

Abstract
Antigenic variation in Trypanosoma brucei has selected for the evolution of a massive archive of silent Variant Surface Glycoprotein (VSG) genes, which are activated by recombination into specialized expression sites. Such VSG switching can occur at rates substantially higher than background mutation and is dependent on homologous recombination, a core DNA repair reaction. A key regulator of homologous recombination is BRCA2, a protein that binds RAD51, the enzyme responsible for DNA strand exchange. Here, we show that T. brucei BRCA2 has undergone a recent, striking expansion in the number of BRC repeats, a sequence element that mediates interaction with RAD51. T. brucei BRCA2 mutants are shown to be significantly impaired in antigenic variation and display genome instability. By generating BRCA2 variants with reduced BRC repeat numbers, we show that the BRC expansion is crucial in determining the efficiency of T. brucei homologous recombination and RAD51 localization. Remarkably, however, this appears not to be a major determinant of the activation of at least some VSG genes.
Introduction
African trypanosomes, in common with many pathogens, survive in the face of host immune responses by undergoing antigenic variation, the continuous changing of pathogen surface antigens. Successive waves of immune response to the variant surface antigens eradicate part but not all of the pathogen population, lengthening the infection and enhancing transmission. A number of strategies for antigenic variation have evolved (Deitsch et al., 1997), but a common approach is the use of gene conversion (Palmer and Brayton, 2007). Trypanosoma brucei antigenic variation involves changes in the composition of a dense, protective coat composed of Variant Surface Glycoprotein (VSG) and can occur at very high rates (Turner and Barry, 1989), a trait shared with other pathogens (Barry and McCulloch, 2001; Criss et al., 2005). T. brucei appears remarkable, however, in the extent to which it has invested in gene conversion (for recent reviews, see Horn and Barry, 2005; Taylor and Rudenko, 2006).
The T. brucei genome contains greater than 1000 VSG genes (Berriman et al., 2005; Marcello and Barry, 2007), an archive of potential variant surface proteins larger than in any other organism described to date. The genome is composed of 100 or so minichromosomes, a few intermediate chromosomes and 11 megabase chromosomes. VSGs are found adjacent to the telomere on all T. brucei chromosome classes, but the majority of the archive is present in subtelomeric arrays on the megabase chromosomes. Each trypanosome cell normally expresses only one VSG at a time, from telomeric VSG expression sites (ESs). The main route of VSG switching is the movement of the silent VSG genes into the ESs (Robinson et al., 1999). Primarily, such recombination reactions involve gene conversion, generating a copy of a silent VSG and displacing the ES VSG. Less common are cross-overs in which chromosome ends are exchanged, activating a new VSG without copying. VSG gene conversions appear to operate in a hierarchy, which is thought to be important in extending the infection (Pays, 1989; Barbet and Kamper, 1993). Telomere-proximal VSG genes are activated early, followed by functional array genes (Morrison et al., 2005). Finally, novel mosaic VSGs are generated by segmental gene conversion reactions involving VSG pseudogenes and gene fragments (Thon et al., 1990; Kamper and Barbet, 1992). Such damaged VSGs represent the huge majority of the VSG archive, and are likely to be a major determinant of the function and success of T. brucei antigenic variation (Kamper and Barbet, 1992; Barbet and Kamper, 1993; Marcello and Barry, 2007). In contrast to a number of other pathogens, where a single expression locus for antigenic variation is found (Criss et al., 2005; Palmer and Brayton, 2007), T. brucei possesses multiple VSG ESs (Becker et al., 2004). Only one ES is fully transcribed at a time, but VSG switches can occur by inactivating the active ES and activating a previously silent ES, though the factors that mediate this process are still being determined (Pays, 2005; Hughes et al., 2007).
