

Abstract
Background
Bast fibres from the phloem tissues of flax are scientifically interesting and economically useful due in part to a dynamic system of secondary cell wall deposition. To better understand the molecular mechanisms underlying the process of cell wall development in flax, we extracted proteins from individually dissected phloem fibres (i.e. individual cells) at an early stage of secondary cell wall development, and compared these extracts to protein extracts from surrounding, non-fibre cells of the cortex, using fluorescent (DiGE) labels and 2D-gel electrophoresis, with identities assigned to some proteins by mass spectrometry.
Results
The abundance of many proteins in fibres was notably different from the surrounding non-fibre cells of the cortex, with approximately 13% of the 1,850 detectable spots being significantly (> 1.5 fold, p ≤ 0.05) enriched in fibres. Following mass spectrometry, we assigned identity to 114 spots, of which 51 were significantly enriched in fibres. We observed that a K+ channel subunit, annexins, porins, secretory pathway components, β-amylase, β-galactosidase and pectin and galactan biosynthetic enzymes were among the most highly enriched proteins detected in developing flax fibres, with many of these proteins showing electrophoretic patterns consistent with post-translational modifications.
Conclusion
The fibre-enriched proteins we identified are consistent with the dynamic process of secondary wall deposition previously suggested by histological and biochemical analyses, and particularly the importance of galactans and the secretory pathway in this process. The apparent abundance of β-amylase suggests that starch may be an unappreciated source of materials for cell wall biogenesis in flax bast fibres. Furthermore, our observations confirm previous reports that correlate accumulation proteins such as annexins, and specific heat shock proteins with secondary cell wall deposition.
Background
Flax (Linum usitatissimum L.) has attracted human attention since the beginning of agriculture [1,2]. This is due in part to the unusual properties of the bast (i.e. phloem) fibres, which because of their great length and high tensile strength have found use in textiles and many other products [3]. Fibre length is achieved almost entirely through intrusive growth, which is a process limited to very few cell types in plants [4,5]. The elongation stage is succeeded by a dynamic process of secondary wall deposition, in which a matrix of galactose-rich polymer in the nascent wall is gradually and centripetally replaced by highly crystalline cellulose [6]. Because secondary wall deposition increases the tensile strength of cells, fibres which have undergone even the very first stages of cell wall thickening can be distinguished mechanically by their resistance to breakage at the "snap-point" of the stem [7]. The snap-point thus defines an important developmental transition from cell elongation to cell wall thickening.
Previously, we and others have produced libraries of cDNAs from fibre-bearing peels of flax and hemp stems [8,9]. In addition to containing bast fibres at various stages of development, these peels also contained many other cell types, including those associated with cambium and transport phloem. Analysis of these libraries by cDNA microarray hybridization and other techniques identified distinct patterns of expression of transcripts of polysaccharide-related enzymes in stem peels during fibre elongation and cell wall deposition. However, due to inherent technical and biological limitations, it is known that in many circumstances, abundance of transcripts and proteins for a given gene may not be highly correlated [10,11]. This well-established limitation on the biological relevance of transcriptome analysis led us to complement our previous work with a survey of the proteins present in developing flax fibres during the onset of secondary wall deposition. This is similar to a proteomics approaches used to study secondary cell wall development of other cell types in other species [12-16]. For this study of the proteome, we also increased the specificity of our analysis by extracting proteins from phloem fibres that had been individually dissected from the snap point of growing stems, and comparing their abundance to proteins in the surrounding, non-fibre cells of the cortex from the same stems. The objective of this study is therefore to identify those proteins that contribute to the interesting pattern of cell wall deposition in flax fibres.
Results and discussion
Separation of fibre and non-fibre proteins
To increase our understanding of the proteins that contribute to the unique properties of flax bast fibres, we extracted proteins from ultimate fibres (i.e. individual cells) dissected from the snap-point region of vegetative stems (21–24 days post germination) (Figure 1). The snap-point is the stem region in which secondary wall deposition begins [7]. We also collected the surrounding non-fibre cells (consisting predominantly of parenchyma, sieve elements, and companion cells) from the cortex of the snap-point. Throughout the remainder of this report, will refer to the ultimate bast fibres we collected from the snap-point as simply "fibres", and the surrounding, non-fibre cells of the cortex as the "non-fibre fraction". By labelling proteins from fibres and the non-fibre fraction with contrasting fluorescent dyes, and separating the mixture of the two samples simultaneously using 2D gel electrophoresis (DiGE), we were able to identify proteins that were more abundant in fibres as compared to the non-fibre fraction (Figure 2).
Figure 1.

A typical flax plant at the time of fibre extraction. (A) The 3 cm region of the stem from which fibres were dissected is indicated by the bracket. (B) Detail of a transverse section of fresh stem tissues at the time of harvest. This hand section was obtained from just below the snap-point to demonstrate the arrangement of tissues within the stem, i.e. transverse sectioning was not used when obtaining tissues for protein analysis. A bracket indicates the region of the cortex from which the fibre and non-fibre fractions would be obtained. The position of representative fibres within the cortex is shown by arrowheads. The scale bar is 100 μm. (C) Stem tissues during dissection. Fibres from which surrounding, non-fibres cells been partially removed are indicated by arrowheads. A fully dissected fibre, comprising a single cell is indicated by the arrow. This fibre is representative of the cells from which proteins were extracted. The scale bar is 100 μm.
Figure 2.

Representative analytical DiGE gel. Proteins extracted from fibre and surrounding non-fibre tissues were fluorescently labeled with red and green dyes, respectively, and were mixed then separated simultaneously using 2D gel electrophoresis. Labels correspond to protein spot numbers used in Table 1 and in the text. The pH range of the first dimension separation is from 3 (left) to 10 (right).
In each of four replicate gels, we detected an average of 1850 distinct protein spots from fibres, and 1695 spots from the non-fibre fraction. In total, 558 protein spots differed in fluorescent signal intensity by at least 1.5 fold (p ≤ 0.05) between the samples, with 246 spots (13% of total detected) enriched in fibres and 312 spots (18% of total) enriched in the non-fibre fraction (Figure 3). The distinctive protein profiles of fibres and the non-fibre fraction were also evident from visual inspection of the DiGE gel image (Figure 2). Phloem fibres therefore appear to express a complement of proteins that is distinct from surrounding cell types in the stem.
Figure 3.
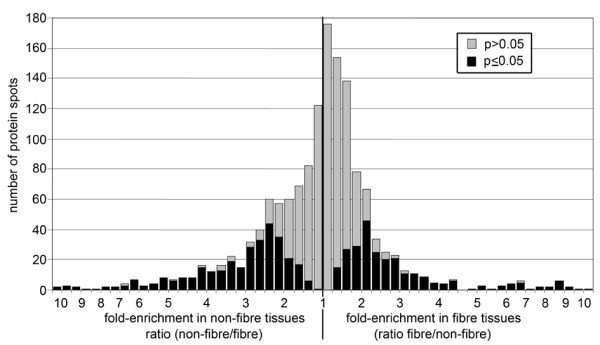
Frequency distribution of mean intensity ratios for all spots. A mean ratio near 1 meant the spot was found in equal abundance in both tissues; spots represented to the right of this point on the axis had higher signal intensity in fibre tissues, while spots represented to the left were more intense in non-fibre tissues. The grey and black regions of each bar show the portion of spots for which p > 0.05 and p ≤ 0.05, respectively, in a t-test of the significance of differences in intensity between fibre and non-fibre tissues.
