HY, Cai JX, Kuhlenkamp J, Kaplowitz N, Lu SC. Hepatology. 1998;27:147–153. doi: 10.1002/hep.510270123. [DOI] [PubMed] [Google Scholar]
- 74.Lu SC, Sun WM, Yi J, Ookhtens M, Sze G, Kaplowitz N. J. Clin. Inv. 1996;97:1488–1496. doi: 10.1172/JCI118571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eisenbrand GS, Golzer P. Chem. Res. Toxicol. 1995;8:40–46. doi: 10.1021/tx00043a005. [DOI] [PubMed] [Google Scholar]
- 76.Weibel ER, Staubli W, Gnagi HR, Hess FA. J. Cell Biol. 1969;42:68–91. doi: 10.1083/jcb.42.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Voehringer DW. Free Radical Biol. Med. 1999;27:945–950. doi: 10.1016/s0891-5849(99)00174-4. [DOI] [PubMed] [Google Scholar]
- 78.Mceligot AJ, Yang S, Meyskens FL. Ann. Rev. Nutr. 2005;25:261–295. doi: 10.1146/annurev.nutr.25.050304.092633. [DOI] [PubMed] [Google Scholar]
- 79.Wibo M, Amar-Costesec A, Berthet J, Beayfay H. J. Cell Biol. 1971;51:52–71. doi: 10.1083/jcb.51.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]