Homologous recombination performs core functions in all cells by reversing genotoxic damage and ensuring the completion of DNA replication (Sung and Klein, 2006). It can also provide for telomere maintenance, and is needed for genetic exchange during meiosis. Homologous recombination appears to act in T. brucei VSG switching, as mutation of RAD51 (McCulloch and Barry, 1999), the central eukaryotic enzyme of homologous strand exchange, impairs the process, as does mutation of a RAD51-related protein, RAD51-3 (Proudfoot and McCulloch, 2005). Such a reliance on a core DNA repair process for the specialized recombination of antigenic variation is true also for Neisseria sp. (Sechman et al., 2005). However, whether specific factors for antigenic variation exist, and whether antigenic variation has imposed adaptations on the homologous recombination machinery, have been little explored. One stage at which such features might be seen is in the regulation of RAD51 function, which is influenced by a wide range of factors (Sung and Klein, 2006). One of these is BRCA2, the protein encoded by one human breast cancer susceptibility gene (Narod and Foulkes, 2004). BRCA2 homologues are widespread in eukaryotes (Warren et al., 2002; Lo et al., 2003), and analyses in the nematode Caenorhabditis elegans (Petalcorin et al., 2007) (CeBRC-2), in the fungus Ustilago maydis (Kojic et al., 2002; Yang et al., 2005) (Brh2) and in the plant Arabidopsis thaliana (Dray et al., 2006) suggest that at least one function is conserved with a role initially described in mammals: RAD51 interaction via BRC repeat motifs in BRCA2 (Bork et al., 1996; Wong et al., 1997; Chen et al., 1998; Marmorstein et al., 1998). At least one BRC repeat has been described in all characterized BRCA2 homologues (Lo et al., 2003). Mammalian BRCA2 has eight non-identical BRC repeats (Lo et al., 2003), at least six of which bind RAD51 in humans (Wong et al., 1997; Chen et al., 1998; Marmorstein et al., 1998). Similarly, A. thaliana BRCA2 has four degenerate BRC motifs, some of which bind RAD51 (Dray et al., 2006). In contrast, C. elegans BRCA2/CeBRC-2 (Martin et al., 2005) and U. maydis BRCA2/Brh2(Kojic et al., 2006) contain only a single RAD51-binding BRC repeat. In addition to binding RAD51 via the BRC repeats, BRCA2 interacts with RAD51 through unrelated sequences in a number of organisms (Mizuta et al., 1997; Sharan et al., 1997; Esashi et al., 2005; Petalcorin et al., 2007; Zhou et al., 2007). In mammals and C. elegans, non-BRC repeat binding appears to be specific for RAD51 filaments (Davies and Pellegrini, 2007; Esashi et al., 2007; Petalcorin et al., 2007), whereas the BRC repeats can bind both RAD51 monomers and filaments (Galkin et al., 2005; Shivji et al., 2006).
Previous work has suggested that a BRCA2 homologue in T. brucei contains a remarkable expansion in BRC repeat number (Warren et al., 2002; Lo et al., 2003). Here, we have examined BRCA2 from T. brucei and related kinetoplastid parasites, and show that this is a recent evolutionary adaptation. To test if this is due to the use of homologous recombination in antigenic variation, we have generated T. brucei BRCA2 mutants and variants of the protein with alterations in conserved motifs, including the BRC repeats. We show, first, that BRCA2 acts in DNA repair, recombination and antigenic variation in T. brucei. Second, we demonstrate that the BRC repeat number or sequence composition of BRCA2 is critical in general repair and recombination but, remarkably, appears not to be important in the activation of early expressed VSG genes.
Results
A recent evolutionary expansion of BRC repeats in T. brucei BRCA2
Trypanosoma brucei and related, evolutionarily diverged kinetoplastid parasites each encode a single BRCA2 homologue (Warren et al., 2002; Lo et al., 2003). These proteins possess significant sequence similarity with BRCA2 homologues from vertebrates, plants and fungi in the DSS1-DNA binding region (Lo et al., 2003), within which most domains are conserved (Yang et al., 2002; C. Hartley and R. McCulloch, submitted). Regulation of the kinetoplastid BRCA2 proteins' function by DSS1 is also likely to be conserved, as a homologue of DSS1 is also identifiable in each kinetoplastid genome and the BRCA2–DSS1 interacting residues (Yang et al., 2002) are conserved in each protein. Finally, it seems likely that the kinetoplast BRCA2 proteins are nuclear, as localization signals can be detected in the T. brucei (Fig. 2A) and T. cruzi polypeptide sequences (data not shown).
Fig. 2.

Analysis of BRC repeat number in T. brucei BRCA2. A. The predicted domain organization of BRCA2 from T. b. brucei strain TREU927 is shown; 15 putative BRC repeats are indicated, as is the DSS1-DNA-binding domain (DBD) and two putative nuclear localization signals (NLSs). Arrows denote oligonucleotide primers used in minisatellite variant repeat (MVR) mapping. B. MVR mapping of BRC repeat number in genomic DNA from T. b. brucei strain Lister 427, T. b. rhodesiense isolate Eliane and T. b. gambiense isolate 222; size markers are indicated. C. A summary of the numbers of BRC repeats in the two alleles of BRCA2 in T. b. brucei (Tbb) strains TREU927, Lister 427 and EATRO795, and in isolates of the T. brucei subspecies T. b. rhodesiense (Tbr; Eliane) and T. b. gambiense (Tbg; 222); BRC numbers were inferred from MVR mapping (above), Southern analysis and sequencing of PCR clones of the BRC repeat regions. D. A Southern blot of restriction-digested genomic DNA from T. b. brucei strain Lister 427, probed with the BRCA2 ORF.