Protein identification by LC/MS
We picked 190 protein spots that were enriched in fibre samples for identification by mass spectrometry. Spots were selected based on criteria of large spot volume, high fold-enrichment of signals, and well-focused spot morphology. For comparison, we also selected an additional 50 spots that were enriched in non-fibre fractions or that were similarly abundant in both types of protein samples. Although the patterns of fold-enrichment that we report were reproducible within the statistical parameters indicated (Table 1), individual ratios should not be extrapolated quantitatively to whole proteins, in part because some proteins may be represented by more than one spot.
Table 1.
Protein identities based on peptide matches to Genbank protein databases
| fold enrich.b | ||||||||
| spot ID# | func. cat.a | protein identity | Genbank ID | fibre | non-fibre | p-valuec | Mowse score | pep. countd |
| 2 | C&E | aconitate hydratase | 4586021 | 1.5 | 0.14 | 64 | 2 | |
| 3 | C&E | aconitate hydratase | 4586021 | 1.5 | 0.08 | 68 | 2 | |
| 17 | C&E | β-amylase | 1771782 | 8.8 | < 0.01 | 46 | 2 | |
| 39 | C&E | ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit | 168312 | 1.5 | 0.25 | 85 | 2 | |
| 40 | C&E | ribulose-1,5-bisphosphate carboxylase, large subunit | 168312 | 2.0e | 0.08 | 180 | 4 | |
| 44 | C&E | ribulose-1,5-bisphosphate carboxylase, large subunit | 1834444 | 6.1 | < 0.01 | 129 | 5 | |
| 45 | C&E | ribulose-1,5-bisphosphate carboxylase, large subunit | 2687483 | 5.4 | < 0.01 | 130 | 4 | |
| 46 | C&E | ribulose-1,5-bisphosphate carboxylase, large subunit | 6983900 | 2.9 | < 0.01 | 232 | 6 | |
| 47 | C&E | ribulose-1,5-bisphosphate carboxylase, large subunit | 1817560 | 3.3 | < 0.01 | 250 | 5 | |
| 48 | C&E | enolase | 9581744 | 1.1 | 0.65 | 265 | 7 | |
| 49 | C&E | enolase | 8919731 | 1.1 | 0.93 | 158 | 3 | |
| 50 | C&E | enolase | 9581744 | 3.4 | 0.02 | 206 | 6 | |
| 51 | C&E | ribulose-1,5-bisphosphate carboxylase, large subunit | 4098530 | 2.8 | 0.04 | 103 | 4 | |
| 57 | C&E | fumarate hydratase | 108708038 | 2.5 | 0.01 | 83 | 2 | |
| 58 | C&E | fumarate hydratase | 15226618 | 1.6 | 0.33 | 100 | 4 | |
| 59 | C&E | 6-phosphogluconate dehydrogenase | 2529229 | 1.5 | 0.19 | 100 | 3 | |
| 63 | C&E | citrate synthase | 11066954 | 3.7 | < 0.01 | 123 | 4 | |
| 68 | C&E | phosphoglycerate kinase | 1161600 | 1.2 | 0.56 | 257 | 4 | |
| 71 | C&E | phosphoglycerate kinase | 92872324 | 1.7 | 0.06 | 426 | 7 | |
| 72 | C&E | ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit | 66735801 | 96 | 3 | |||
| 73 | C&E | rubisco activase | 13430332 | 6.1 | < 0.01 | 70 | 3 | |
| 74 | C&E | rubisco activase | 170129 | 5.2 | < 0.01 | 61 | 3 | |
| 75 | C&E | phosphoglycerate kinase | 3328122 | 2.9 | 0.02 | 250 | 6 | |
| 77 | C&E | fructose-bisphosphate aldolase | 15227981 | 1.1 | 0.82 | 155 | 3 | |
| 78 | C&E | fructose-bisphosphate aldolase | 20204 | 2.4 | 0.03 | 102 | 2 | |
| 79 | C&E | fructose-bisphosphate aldolase | 15227981 | 1.1 | 0.6 | 116 | 2 | |
| 80 | C&E | fructose-bisphosphate aldolase | 20204 | 1.3 | 0.04 | 177 | 3 | |
| 81 | C&E | rubisco activase | 4261547 | 2.2 | 0.03 | 60 | 2 | |
| 82 | C&E | succinate-CoA ligase | 15225353 | 2.3 | 0.02 | 253 | 5 | |
| 83 | C&E | glyceraldehyde-3-phosphate dehydrogenase | 120666 | 2.6 | 0.01 | 76 | 2 | |
| 86 | C&E | glyceraldehyde-3-phosphate dehydrogenase | 3023813 | 1.1 | 0.49 | 71 | 3 | |
| 87 | C&E | glyceraldehyde-3-phosphate dehydrogenase | 74419004 | 3.8 | < 0.01 | 215 | 6 | |
| 90 | C&E | malate dehydrogenase | 18297 | 1.6 | 0.17 | 241 | 4 | |
| 91 | C&E | malate dehydrogenase | 18297 | 1.4 | 0.26 | 138 | 4 | |
| 92 | C&E | malate dehydrogenase | 10334493 | 3.3 | < 0.01 | 296 | 7 | |
| 93 | C&E | fructokinase | 31652274 | 6.7 | < 0.01 | 142 | 5 | |
| 94 | C&E | fructokinase | 31652274 | 2.2 | < 0.01 | 154 | 3 | |
| 96 | C&E | kinase/ribokinase, potential fructokinase | 15224669 | 2 | 0.01 | 208 | 8 | |
| 1 | ATP | AAA-ATPase | 86212372 | 1.6 | 0.24 | 322 | 10 | |
| 7 | ATP | ATPase, transitional endoplasmic reticulum | 7378614 | 1.2e | 0.65 | 101 | 4 | |
| 24 | ATP | vacuolar proton-ATPase | 50251203 | 2.6 | 0.02 | 585 | 13 | |
| 31 | ATP | ATP binding | 15221770 | 1 | 0.87 | 100 | 4 | |
| 35 | ATP | F1 ATPase | 12986 | 1.6 | 0.05 | 143 | 6 | |
| 40 | ATP | ATP synthase β subunit | 21684923 | 2.0e | 0.08 | 192 | 4 | |
| 42 | ATP | ATP synthase β subunit | 19685 | 1 | 0.99 | 675 | 12 | |
| 43 | ATP | ATP synthase β subunit | 56784991 | 1.8 | 0.06 | 307 | 7 | |
| 99 | ATP | F1-ATPase gammma subunit | 303626 | 1 | 0.66 | 84 | 3 | |
| 105 | ATP | vacuolar V-H+ATPase subunit E | 5733660 | 1.8 | 0.01 | 53 | 2 | |
| 106 | ATP | vacuolar V-H+ATPase subunit E | 5733660 | 1.1 | 0.82 | 100 | 4 | |
| 12 | CWP | β-galactosidase | 115437888 | 8.4 | < 0.01 | 43 | 3 | |
| 13 | CWP | β-galactosidase | 3641863 | 8.9 | < 0.01 | 42 | 2 | |
| 14 | CWP | β-galactosidase | 3641863 | 5.4 | < 0.01 | 105 | 5 | |
| 15 | CWP | β-galactosidase | 3641863 | 8.8 | < 0.01 | 96 | 5 | |
| 16 | CWP | β-galactosidase | 34913072 | 9.3 | < 0.01 | 72 | 4 | |