The only other clear similarity between kinetoplastid and other eukaryotic BRCA2 proteins lies in the BRC repeats (Bork et al., 1996; Bignell et al., 1997). However, the BRC repeat number and organization in T. brucei are highly unusual (Fig. 1 and Table S1). In general terms, it appears that the simpler the organism, the smaller the number of BRC repeats in BRCA2. In illustration, of 14 BRCA2 proteins identified in the genomes of a diverse range of unicellular organisms (Table S1), nine have between one and three BRC repeats. Conversely, of eight multicellular organisms, seven have three or more BRC repeats: most vertebrate BRCA2 proteins contain eight repeats, the plant A. thaliana has four, and the insects Drosophila melanogaster and Anopheles gambiae have three and 10 respectively. It seems plausible therefore that developmental complexity and/or increased genome size exerts an evolutionary pressure for greater numbers of repeats due to greater demands for control of homologous recombination. C. elegans BRCA2/CeBRC-2 appears to be an exception, however, with only one BRC repeat, although the reasons for this are unclear. Some protists are also exceptions, displaying greater BRC repeat numbers. Trichomonas vaginalis BRCA2 has 14 predicted BRC repeats, which may be related to the large genome size of this parasite (Carlton et al., 2007). However, this does not always provide an explanation, as Apicomplexan BRCA2 proteins, such as in Cryptosporidium parvum, Plasmodium falciparum and Toxoplasma gondii, have between six and eight predicted BRC repeats and T. brucei BRCA2 is predicted to contain 15.
Fig. 1.

Conservation of BRC repeats in eukaryotic BRCA2. The upper diagrams show the number and location of BRC repeat motifs (black bars) in BRCA2 proteins from a number of eukaryotes; the sizes of the BRCA2 polypeptides (in amino acids) are indicated. The boxed diagram shows conservation of BRC repeats in trypanosomatid BRCA2 proteins relative to H. sapiens; critical residues for RAD51 binding inferred by Lo et al. (2003) are indicated as a BRC fingerprint (O, polar; +, positively charged; I, hydrophobic; i, small hydrophobic).
The BRC repeats of T. brucei BRCA2 are unusual in two further ways. First, unlike the broad conservation of increased BRC repeat number in the apicomplexans, BRCA2 of T. brucei is distinct from closely related kinetoplastids (Fig. 1). T. cruzi and Leishmania major diverged from T. brucei 200–300 million years ago (Overath et al., 2001) and each BRCA2 has two non-identical BRC repeats. Even more strikingly, two Trypanosoma species, T. vivax and T. congolense, that belong to the same salivarian clade as T. brucei (Cortez et al., 2006) are predicted to possess BRCA2 proteins with only one and three BRC repeats respectively. The second unusual feature of T. brucei BRCA2 is that the 15 predicted BRC repeats are arranged as a tandem array in which 14 are identical in sequence [in fact, each repeat of the array is 44 amino acids (132 bp) in length, longer than the 35-amino-acid BRC motif; Fig. 1]. In all other organisms in which BRCA2 has more than one BRC repeat, with the notable exception of T. congolense, they are distributed unevenly in the polypeptide sequence and are degenerate in sequence outwith the key functional residues (Lo et al., 2003).
Before examining why T. brucei BRCA2 has such unusual structural organization, we checked that the above predictions do not arise from genome sequence compilation errors. We first performed minisatellite variant repeat (MVR) mapping on clones of a number of T. b. brucei strains and T. brucei subspecies (Fig. 2). In all cases, MVR PCR yielded the expected ladder of products. In T. b. brucei strain Lister 427, the largest product was equivalent to 12 BRC repeats and a pronounced, smaller band was generated with 10 repeats, suggesting two distinct BRCA2 alleles (Fig. 2A). In the T. b. brucei genome sequence strain TREU927, which was predicted to have 15 BRC repeats, each allele contained 11 repeats, while a further T. b. brucei strain, EATRO795, contained two alleles with 12 BRC repeats (Fig. 2C). MVR PCR of isolates from T. b. rhodesiense and T. b. gambiense suggested a lower number of BRC repeats than in T. b. brucei: each contained a larger allele with eight repeats and a smaller allele with five or six repeats respectively (Fig. 2B and C). To confirm these findings, we performed Southern mapping of BRCA2, demonstrating two distinct BRCA2 alleles of the expected size in both T. b. brucei Lister 427 (Fig. 2D) and T. b. gambiense (data not shown). Next, we PCR-amplified the BRC-repeat region from BRCA2 from each of the above T. brucei strains and subspecies, as well as from T. vivax and T. congolense, and sequenced the products after cloning. For T. brucei, this confirmed the predicted BRC repeat organization: in all cases, the most C-terminal coding repeat (‘BRC repeat 15’, Fig. 1) was degenerate in sequence relative to all the upstream repeats, which were virtually identical at the nucleotide level (< 1 base change per repeat, on average) both within a given BRCA2 gene and between strains/subspecies (data not shown). BRCA2 from T. vivax was confirmed as containing a single BRC repeat, while the T. congolense strain used here (TREU1457) contained two very similar (5 bp differences) repeats, rather than the three predicted from genome sequencing (strain IL3000). Taken together, these data indicate that the BRC repeat organization in T. brucei BRCA2 is a recent evolutionary expansion, generating numbers that, while variable between isolates, are significantly higher than BRCA2 orthologues in closely related kinetoplastid parasites.