| 18 | CWP | MUCILAGE-MODIFIED 4 | 42562732 | 4.1 | < 0.01 | 57 | 2 | |
| 19 | CWP | rhamnose biosynthetic enzyme | 108707484 | 6.6 | < 0.01 | 100 | 6 | |
| 27 | CWP | phosphoglucomutase | 12585309 | 1.8 | 0.15 | 170 | 4 | |
| 28 | CWP | phosphoglucomutase | 6272281 | 3.7 | 0.02 | 122 | 5 | |
| 36 | CWP | UDP-glucose pyrophosphorylase | 6136112 | 1.4 | 0.37 | 82 | 3 | |
| 38 | CWP | UDP-glucose pyrophosphorylase | 82659609 | 3.5 | 0.01 | 166 | 6 | |
| 41 | CWP | UDP-glucose pyrophosphorylase | 9280626 | 1.6e | 0.1 | 129 | 6 | |
| 64 | CWP | β-galactosidase | 3641863 | 1.2 | 0.51 | 72 | 2 | |
| 76 | CWP | NAD-dependent epimerase/dehydratase (UXS6) | 15226950 | 6.1 | < 0.01 | 109 | 4 | |
| 88 | CWP | UDP-glucose 4-epimerase | 12643850 | 1.1 | 0.84 | 60 | 2 | |
| 101 | CWP | GDP-4-keto-6-deoxy-D-mannose-3,5-epimerase-4-reductase | 12324315 | 2.3 | < 0.01 | 155 | 3 | |
| 104 | CWP | dTDP-D-glucose 4,6-dehydratase-like | 50253123 | 3 | < 0.01 | 56 | 2 | |
| 9 | 1C | Met synthase | 77556633 | 2 | < 0.01 | 222 | 6 | |
| 10 | 1C | Met synthase | 8439545 | 2.2 | < 0.01 | 105 | 3 | |
| 41 | 1C | S-adenosyl-L-homocysteine hydrolase | 1710838 | 1.6e | 0.1 | 174 | 5 | |
| 53 | 1C | serine hydroxymethyltransferase | 11762130 | 2.2 | 0.02 | 129 | 4 | |
| 60 | 1C | Met adenosyltransferase | 37051117 | 2.1 | 0.02 | 94 | 4 | |
| 55 | MemT | GDP dissociation inhibitor | 8439465 | 2 | 0.13 | 212 | 5 | |
| 56 | MemT | GDP dissociation inhibitor | 8439465 | 1.9 | 0.08 | 158 | 4 | |
| 95 | MemT | K+ channel β-subunit | 15219795 | 8.6 | 0.01 | 132 | 4 | |
| 102 | MemT | 34 kDa outer mitochondrial membrane porin-like protein | 83283993 | 1.7 | 55 | 2 | ||
| 103 | MemT | 36kDa porin I | 515358 | 3.9 | < 0.01 | 104 | 4 | |
| 5 | C&S | myosin heavy chain | 108710464 | 2.5 | 0.05 | 46 | 2 | |
| 6 | C&S | myosin heavy chain | T00727 | 3.6 | 0.01 | 48 | 2 | |
| 22 | C&S | dynamin central region | 92891191 | 3.1 | 0.09 | 83 | 3 | |
| 25 | C&S | dynamin-like | 21593776 | 1 | 0.77 | 143 | 4 | |
| 37 | C&S | β-tubulin | 295851 | 1.8 | 0.06 | 161 | 6 | |
| 52 | C&S | tubulin/FtsZ family, GTPase domain | 62734655 | 1.7 | 0.06 | 367 | 12 | |
| 69 | C&S | actin | 32186910 | 3.1 | 0.01 | 281 | 8 | |
| 70 | C&S | actin | 15242516 | 1.5 | 0.21 | 459 | 12 | |
| 4 | P&AA | elongation factor EF-2 | 6056373 | 2.5 | 0.02 | 40 | 3 | |
| 7 | P&AA | ClpC protease | 4105131 | 1.2e | 81 | 4 | ||
| 8 | P&AA | ClpC protease | 18423214 | 1.7 | 0.06 | 286 | 11 | |
| 11 | P&AA | HSP 90 | 1708314 | 1.7 | 0.01 | 312 | 10 | |
| 20 | P&AA | HSP 70-3 | 38325815 | 1.9 | 0.08 | 404 | 11 | |
| 21 | P&AA | HSP 70 | 62733235 | 1.7 | 0.11 | 612 | 13 | |
| 23 | P&AA | HSP 70 | 22636 | 2 | 0.04 | 100 | 3 | |
| 29 | P&AA | chaperonin CPN60-1 | 108706134 | 2.7 | 0.04 | 139 | 6 | |
| 30 | P&AA | chaperonin CPN60-1 | 108706134 | 1.5 | 0.04 | 327 | 7 | |
| 32 | P&AA | HSP 60 | 16221 | 2.1 | 0.02 | 140 | 4 | |
| 54 | P&AA | eukaryotic elongation factor 1A | 24371059 | 2.3 | 0.02 | 227 | 7 | |
| 61 | P&AA | 26S protease regulatory subunit | 1709798 | 2.1 | < 0.01 | 85 | 3 | |
| 62 | P&AA | translation initiation factor eIF-4A | 475221 | 1.6 | 0.09 | 262 | 9 | |
| 65 | P&AA | 26S proteasome subunit P45 | 92870338 | 1.9 | 0.11 | 90 | 3 | |
| 66 | P&AA | aminomethyltransferase | 3334196 | 3.7 | < 0.01 | 67 | 2 | |
| 67 | P&AA | elongation factor-1 alpha | 396134 | 1.2 | 0.5 | 54 | 3 | |
| 72 | P&AA | glutamine synthetase | 121341 | 1.7e | 0.26 | 119 | 4 | |
| 84 | P&AA | P0 ribosomal protein | 1143507 | 2.5 | < 0.01 | 155 | 3 | |
| 89 | P&AA | glutamate-ammonia ligase | 99698 | 1.2e | 0.5 | 65 | 3 | |
| 114 | P&AA | eukaryotic translation initiation factor 5A | 8778393 | 2 | 0.04 | 91 | 2 | |
| 26 | misc | nucleolar protein NOP5 | 108708132 | 1.4 | 0.37 | 47 | 2 | |
| 33 | misc | ferric leghemoglobin reductase | 5823556 | 1.6 | 0.2 | 124 | 4 | |
| 34 | misc | calreticulin | 3288109 | 1 | 0.9 | 78 | 3 | |
| 85 | misc | peroxidase | 1389835 | 2.4 | 0.03 | 214 | 7 | |
| 89 | misc | type IIIa membrane protein cp-wap13 | 2218152 | 58 | 3 | |||
| 97 | misc | annexin | 1429207 | 2.2 | 0.01 | 146 | 4 | |
| 98 | misc | annexin | 1429207 | 4.1 | 0.03 | 71 | 2 | |
| 100 | misc | enoyl-ACP reductase | 2204236 | 2.1 | 0.01 | 44 | 2 | |
| 107 | misc | protein kinase C inhibitor | 20062 | 2.8 | < 0.01 | 97 | 5 | |
| 108 | misc | 14-3-3 protein | 695767 | 2.7 | 0.01 | 44 | 3 | |
| 109 | misc | guanine nucleotide regulatory protein | 395072 | 1.5 | 0.31 | 64 | 2 | |
| 110 | misc | NAD(P)H dependent 6'-deoxychalcone synthase | 18728 | 1.1 | 0.82 | 56 | 3 | |
| 111 | misc | inorganic pyrophosphatase | 48927683 | 2.8 | 0.02 | 148 | 3 | |
| 112 | misc | maturase K | 33332553 | 3.4 | 0.01 | 55 | 2 | |
| 113 | misc | CBS (cystathionine β-synthase) domain-containing | 15238284 | 1.6 | 0.1 | 92 | 2 | |
a) Functional category: ATPases (ATP); Cell wall polysaccharide metabolism (CWP); Cytoskeleton and secretion (C&S); Membrane transport (MemT); Miscellaneous (misc); One-carbon metabolism (1C); Primary carbon and energy metabolism (C&E); Protein and amino acid metabolism (P&AA). Only the highest scoring protein for each spot is categorized.
b) Fold enrichment in fibre tissues or non-fibre tissues as compared to the other tissue type, expressed as linear ratio of mean signal intensities.
c) P-value for a t-test of significant differences in mean signal intensities between fibre and non-fibre tissues.
d) Peptide count, i.e. the number of peptides per spot that match the Genbank ID shown.
e) Spots in which multiple proteins were identified. The intensity ratios shown may be due to differences in abundance of more than one protein.
Protein identities are sorted by functional category, in the order in which each category is presented in the text, and then alphabetically within each functional category. Additional data (including peptide sequences) is provided in Additional File 1.
We subjected a total of 240 spots to analysis by LC/MS. Of these, 126 spots produced spectra that could not be assigned to existing sequences, while spectra from the remaining 114 spots produced significant matches (i.e. MOWSE scores 40–675; two or more peptides matched per spot) to predicted spectra from Genbank protein databases (Table 1). Four spots (#7, #41, #72, #89) contained predicted peptides that matched more than one distinct protein, indicating the presence of multiple proteins in some spots on the gel. Of the spots to which we assigned protein identities, 76 were enriched by at least 1.5 fold (i.e. 1.5×) in fibre samples, and 51 of these were statistically more abundant (p ≤ 0.05) in fibres than the non-fibre fraction. Conversely, we were able to assign identity to 17 spots enriched 1.5-fold or more in the non-fibre fraction; at least seven of these were associated with photosynthesis (spots #44–#47, #73, #74, #81). Because photosynthesis is a process expected to dominate metabolism in the non-fibre fraction, these observations are consistent with the physical separation of fibre and non-fibre tissues we hoped to achieve by dissection. We will focus the remainder of this report on the spots that were enriched in fibres.
The fibre-enriched proteins to which we were able to assign putative identities were classified into eight functional categories (Figure 4). Aside from the category we called "miscellaneous", which represented a diverse set of functions, most of the proteins that were identified in fibre samples could be assigned to one of three categories related to the conversion of carbohydrates for energy or glycan biosynthesis, namely: primary carbon and energy metabolism; one-carbon metabolism; and cell wall and polysaccharide metabolism (Figure 4). The predominance of these proteins for the metabolism of carbohydrates and related compounds is consistent with the major biochemical activities we expected to observe within cells active in secondary wall biogenesis. In addition, we assigned a smaller number of proteins to each of the remaining categories: membrane transport; cytoskeleton & secretion; ATPases; and protein & amino acid metabolism. The membership of proteins assigned to spots in each of the eight functional categories is shown in Table 1, and is discussed in more detail in the following sections.
Figure 4.

Functional categorization of fibre-enriched proteins. All spots for which signal intensity was at least 1.5-fold greater in fibres as compared to non-fibres, and for which identity could be assigned by MS, were assigned to one of the categories shown. The grey and black regions of each bar show the portion of spots for which p > 0.05 and p ≤ 0.05, respectively, in a t-test of the significance of differences in intensity between fibre and non-fibre tissues. ATPases (ATP); Cell wall polysaccharide metabolism (CWP); Cytoskeleton and secretion (C&S); Membrane transport (MemT); Miscellaneous (misc); One-carbon metabolism (1C); Primary carbon and energy metabolism (C&E); Protein and amino acid metabolism (P&AA).
Primary carbon and energy metabolism
The conversion of monosaccharides and starch into energy is the inferred function of the largest proportion of proteins that were enriched (> 1.5 fold) in fibres, as compared to the non-fibre fraction at the stem snap point (Figure 4). These reactions are also summarized in Figure 5. Two of the most highly enriched proteins we detected in any functional category were β-amylase (spot #17; 8.8× fold enriched in fibres), and fructose kinase (#93, 6.7×; #94, 2.2×; #96, 2.0×), which catalyze the first steps in the catabolism of starch and fructose, respectively (Table 1). The increased relative abundance of these enzymes in fibres provides some insight into the immediate sources of carbon and energy for secondary wall biogenesis. We also detected the statistically significant (p ≤ 0.05) enrichment of enzymes of glycolysis and related processes, namely fructose-bisphosphate aldolase (#78, 2.4×), glyceraldehyde 3-phosphate dehydrogenase (#83, 2.6×; #87, 2.8×), and phosphoglucomutase (#27, 1.8×; #28, 3.7×), as well as the presence of phosphoglycerate kinase (#68, #71). Finally, we identified fibre-enriched protein spots putatively representing 5 of 8 enzymes of the tricarboxylic acid cycle, where further energy and metabolic precursors are generated from the products of glycolysis. The tricarboxylic acid cycle -associated proteins that were significantly enriched in fibres and included citrate synthase (#63, 3.7×), succinyl coA-ligase (#82, 2.3×), fumarase (#57, 2.5×), and malate dehydrogenase (#92, 3.3×). Aconitate hydratase (#2, #3) was also detected, although its fold-enrichment was not statistically significant (p > 0.05).
Figure 5.

Relative abundance of fibre-enriched proteins identified as enzymes in selected reactions of carbohydrate and one-carbon metabolism. Numbers following the symbol '#' are the unique spot identifiers used in Table 1 and throughout the text. Values in boxes show the fold-enrichment (i.e. signal intensity in fibres/non-fibres). Grey and black filled boxes indicate spots for which p > 0.05 and p ≤ 0.05, respectively, in a t-test of the significance of differences in intensity between fibre and non-fibre tissues. No intensity ratio is shown for #41, because multiple proteins were identified within this spot. Pathways shown are based on data from KEGG and AraCyc [37, 38]. Not all reactants or co-factors are shown. Abbreviations used in names of substrates include fructose (Fru), galactose (Gal), glucose (Glc), glyceraldehyde-3-phosphate (G3P), homocysteine (HCys), maltose (Mal), phosphoglycerate (PG), phosphoenolpyruvate (PEP), rhamnose (Rha), S-adenosyl homocysteine (SAH), tetrahydrofolate (THF).
ATPases
Many subunits of the ATPase/synthase complex were identified in either fibres or the non-fibre fraction, including an α-subunit (#35), β-subunits (#42, #43), and a γ-subunit (#99). The tissue-specific abundance patterns of these various subunits were surprisingly complex: the γ-subunit and one β-subunit (#42) were associated with equal spot intensities in both sample types, while the other ATP synthase β-subunit (#44), was 1.8× more abundant in the non-fibre fraction. Only the α-subunit was more abundant (1.6×) in fibres.