T. brucei BRCA2 acts in homologous recombination and antigenic variation
To examine the functions of BRCA2 in T. brucei we generated null mutants in bloodstream stage cells, deleting the entire BRCA2 ORF (Fig. 3) by two rounds of transformation, replacing the two alleles with constructs encoding resistance to blasticidin and puromycin. This was performed in both the Lister 427 T. brucei strain and in a transgenic derivative named 3174 (McCulloch et al., 1997; McCulloch and Barry, 1999; Proudfoot and McCulloch, 2005), which allows analysis of switching mechanisms. In each strain, we independently derived two lines of BRCA2 heterozygous (+/−) and homozygous (−/−) mutants, and re-expressed BRCA2 in one of the −/− mutants (−/−/+). In all cases, the mutants (Fig. 3B) and re-expressers were confirmed by Southern mapping and reverse transcription polymerase chain reaction (RT-PCR) (data not shown). Consistent with a role for BRCA2 in DNA repair, the −/− mutants displayed increased sensitivity to methyl methanesulphonate (MMS) or bleomycin-induced DNA damage relative to wild type (WT), +/− or −/−/+ cells (C. Hartley and R. McCulloch, submitted). In addition, the brca2−/− mutants displayed replication or cell division defects not observed in mutants of other T. brucei DNA repair and homologous recombination genes (C. Hartley and R. McCulloch, submitted).
Fig. 3.

Assaying the role of T. brucei BRCA2 in homologous recombination. A. The complete BRCA2 ORF was deleted (triangle) in wild type (WT) bloodstream stage T. brucei cells and replaced by cassettes encoding resistance to blasticidin (ΔBRCA2::BSD) or puromycin (ΔBRCA2::PUR). B. A Southern blot of SacII- and StuI-digested genomic DNA from WT cells (two distinct BRCA2 alleles can be seen), and from two independently generated heterozygous (+/−1, 2) and homozygous mutants (−/− 1, 2). Size markers (kb) and the positions of intact and disrupted BRCA2 alleles are indicated. C. To assay recombination, the number of transformants recovered (per 106 cells put on antibiotic selection) when the construct tub-HYG-tub was electroporated into the parasite cell lines was measured; values shown are averages from three experiments, and bars indicate standard errors. BRCA2 re-expressers (−/−/+) were generated by re-inserting the BRCA2 ORF into one of the −/− mutants.
To examine the contribution of BRCA2 to T. brucei homologous recombination, the transformation efficiency of WT, +/−, −/− and −/−/+ cells was compared following electroporation with targeting constructs. In the Lister 427 mutants, we used linearized tub-HYG-tub plasmid, which targets a hygromycin resistance gene to the tubulin array, replacing an α tubulin ORF by homologous recombination on terminal flanking sequence (240 bp of tubulin β–α intergenic sequence upstream, and 330 bp of α–β intergenic sequence downstream). Transformants were generated in the brca2−/− mutants 12.5- to 22.5-fold less efficiently than the other cells (Fig. 3C). Very similar results were found in 3174 mutants using an equivalent plasmid in which HYG was replaced by a phleomycin resistance gene (data not shown). In all cases, examples of the brca2−/− transformants were characterized by Southern blotting, which showed that all integrations had targeted the tubulin array via homology (data not shown). These results are comparable with mutants of other T. brucei homologous recombination factors (RAD51, RAD51-3, RAD51-5 and MRE11) (McCulloch and Barry, 1999; Conway et al., 2002; Robinson et al., 2002; Proudfoot and McCulloch, 2005) and indicate that BRCA2 acts in homologous plasmid integration, but BRCA2-independent pathways also operate.
We next used the mutant cell lines made in the 3174 strain to examine the ability of the brca2−/− mutants to undergo antigenic variation. Measuring the efficiency at which VSG-switched variants arose (Fig. 4A) indicated that ablation of BRCA2 reduced the VSG-switching frequency around 8- to 11-fold relative to the WT, +/− or −/−/+ cells. The extent of this switching impairment is very similar to that seen in RAD51 and RAD51-3 mutants (McCulloch and Barry, 1999; Proudfoot and McCulloch, 2005), confirming that much of VSG switching involves homologous recombination. Characterization of the reactions that gave rise to VSG-switched variants indicated that gene conversion and transcriptional switching events could still be detected in the brca2−/− cells (Fig. 4B), indicating that the reduced VSG switch frequency was accounted for by impairment of both reaction pathways. This surprising result is equivalent to VSG-switching phenotypes observed in RAD51 and RAD51-3 mutants (McCulloch and Barry, 1999; Proudfoot and McCulloch, 2005) and may indicate that BRCA2 acts in conjunction with these factors in this process.
Fig. 4.