In addition to the ATPase/synthases described above, we identified peptides from several other types of putative ATPases, including three protein spots containing vacuolar-type ATPase (v-ATPase), of which, two spots (#24, 2.6×; #105, 1.8×) were significantly (p ≤ 0.05) enriched in fibres. v-ATPases are some of the most abundant membrane proteins in the vacuole and endomembrane system, and their enrichment may reflect increased relative abundance of these organellar structures in fibres [17]. We also detected a putative plasma membrane-associated AAA-ATPase (#1, 1.6×) in fibres, although this was not deemed to be more abundant in fibres by our usual statistical criteria. Both v-ATPases and AAA-ATPases have been previously demonstrated to be essential for vesicle transport, and might therefore be active in secondary wall-specific processes in developing fibres [17,18].
Cell wall and polysaccharide metabolism
Cell walls consist of many types of polymers, including cellulose, hemicellulose, and pectins. However, with the possible exception of an NAD-dependent epimerase/dehydratase with similarity to UDP-xylose synthases (#76, 6.1×), and GDP-4-keto-6-deoxy-D-mannose-3,5-epimerase-4-reductase (GME, #101, 2.3×) almost all of the fibre-enriched, cell wall-related enzymes we identified were most likely associated with the metabolism of pectin-like substances. For example, we identified proteins from six spots as β-galactosidases. Five of these (#12–#16) were co-located in a charge train and the sixth (#64) was an isolated spot of lower apparent molecular weight. The five spots in the charge train were significantly more intense in fibres (5.4–9.3×), while the lower molecular weight spot was nearly similar in abundance in both types of tissues (1.15×). Within the charge train, peptides from three spots aligned with a chickpea β-galactosidase as the highest scoring match. This chickpea β-galactosidase has previously demonstrated exo- and endo- cleavage activity towards the side-chains of pectins and is found in elongating hypocotyls [19,20]. In developing flax fibres, the deposition of a rhamnogalactan-type pectin consisting of 55–85% galactose is known to precede establishment of the crystalline, cellulosic fibrils that characterize the mature secondary wall [6]. Because the galactose residues of rhamnogalactans are one of the putative substrates for β-galactosidase, we speculate that the abundance of this enzyme in developing fibres is evidence of an important role for it in remodeling, removing, or recycling of galactans as part a dynamic process of cell wall deposition. However, it is also possible that the β-galactosidase we detected hydrolyzes other galactosyl bonds, such as those that decorate arabinogalactan proteins [21]. Finally, the appearance of the β-galactosidase spots in a train along the axis of the first dimension separation of our electrophoretic gels is consistent with extensive post-translational modification of this abundant protein.
In addition to β-galactosidase, we also identified other spots representing one or more enzymes with possible roles in the metabolism of pectic polysaccharides. Three spots (#18, 4.1×; #19, 6.6×; #104, 3.0×) were more enriched in fibres as compared to the non-fibre fraction and share homology with UDP-rhamnose synthase. Because these enzymes would normally be expected to contribute to the growth of rhamnogalactans, it is interesting to observe their enrichment in the same cells in which β-galactosidase might hydrolyze galactosidic bonds within these polymers. The potential co-existence of both catabolic and anabolic processes of galactan metabolism is consistent with a rapid turnover of these polymers during cell wall deposition, although the existence of the inferred enzymatic activities must still be confirmed experimentally.
One-carbon metabolism
Four enzymes associated with one-carbon (1C) metabolism were identified among the fibre-enriched protein spots in our study. Three of these: methionine synthase (#9, #10; 2.0×, 2.2× respectively), methionine adenosyltransferase (#60; 2.1×), and adenosylhomocysteinase (#41; 1.6×) are components of the S-adenosyl methionine (SAM) cycle, while the remaining protein, serine hydroxymethyltransferase (#53; 2.2×), catalyzes the transfer of carbon into the SAM cycle, via folate. Because the cumulative function of these enzymes is to provide activated methyl groups for transfer to acceptors, the identity of the major methyl transferases and their substrates in fibres is an obvious question. In plants, potential acceptors of activated methyl groups include a wide variety of molecules, among them components of pectin or lignin [22]. Because the amount of lignin present in flax fibres is low in comparison to other types of schlerenchyma, particularly at the early stage of cell wall development associated with the snap point, [23,24], it seems unlikely that lignin is the major sink for methyl flux through the SAM cycle. Thus, pectin or other actively accumulating substances may be targets for SAM-mediated methylation in developing fibres.
Membrane transport
Only a few proteins related to transport across membranes were detected in our study. This may be due in part to the difficulty of extracting and resolving certain membrane-associated proteins. Nevertheless, we identified a K+ channel β-subunit was highly enriched (#97; 8.6×) in fibres, as well as two porins (#102, #102; 1.7×, 3.9×, respectively). The biological significance of the porins is unclear, however, increased expression of K+ channels has been previously correlated with sucrose uptake in developing cotton fibres. Thus the strong enrichment of K+ channel proteins we observed may reflect a similar process of the uptake of reduced carbon in flax fibres [25,26].
Cytoskeleton and secretion
Structural components of the cytoskeleton, as well as proteins related to vesicle traffic, were also relatively more abundant in fibre protein extracts as compared to surrounding tissues. We observed relative enrichment of at least 1.5-fold of actin (#69, #70) and tubulin (#37) in fibres. These proteins may be enriched in fibres, as compared to cells of the non-fibre fraction, due in part to the differences in architecture and surface/volume ratios of these cells. Additionally, increased relative abundance of cytoskeleton proteins in fibres undergoing cell wall thickening may reflect the role of the cytoskeleton in deposition of cellulose and other cell wall components. An active secretory system, which delivers non-cellulosic polysaccharide components to the cell wall, is also expected to be present in developing flax fibres; the enrichment of myosin (#5, 2.5×; #6, 3.6×), dynamin-like proteins (#22, 3.1×), and GDP-dissociation inhibitor (#55, 2.0×; #56, 1.9×) in these cells is therefore consistent with developmental processes presumed to be active in the cells we sampled. We also note that other components of the cytoskeleton mentioned in a structural context above (i.e. actin and tubulin) may have additional functions specifically related to secretion and other aspects of secondary wall deposition [27-29].
Protein and amino acid metabolism
Enzymes related to protein metabolism (e.g. protein synthesis and folding) were moderately enriched (1.5× – 2.7×) in fibres as compared to the non-fibre fraction. Two translation initiation factors were more abundant in the fibre sample: eIF-4A (#62, 1.6×) and eIF-5A (#114, 2.0×). Proteins in the eIF-4A family form part of the ribosomal machinery and are involved in binding and unwinding mRNA for translation, while some eIF-5A isoform family members have more diverse functions in cell division and related processes [30]. A translational elongation factor EF2 (#4, 2.5×) was also more abundant in fibres, while spots containing EF1á were similarly abundant (#67, 1.2×) or 2.3× fold less abundant (#54) in fibres as compared to the non-fibre fraction.
Heat shock proteins HSP60 (#29, 2.7×; #30, 1.5×; #32, 2.1×), HSP70 (#20, 1.9×; #21, 1.7×; #23, 2.0×), and HSP90 (#11, 1.7×) were also enriched in fibres. These proteins may function in the processes of cytosolic protein folding and protein import into mitochondria and chloroplasts, which are commonly associated with members of the HSP60, HSP70, and HSP90 families [31]. Additionally, because HSP70s have been shown to have specific functions in cell wall development in yeast, we cannot exclude the possibility that some of these proteins are active at the plasma membrane during the deposition of the flax fibre secondary wall [32,33].
Miscellaneous
Several of the proteins we identified could not be classified into any of the larger functional categories we have already described. Eight of these proteins were enriched by 1.5× (p ≤ 0.05) or more in fibres, and may accordingly have specific roles in fibre development. These included annexins (#97, 2.2×; #98, 4.1×), enoyl-ACP reductase (#100, 2.1×), maturase K (#112, 3.4×), a 14-3-3 protein (#108, 2.6×), peroxidase (#85, 2.4×), and a protein kinase C inhibitor (#107, 2.8×). Among these, the enrichment of annexin in developing fibres is particularly interesting, given its previous association with cellulose synthase in structural and proteomic studies of cotton fibres [16,34].
Comparison to transcriptomic analysis
The experimental approach used in the present study differs in many ways from our previously reported microarray analysis of flax stems [8]. Importantly, in the previous report, we did not dissect fibres away from other stem tissues; rather we compared transcript abundance in stem segments containing fibres at different stages of development. Therefore, a global comparison of these datasets is not warranted. Notwithstanding these limitations, we noted that three carbohydrate-related enzymes were detected both as proteins enriched in fibres from the snap-point region of the stem, and previously as transcripts expressed in the region of the stem containing the snap-point, including β-galactosidase (#12–16, #64), fructokinase (#93, #94), and GME (#101) (Table 1). In the transcriptomic data, β-galactosidase and fructokinase were significantly more abundant in the region of the snap-point as compared to segments from nearer either the apex or base of the stem, while GME showed highest transcript abundance in the apical-most segment, which may be due to differences in the turnover of these various gene products. On the other hand, our previous work also identified many other snap-point enriched transcripts that were not detected as proteins in the previous study. These include arabinogalactan proteins and lipid transfer proteins that were further demonstrated by qRT-PCR to be enriched specifically in the phloem tissues of the snap-point, as compared to leaves or the xylem of stems. Discrepancies between transcriptomic and proteomic analyses have been previously documented by ourselves and others, and are presumably due to differences in efficiencies of extraction and detection of various proteins, among many other technical and biological factors [35]. For example, Bayer et al. specifically noted under representation of AGPs and other cell wall proteins within their proteomic analysis, due possibly to the high degree of glycosylation of these proteins [12]. Thus, it appears likely that a comprehensive description of gene expression within developing flax fibres cannot be provided by either transcript or protein profiling, alone, but instead the results of many different experimental approaches must be considered together.
Conclusion
We have described a differential proteomic profile of a single plant cell type at a well-defined developmental stage, during which secondary cell wall biogenesis is occurring. The fibre-enriched proteins we identified are consistent with the dynamic process of secondary wall deposition previously suggested by histological and biochemical analyses, and particularly the importance of galactans and the secretory pathway in this process [6]. The apparent abundance of amylase suggests that starch may be an unappreciated source of materials for cell wall biogenesis. Furthermore, our observations confirm previous reports that correlate accumulation of proteins such as annexins, and specific heat shock proteins with secondary cell wall deposition [6,16,33]. Together, the proteins we have identified in this study provide a basis for better understanding the unique properties of phloem fibre secondary cell walls, and define targets for detailed genetic and biochemical analyses in future.
Methods
Plant material
Fibres (i.e. individual cells) and surrounding, non-fibre cells of the cortex were isolated from the stems of Linum usitatissimum L., var. Norlin. A total of 495 plants were harvested from four independently grown populations. Seeds were sown two per 10 cm pot and grown as previously described [8]. After 3 weeks of growth, the mean distance from the apex to snap-point was 5.9 cm, with mean plant height of 19 cm. A 3 cm segment of stem, spanning from 2 cm to 5 cm below the snap-point, was further dissected to separate the individual fibres and surrounding non-fibre cells of the cortex (i.e. "the non-fibre fraction", consisting predominantly of parenchyma, sieve elements, and companion cells, but excluding epidermis, xylem and pith) for proteomic analysis. After dissection, fibres and surrounding tissues were rinsed in deionized water, blotted, then frozen in liquid nitrogen, and stored at -80°C.
Protein isolation from tissues
Tissues were ground to a powder in liquid nitrogen and then further ground for one minute in 1 mL cold TCA/acetone buffer (20 mM DTT, 10% trichloroacetic acid in cold acetone). Homogenates were transferred with an additional 1 mL of buffer to microcentrifuge tubes and were allowed to precipitate overnight at -20°C. After centrifugation (13000 rpm, 10°C, 15 minutes), pellets were rinsed once with 1 mL 20 mM DTT in acetone for 1 h at -20°C, then pellets were left to dry at -20°C for 2 h, and dissolved in 200 μL of urea/thiourea buffer (7 M urea, 2 M thiourea, 4% (w/v) CHAPS, 30 mM Tris-Cl) by vortexing at room temperature for 30 minutes. The solution was clarified by centrifugation (13000 rpm, 17°C, 5 minutes) and supernatants were further processed by using the 2D Clean-Up Kit (Amersham Biosciences). Precipitates were re-dissolved in 60 μl of the urea/thiourea buffer, and concentrations of the protein samples were determined using the 2D Quant Kit (Amersham Biosciences) and NanoDrop® ND-1000 spectrophotometer (NanoDrop Technologies) against a BSA standard curve.
Fluorescent labeling of proteins
Four independent pools of approximately 125 plants each were grown in nominally identical conditions that were spatially and temporally separated from each other. Proteins were isolated separately from tissues dissected from each pool of plants, to produce four paired protein samples from fibres and the non-fibre fraction, where each pair of samples was biologically independent from every other pair. We labeled each 30 μg protein sample (pH adjusted to 8.5) with 240 pmol of Cy2, Cy3 or Cy5 fluorescent dyes, using the CyDye™ DiGE fluors (minimal dyes) labeling kit (Amersham Biosciences). Labeling reactions were stopped by the addition of 1 μl of 10 mM lysine to each tube, and after a further 10-minute incubation on ice, the volume of each sample was doubled with the addition of a sample buffer (7 M urea, 2 M thiourea, 2% (v/v) ampholyte, 2% (w/v) DTT, 4% (w/v) CHAPS) to ready the samples for IEF. Labeled samples were mixed together as stated in Table 2 to create four analytical gels, with each gel containing an internal standard and both tissue samples. The internal standard is prepared by mixing equal masses of protein extracts from fibre and non-fibre fractions of each biologically independent harvest.
Table 2.
Experimental design relative to labeling and sample loading of analytical gels.
| gel | Cy2 labeled | Cy3 labeled | Cy5 labeled |
| 1 | internal standard #1 30 μg | fibre sample #1 30 μg | non-fibre sample #1 30 μg |
| 2 | internal standard #2 30 μg | fibre sample #2 30 μg | non-fibre sample #2 30 μg |
| 3 | internal standard #3 30 μg | non-fibre sample #3 30 μg | fibre sample #3 30 μg |
| 4 | internal standard #4 30 μg | non-fibre sample #4 30 μg | fibre sample #4 30 μg |
Note: each gel contains proteins from a unique pool (#1–#4) of independently grown plants. The Cy2-labeled internal standard is a mixture of equal masses of proteins from fibre and non-fibre samples.