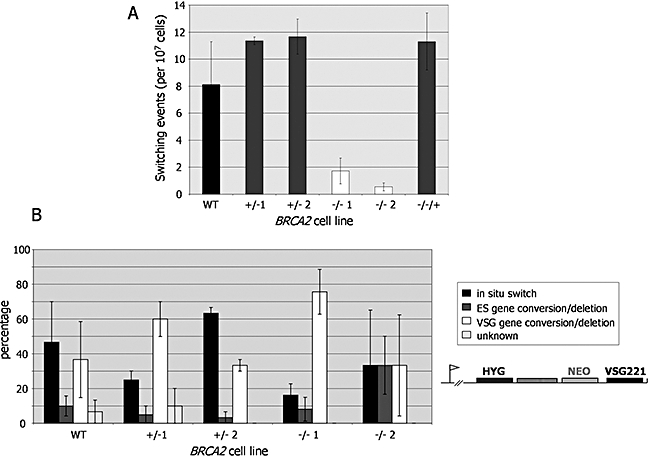
Assaying the role of T. brucei BRCA2 in VSG switching. A. VSG-switching frequencies for wild-type T. brucei cells (3174.2; WT) were compared with two independently generated heterozygous (+/−1, 2) and homozygous (−/− 1, 2) BRCA2 mutants, in addition to a cell line in which BRCA2 was re-expressed from the tubulin locus (−/−/+). The values shown are the means of at least three independent experiments, and bars represent standard errors. B. The mechanism of VSG switching used in each cell line was determined by assaying for the expression of hygromycin and G418 resistance from the antibiotic marker genes (HYG and NEO respectively) inserted around VSG-associated 70 bp repeat sequences (hatched box) in the active VSG221 expression site (ES) in strain 3174 (diagram shows this gene arrangement; The flag denotes promoter), as well as using PCR to assess whether HYG, NEO or VSG221 had been removed by gene conversion or DNA deletion. The percentage of clones that had undergone VSG switching by transcriptional (in situ) switching, gene conversion/deletion of HYG, NEO and VSG221 (ES gene conversion/deletion) or gene conversion/deletion of only NEO and VSG221 (VSG gene conversion/deletion) for each cell line is shown; bars denote standard errors, and switched variants that do not conform to the above mechanisms are indicated as ‘unknown’.
The BRC repeat organization of T. brucei BRCA2 is an important determinant of homologous recombination efficiency
To examine why T. brucei BRCA2 has evolved such an unusual arrangement of BRC repeats, we generated a number of BRCA2 variants (Fig. 5A). To test if BRCA2 with a single BRC repeat is competent in DNA repair and recombination we examined BRCA2 from T.vivax (TvBRCA2), which naturally possesses a single BRC repeat (Fig. 1). TvBRCA2 and T. brucei BRCA2 (TbBRCA2) share 26% overall sequence identity, rising to 42% C-terminal to the BRC repeats. Sequence comparisons predict that TvBRCA2 will bind T. brucei RAD51: the BRC repeat retains all the key residues for RAD51 interaction predicted by Lo et al. (2003) (Fig. 1); and the single RAD51 orthologue in each Trypanosoma species share 79% sequence identity (87% similarity) with each other, including conservation of all residues predicted to bind BRC repeats (data not shown) (Lo et al., 2003). We also manipulated the T. brucei BRCA2 gene, reducing the BRC repeat number to just one. In this protein (1BRC) only the variant, C-terminal BRC repeat (‘repeat 15’; Fig. 1) is retained, but all other parts of BRCA2 are unaltered. Despite being distinct from the upstream repeats, the C-terminal BRC peptide retains residues predicted to be critical for binding RAD51 (Lo et al., 2003). Next, a truncated version of T. brucei BRCA2 was generated (BRCrep) that encodes only the BRC repeat region plus a C-terminal predicted nuclear localization signal (NLS); all C-terminal sequence (including the DSS1-DNA-binding domain) was deleted, as was 80-amino-acid N-terminal to the BRC repeats. Finally, a BRCA2 variant was generated in which the BRCrep polypeptide was translationally fused to the T. brucei Replication Protein A (RPA) 50 kDa subunit, which is homologous to the 70 kDa RPA-1 protein of other eukaryotes and binds single-stranded DNA via a conserved oligonucleotide/oligosaccharide binding fold domain in Leishmania (Neto et al., 2007). This BRC–RPA fusion is functionally equivalent to similar polypeptides examined in U. maydis (Kojic et al., 2005) and mammalian cells (Saeki et al., 2006).
Fig. 5.

BRCA2 variants and polypeptide fusions to assess the function of the BRC repeats in T. brucei recombination and VSG switching. A. Functional domains of BRCA2 variants and polypeptide fusions analysed by expression in bloodstream stage brca2−/− mutants are shown. Full-length T. brucei BRCA2 may contain up to 15 BRC repeats, a conserved DSS1-DNA-binding domain (DBD) and two putative nuclear localization signals (NLSs). T. vivax BRCA2 displays conservation of the DBD, but has only one BRC repeat and NLSs have not been predicted. 1BRC differs from full-length T. brucei BRCA2 only in reduction of the BRC array to a single repeat. BRCrep is a polypeptide fragment of T. brucei BRCA2 encompassing the BRC repeats and 33 downstream amino acids, including a bipartite NLS. BRC–RPA is a fusion of the BRCrep polypeptide to the 50 kDa T. brucei replication protein A subunit. B. Homologous recombination efficiency was assayed by determining the number of transformants recovered (per 106 cells put on antibiotic selection) when the construct tub-HYG-tub was electroporated into wild-type (WT) cells, brca2−/− mutants and −/− cells expressing the BRCA2 variant polypeptides detailed above (−/−/+). C. VSG-switching frequencies of WT (Lister 427) T. brucei cells, brca2−/− mutants and −/− cells expressing the BRCA2 variant polypeptides are shown. Values are the means of at least three independent experiments, and bars represent standard error.