2DE of CyDye labeled protein mixtures
All subsequent handling and separation steps for 2DE were conducted away from light. 24 cm, 3–10 NL Immobiline™ drystrips (Amersham Biosciences) were passively re-hydated for 10 h in (8 M urea, 4% (w/v) CHAPS, 1% (v/v) ampholytes 3–10, 13 mM DTT, trace bromophenol blue). A total of 56 kVh at 20°C was used to focus the proteins using an IPGphor™ II (Amersham Biosciences). Paper wicks on the basic end were spiked with 13 mM DTT and were changed three times during the run. Following IEF, strips were equilibrated for SDS-PAGE separation by gentle agitation for 15 minutes in 6 M urea, 50 mM tris-Cl (pH 8.8), 30% (v/v) glycerol, 2% (w/v) SDS, trace bromophenol blue plus 0.5% (w/v) DTT, followed by 15 minutes in the same solution with 4.5% (w/v) IAA instead of DTT. After equilibration, the strips were sealed onto the top edge of self-cast, large-format, 12.5% acrylamide gels using sealing solution (1% low-melt agarose, trace bromophenol blue in 1X running buffer). The four analytical gels were separated by molecular weight during SDS-PAGE, simultaneously, using the Ettan™ Dalt six (Amersham Biosciences). The gels were run at 2 W/gel for 30 minutes then 8 W/gel until the bromophenol blue dye front just touched the end of the gels.
Imaging and analysis
Fluorescently labeled gels were imaged at 100 μm resolution with PMT voltage between 50000 and 63558 V. DeCyder™ 6.5 (Amersham Biosciences) was used to match, normalize, and statistically analyze spots. After in-gel normalization using Differential In-gel Analysis (DIA), the Biological Variation Analysis (BVA) module was used for statistical analysis and normalization across all analytical gels.
Spot-picking and tryptic digestion of proteins
Preparative gels, loaded with about 125 μg of protein, were post-stained with Deep Purple™ total protein stain (Amersham Biosciences) and spot-matched to the analytical gels. Gel spot excision and subsequent tryptic digestion were conducted using an Ettan™ Spot-picker (Amersham Biosciences) and ProteomeWorks™ MassPREP™ robotic handling station (Bio-Rad Laboratories and Waters corporations), resulting in peptides in a final extraction solution of 2% ACN, 0.1% formic acid in H20.
Protein identification
LC MS/MS analysis was performed using an online 1100 series XCT Ion trap (Agilent Technologies). The autosampler injected 18 μL of each sample onto an enrichment column (Zorbax 300SB-C18 5 μm 5 × 0.3 mm) that connected to a second column (Zorbax 300SB-C18 5 μm 150 × 0.3 mm) in a peptide-separation gradient that started at 85% solvent A (0.1% formic acid in H2O) and ended at 55% solvent B (0.1% formic acid, 5% H2O in ACN) over a 42 minute span. This was followed by 10 minutes of 90% solvent B to cleanse the columns before returning to 97% solvent A for the next sample. The MS ran a 300–2200 m/z scan followed by MS/MS analysis of the most intense ions. Raw spectral data was processed into Mascot Generic File (.mgf) format using the default method in the ChemStation Data Analysis module and ion searches were completed in MASCOT [36] with the search parameters of: peptide tolerance of 2 Da, parent ion tolerance of 0.8 m/z, ion charge of +1, +2 and +3.
List of abbreviations
1C: one-carbon; DiGE: differential gel electrophoresis; dTDP: thymine diphosphate deoxynucleotide; EF: translational elongation factor; eIF: translational initiation factor; GDP: guanine diphosphate; SAM: S-adenosyl methionine; UDP: uridine diphosphate.
Authors' contributions
NSCH designed and conducted all experiments, including operation of the mass spectrometer and interpretation of mass spectra, and wrote the original draft of this manuscript. MKD supervised all research, and contributed to writing and editing of the manuscript.
Supplementary Material
Additional information (e.g. peptide sequences; Mr, pI of database matches, PFAM domains) on spot identifications that was not otherwise conveyed in Table 1.
Acknowledgments
Acknowledgements
We thank Anthony Cornish, Ana Lopez-Campistrous, and Paul Semchuk for technical advice. This work was funded by a Discovery Grant from NSERC (Natural Sciences and Engineering Research Council), an Alberta Ingenuity New Faculty Grant, and an Alberta Innovation and Science Acceleration Grant.
Contributor Information
Naomi SC Hotte, Email: nhotte@ualberta.ca.
Michael K Deyholos, Email: deyholos@ualberta.ca.
References
- McCorriston J. The fiber revolution: Textile extensification, alienation, and social stratification in ancient Mesopotamia. Current Anthropology. 1997;38:517–549. doi: 10.1086/204643. [DOI] [Google Scholar]
- Vanzeist W, Bakkerheeres JAH. Evidence for Linseed Cultivation before 6000 Bc. Journal of Archaeological Science. 1975;2:215–219. doi: 10.1016/0305-4403(75)90059-X. [DOI] [Google Scholar]
- Mohanty AK, Misra M, Hinrichsen G. Biofibres, biodegradable polymers and biocomposites: An overview. Macromolecular Materials and Engineering. 2000;276:1–24. doi: 10.1002/(SICI)1439-2054(20000301)276:1<1::AID-MAME1>3.0.CO;2-W. [DOI] [Google Scholar]
- Esau K. Vascular differentiation in the vegetative shoot of Linum III. - The origin of the bast fibers. American Journal of Botany. 1943;30:579–586. doi: 10.2307/2437468. [DOI] [Google Scholar]
- Lev-Yadun S. Intrusive growth - the plant analog of dendrite and axon growth in animals. New Phytologist. 2001;150:508–512. doi: 10.1046/j.1469-8137.2001.00143.x. [DOI] [Google Scholar]
- Gorshkova TA, Morvan C. Secondary cell-wall assembly in flax phloem fibres: role of galactans. Planta. 2006;223:149–158. doi: 10.1007/s00425-005-0118-7. [DOI] [PubMed] [Google Scholar]
- Gorshkova TA, Sal'nikova VV, Chemikosova SB, Ageeva MV, Pavlencheva NV, van Dam JEG. The snap point: a transition point in Linum usitatissimum bast fiber development. Industrial Crops and Products. 2003;18:213–221. doi: 10.1016/S0926-6690(03)00043-8. [DOI] [Google Scholar]
- Roach M, Deyholos M. Microarray analysis of flax ( Linum usitatissimum L.) stems identifies transcripts enriched in fibre-bearing phloem tissues. Molecular Genetics and Genomics. 2007;278:149–165. doi: 10.1007/s00438-007-0241-1. [DOI] [PubMed] [Google Scholar]
- Day A, Addi M, Kim W, David H, Bert F, Mesnage P, Rolando C, Chabbert B, Neutelings G, Hawkins S. ESTs from the fibre-bearing stem tissues of flax (Linum usitatissimum L.): Expression analyses of sequences related to cell wall development. Plant Biology. 2005;7:23–32. doi: 10.1055/s-2004-830462. [DOI] [PubMed] [Google Scholar]
- Mooney BP, Miernyk JA, Greenlief CM, Thelen JJ. Using quantitative proteomics of Arabidopsis roots and leaves to predict metabolic activity. Physiologia Plantarum. 2006;128:237–250. doi: 10.1111/j.1399-3054.2006.00746.x. [DOI] [Google Scholar]