Each of the above variants, plus full-length TbBRCA2, were expressed in one of the brca2−/− Lister 427 cell lines by integration of the ORFs into the T. brucei tubulin array. All were expressed using the same integration construct, meaning that their level of expression, assuming equal translation efficiencies, should be comparable and correct integration was confirmed by Southern analysis (data not shown). Expression of most of the proteins could be demonstrated by complementation of phenotypic defects: BRC–RPA and TbBRCA2 reverted the MMS sensitivity of the brca2−/− cells (C. Hartley and R. McCulloch, submitted), TbBRCA2, TvBRCA2 and 1BRC reverted the replication/cell division defect of the mutants (C. Hartley and R. McCulloch, submitted) and all but BRCrep was active in VSG switching (see below). To examine if the BRCA2 variants function in homologous recombination, the transformation efficiency of tub-HYG-tub was compared in the different expresser cell lines (Fig. 5B). As we have shown already (Fig. 3), full-length TbBRCA2 caused reversion of the brca2−/− integration defect, with transformation frequencies equivalent to WT and +/− cells. In contrast, none of the variant proteins functioned efficiently in this assay. Although transformants arose in each expresser cell line at slightly higher rates than −/− cells, in each case this was significantly lower (around 4–5) than the frequencies seen in the WT or TbBRCA2 −/−/+ cell lines (P-values < 0.001 for all variants; two-sample t-test). This indicates that each is impaired in this pathway of homologous recombination.
T. brucei BRCA2 with a single BRC repeat is impaired in RAD51 focus formation
Phleomycin treatment and induction of a DNA double-strand break in T. brucei causes re-localization of RAD51 to discrete subnuclear foci (Proudfoot and McCulloch, 2005; Glover et al., 2008). Formation of such foci appears to be a conserved response to DNA damage that is controlled by a number of factors (see Lisby and Rothstein, 2004; Tarsounas et al., 2004 for reviews), including BRCA2 in other eukaryotes (Tarsounas et al., 2003; Yu et al., 2003; Kojic et al., 2005; Martin et al., 2005). To examine if this is true also in T. brucei, we examined RAD51 localization by indirect immunofluorescence, before and after phleomycin treatment, in the BRCA2 mutants (Fig. 6). As we have described before, very few RAD51 foci were detectable in the absence of damage (only around 3–10% of cells), whereas treatment with 1.0 μg ml−1phleomycin for 18 h resulted in detectable foci in > 75% of WT, +/− and full-length TbBRCA2 −/−/+ cells (Fig. 6B). In contrast, no evidence was found for RAD51 foci induction in the brca2−/− cells, suggesting that BRCA2 has a profound role in this response (Fig. 6B). The −/− cells in which either TvBRCA2, the 1BRC variant or the BRCrep truncation was expressed displayed a similar deficiency in RAD51 foci formation (Fig. 6). In contrast, BRC–RPA was capable of supporting foci formation (Fig. 6A), despite being impaired in homologous integration. Quantification of the response of the −/−/+ BRC–RPA cells showed they were, in fact, somewhat less efficient at making RAD51 foci than WT cells (Fig. 6B), which correlates the partially increased sensitivity to phleomycin we have observed (C. Hartley and R. McCulloch, submitted). Nevertheless, as has been described in mammalian cells (Saeki et al., 2006) and in U. maydis (Kojic et al., 2005), a fusion of the T. brucei BRC repeats with a heterologous DNA-binding domain is capable of functioning in DNA damage repair. Why, then, the T. brucei BRC–RPA fusion is non-functional in homologous recombination, unlike the equivalent protein in mammalian cells (Saeki et al., 2006), is not clear. This may reflect differences in the assays used in Saeki et al. (2006) compared with here, or distinct activities of the parasite and host BRCA2 proteins.
Fig. 6.
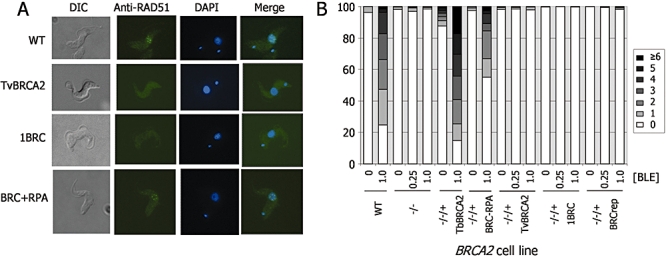
RAD51 subnuclear foci in T. brucei bloodstream stage cells. A. An example of a wild-type (WT) T. brucei cell with discernible subnuclear RAD51 foci following 18 h growth in 1.0 μg ml−1 phleomycin is shown (top). Below are examples of brca2−/− cells expressing either T. vivax BRCA2, T. brucei BRCA2 with only one BRC repeat (1BRC) or a fusion of the T. brucei BRC repeats of BRCA2 and RPA following the same treatment. Cells were visualized by differential interference contrast (DIC; left), the DNA was stained with DAPI (right centre) and RAD51 (left centre) was visualized by indirect immunofluorescence. Merged DAPI and RAD51 images are also shown (right). B. Quantification of the number of RAD51 foci detectable after 18 h growth in differing concentrations of phleomycin (BLE; μg ml−1); BRCA2 variants expressed in the brca2−/− cells (indicated by −/−/+) are detailed in Fig. 5.
T. brucei BRCA2 with a single BRC repeat is functional in VSG switching
To examine if the BRCA2 variants support VSG switching, we used the expresser cell lines in Lister 427, rather than generating equivalent cell lines in strain 3174, thereby allowing direct comparison with the phenotypic analyses discussed above. Strain 3174 is useful in analysing VSG switching as it contains antibiotic resistance markers in the active VSG ES (encoding VSG221) (McCulloch et al., 1997), meaning that singular VSG expression in a population can be selected by antibiotic pressure, and switched variants allowed to arise over a defined number of generations by removing that selection. The antibiotic markers can also be used to characterize the switching events that gave rise to clonal switched variants, which are recovered by immune selection in vivo. Lister 427 also expresses the VSG221 ES, but it is not genetically marked, so exclusive expression cannot be maintained and examination of VSG switching is limited to measuring VSG-switching frequency. Before analysing VSG switching, we first confirmed, by Western analysis, that VSG221 continued to be expressed in each expresser cell line (data not shown). We then followed an abridged version of the strain 3174 VSG-switching assay described previously (McCulloch et al., 1997; McCulloch and Barry, 1999). Although there was greater variation, the VSG-switching frequency determined for WT Lister 427 cells (Fig. 5C) was comparable with WT 3174 (see Fig. 4), and the brca2−/− mutants were still seen to be impaired (Fig. 5C). Strikingly, both cell lines expressing a BRCA2 variant with a single BRC repeat, and the cells expressing the BRC–RPA fusion, were unaltered in their capacity to undergo VSG switching relative to either the WT cells or the −/−/+ TbBRCA2 cells (Fig. 5C). Only the cells expressing the isolated BRC repeat domain were impaired in VSG switching. These results indicate that the tandem array of identical BRC repeats in T. brucei BRCA2, despite being crucial for efficient plasmid integration, re-localization of RAD51 following phleomycin damage and DNA repair (C. Hartley and R. McCulloch, unpublished), is not a determinant of VSG switching efficiency during an acute infection.
T. brucei BRCA2 mutants display gross chromosomal rearrangements in their megabase chromosomes
BRCA2 mutation in mammalian cells causes accumulation of aberrant chromosomes, including chromosome loss, breakage and translocations (Patel et al., 1998; Yu et al., 2000). Similar gross chromosomal rearrangements (GCRs) have been described in U. maydis BRCA2/Brh2 mutants (Kojic et al., 2002). To examine this in T. brucei, each of the +/− and −/−BRCA2 cells lines, as well as WT cells, were cloned and grown for ∼290 generations in vitro. The cells were then re-cloned, and the karyotypes of a number of clones were analysed by Pulsed Field Gel (PFG) electrophoresis (Fig. 7A). The megabase chromosomes of T. brucei range in size from ∼1 to 6 Mb in Lister 427 strain (Melville et al., 2000), and in this gel separation the greatest size discrimination is seen from ∼1 to 2.5 Mb. Considerable variation in chromosome size between brca2−/− clones, indicative of GCR, was apparent even from ethidium bromide staining (particularly mutant 2), in contrast to the relative stability in the +/− or WT clones. However, closer examination suggests two things. First, most such GCR involved reduction in chromosome size. Second, a trend towards reduction in chromosome size, though not as severe as the −/− clones, was also apparent in the BRCA2+/− cells, perhaps indicative of haploinsufficiency. Probing the PFGs with VSG121 (a five-gene family), VSG221 (a single-copy VSG in the active VSG ES) or GPI (a single-copy gene encoding glucose 6-phosphate isomerase) appears to confirm these findings. Only in two clones (lanes 19 and 21), when probed with VSG221, did we find evidence for an increase in chromosome size. VSG121 hybridizes to two chromosomes of approximately 2.1 and 2.3 Mb, and both appear to be smaller in all the brca2−/− clones relative to the WT cells (by as much as 100 kb in some cases). The same chromosomes also appear to have become smaller in two to three of the five BRCA2+/− clones, though to a lesser extent. The VSG221 and GPI blots show that the extent of size change in the chromosomes of the brca2−/− cells can be very severe (for instance, around 500 kb in three brca2−/− clones probed with VSG221). In addition, the smaller homologue of chromosome 1 that harbours GPI (around 2.3 Mb) appears to have reduced in size in three to four of the five BRCA2+/− clones. A Southern blot of XmnI-digested genomic DNA from all of the above clones, probed with VSG121, confirms that the chromosomal changes can involve loss of genetic material, as 11 of the 12 brca2−/− clones had lost at least one of the five gene copies (Fig. 7B). Notably, in no case was the telomeric copy lost, despite the gene being silent in this strain. Indeed, all clones continued to express VSG221 (data not shown), consistent with telomeric sequences being relatively stable.