- Tian Q, Stepaniants SB, Mao M, Weng L, Feetham MC, Doyle MJ, Yi EC, Dai HY, Thorsson V, Eng J, Goodlett D, Berger JP, Gunter B, Linseley PS, Stoughton RB, Aebersold R, Collins SJ, Hanlon WA, Hood LE. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Molecular & Cellular Proteomics. 2004;3:960–969. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- Bayer EM, Bottrill AR, Walshaw J, Vigouroux M, Naldrett MJ, Thomas CL, Maule AJ. Arabidopsis cell wall proteome defined using multidimensional protein identification technology. Proteomics. 2006;6:301–311. doi: 10.1002/pmic.200500046. [DOI] [PubMed] [Google Scholar]
- Blee KA, Wheatley ER, Bonham VA, Mitchell GP, Robertson D, Slabas AR, Burrell MM, Wojtaszek P, Bolwell GP. Proteomic analysis reveals a novel set of cell wall proteins in a transformed tobacco cell culture that synthesises secondary walls as determined by biochemical and morphological parameters. Planta. 2001;212:404–415. doi: 10.1007/s004250000407. [DOI] [PubMed] [Google Scholar]
- Vander Mijnsbrugge K, Meyermans H, Van Montagu M, Bauw G, Boerjan W. Wood formation in poplar: identification, characterization, and seasonal variation of xylem proteins. Planta. 2000;210:589–598. doi: 10.1007/s004250050048. [DOI] [PubMed] [Google Scholar]
- Watson BS, Lei ZT, Dixon RA, Sumner LW. Proteomics of Medicago sativa cell walls. Phytochemistry. 2004;65:1709–1720. doi: 10.1016/j.phytochem.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Yao Y, Yang YW, Liu JY. An efficient protein preparation for proteomic analysis of developing cotton fibers by 2-DE. Electrophoresis. 2006;27:4559–4569. doi: 10.1002/elps.200600111. [DOI] [PubMed] [Google Scholar]
- Schumacher K. Endomembrane proton pumps: connecting membrane and vesicle transport. Current Opinion in Plant Biology. 2006;9:595–600. doi: 10.1016/j.pbi.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Haas TJ, Sliwinski MK, Martinez DE, Preuss M, Ebine K, Ueda T, Nielsen E, Odorizzi G, Otegui MS. The Arabidopsis AAA ATPase SKD1 is involved in multivesicular endosome function and interacts with its positive regulator LYST-INTERACTING PROTEIN5. Plant Cell. 2007;19:1295–1312. doi: 10.1105/tpc.106.049346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban R, Labrador E, Dopico B. A family of beta-galactosidase cDNAs related to development of vegetative tissue in Cicer arietinum. Plant Science. 2005;168:457–466. doi: 10.1016/j.plantsci.2004.09.009. [DOI] [Google Scholar]
- Esteban R, Dopico B, Munoz FJ, Romo S, Martin I, Labrador E. Cloning of a Cicer arietinum beta-galactosidase with pectin-degrading function. Plant and Cell Physiology. 2003;44:718–725. doi: 10.1093/pcp/pcg087. [DOI] [PubMed] [Google Scholar]
- Kotake T, Dina S, Konishi T, Kaneko S, Igarashi K, Samejima M, Watanabe Y, Kimura K, Tsumuraya Y. Molecular Cloning of a {beta}-Galactosidase from Radish That Specifically Hydrolyzes {beta}-(1->3)- and {beta}-(1->6)-Galactosyl Residues of Arabinogalactan Protein. Plant Physiol. 2005;138:1563–1576. doi: 10.1104/pp.105.062562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt BA, Weretilnyk EA. Sustaining S-adenosyl-l-methionine-dependent methyltransferase activity in plant cells. Physiologia Plantarum. 2001;113:435–442. doi: 10.1034/j.1399-3054.2001.1130401.x. [DOI] [Google Scholar]
- Day A, Ruel K, Neutelings G, Cronier D, David H, Hawkins S, Chabbert B. Lignification in the flax stem: evidence for an unusual lignin in bast fibers. Planta. 2005;222:234–245. doi: 10.1007/s00425-005-1537-1. [DOI] [PubMed] [Google Scholar]
- Gorshkova TA, Salnikov VV, Pogodina NM, Chemikosova SB, Yablokova EV, Ulanov AV, Ageeva MV, Van Dam JEG, Lozovaya VV. Composition and distribution of cell wall phenolic compounds in flax (Linum usitatissimum L.) stem tissues. Annals of Botany. 2000;85:477–486. doi: 10.1006/anbo.1999.1091. [DOI] [Google Scholar]
- Ruan YL. Goldacre paper: Rapid cell expansion and cellulose synthesis regulated by plasmodesmata and sugar: insights from the single-celled cotton fibre. Functional Plant Biology. 2007;34:1–10. doi: 10.1071/FP06234. [DOI] [PubMed] [Google Scholar]
- Ruan YL, Llewellyn DJ, Furbank RT. The control of single-celled cotton fiber elongation by developmentally reversible gating of plasmodesmata and coordinated expression of sucrose and K+ transporters and expansin. Plant Cell. 2001;13:47–60. doi: 10.1105/tpc.13.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutte Y, Vernhettes S, Satiat-Jeunemaitre B. Involvement of the cytoskeleton in the secretory pathway and plasma membrane organization of higher plant cells. Cell Biology International. 2007;31:649–654. doi: 10.1016/j.cellbi.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Oda Y, Hasezawa S. Cytoskeletal organization during xylem cell differentiation. Journal of Plant Research. 2006;119:167–177. doi: 10.1007/s10265-006-0260-8. [DOI] [PubMed] [Google Scholar]
- Roberts AW, Frost AO, Roberts EM, Haigler CH. Roles of microtubules and cellulose microfibril assembly in the localization of secondary-cell-wall deposition in developing tracheary elements. Protoplasma. 2004;224:217–229. doi: 10.1007/s00709-004-0064-4. [DOI] [PubMed] [Google Scholar]
- Thompson JE, Hopkins MT, Taylor C, Wang TW. Regulation of senescence by eukaryotic translation initiation factor 5A: implications for plant growth and development. Trends in Plant Science. 2004;9:174–179. doi: 10.1016/j.tplants.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nature Reviews Molecular Cell Biology. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Lopez-Ribot JL, Chaffin WL. Members of the Hsp70 family of proteins in the cell wall of Saccharomyces cerevisiae. J Bacteriol. 1996;178:4724–4726. doi: 10.1128/jb.178.15.4724-4726.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nombela C, Gil C, Chaffin WLJ. Non-conventional protein secretionin yeast. Trends in Microbiology. 2006;14:15–21. doi: 10.1016/j.tim.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Hofmann A, Delmer DP, Wlodawer A. The crystal structure of annexin Gh1 from Gossypium hirsutum reveals an unusual S-3 cluster - Implications for cellulose synthase complex formation and oxidative stress response. European Journal of Biochemistry. 2003;270:2557–2564. doi: 10.1046/j.1432-1033.2003.03612.x. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Yang B, Harris NS, Deyholos MK. Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. Journal of Experimental Botany. 2007;58:3591–3607. doi: 10.1093/jxb/erm207. [DOI] [PubMed] [Google Scholar]
- http://www.matrixscience.com
- Zhang PF, Foerster H, Tissier CP, Mueller L, Paley S, Karp PD, Rhee SY. MetaCyc and AraCyc. Metabolic pathway databases for plant research. Plant Physiology. 2005;138:27–37. doi: 10.1104/pp.105.060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional information (e.g. peptide sequences; Mr, pI of database matches, PFAM domains) on spot identifications that was not otherwise conveyed in Table 1.