Fig. 7.

Karyotype changes in BRCA2 mutants. A. The top and bottom panels to the left show ethidium bromide-stained PFG electrophoretic separations of intact chromosomes from clones of wild-type (WT) cells and from two independently generated BRCA2 heterozygous (+/−) and homozygous (−/−) mutants that had been grown in vitro for ∼290 generations; size markers (Mb) are shown, and lane numbers are indicated. To the right of this are three Southern blots in which the PFGs were probed, successively, with VSG121, VSG221 and glucose 6-phosphate isomerase (GPI). B. A Southern blot of XmnI-digested genomic DNA from samples of the WT, +/− and −/− mutant clones (above). The DNA was separated on a 0.8% agarose gel and the blot probed with VSG121, of which one telomeric (TEL) and four putative subtelomeric array copies are present (Ii–iv). Size markers (kb) and lane numbers are indicated, and clones in which at least one gene copy had been deleted are indicated by asterisks.
Remarkably, the same GCRs were not observed in karyotyping of either the intermediate or minichromosomes (Fig. 8). For both these classes of chromosome, which contain primarily VSG and VSG ES sequences as the main genetic material that can be expressed, the karyotype was substantially more stable than the megabase chromosomes in the brca2−/− mutants, with no severe differences in chromosome size detectable compared with the BRCA2+/− and WT cells. This indicates that although BRCA2 mutation in T. brucei results in GCRs, this is limited, at least predominantly, to the megabase chromosomes.
Fig. 8.
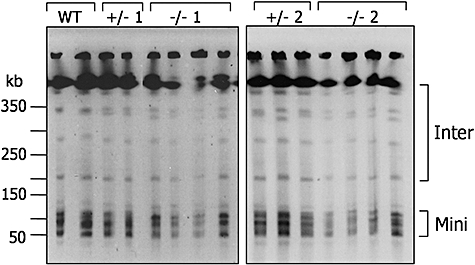
T. brucei intermediate and minichromosomes in BRCA2 mutants. The panels show ethidium bromide-stained PFG electrophoretic separations of intermediate chromosomes (Inter) and minichromosomes (Mini) from clones of wild-type (WT) cells and from two independently generated BRCA2 heterozygous (+/−) and homozygous (−/−) mutants that had been grown in vitro for ∼290 generations; size markers (kb) are shown.
Discussion
In this report we have characterized a homologue of BRCA2 in the parasite T. brucei and document the function of the protein in the maintenance of genome stability, in recombination and in antigenic variation. Although BRCA2 is widely conserved in the eukaryotic kingdom (Warren et al., 2002; Lo et al., 2003), T. brucei is arguably the most diverged eukaryote in which its cellular functions have been examined. The structural organization of T. brucei BRCA2 is highly unusual, possessing an expansion in the number of BRC repeat motifs, which are critical in the interaction of BRCA2 with the key enzyme of eukaryotic homologous recombination, RAD51 (Pellegrini and Venkitaraman, 2004; Shivji and Venkitaraman, 2004; Sung and Klein, 2006). Most characterized single-celled eukaryotes encode BRCA2 homologues with one to three BRC repeats, in contrast with the majority of multicellular eukaryotes, where BRCA2 normally has three or more BRC repeats (Lo et al., 2003). The BRC repeat number in T. brucei BRCA2 is variable between different strains and subspecies of the parasite, but is always between three- and sixfold greater than found in most single-celled organisms (with the exception of apicomplexans and T. vaginalis; see below), and in some strains is greater than has been described anywhere else in nature. Given that this expansion appears not to be present in closely related kinetoplastid organisms, and that the BRC repeat organization takes the form of a tandem array of repeats that are virtually identical in sequence (which contrasts with the dispersed, diverged repeats found in most other organisms) this appears to be a recent evolutionary adaptation. The high-sequence homology of the BRC repeats most likely results in ongoing array expansion and contraction, explaining the variation in repeat number we have documented.
No work to date has examined why some BRCA2 molecules require multiple BRC repeats while others function with as few as one repeat. The simplest explanation is that, in general, the evolution of eukaryotes with increasing genome size and developmental complexity requires greater control or greater overall activity of homologous recombination, which is reflected in more extensive interactions between BRCA2 and RAD51. For example, a larger number of BRC motifs might be needed to sequester the putatively greater amount of RAD51 until it is needed for repair, perhaps preventing uncontrolled recombination. This would be compatible with the demonstration that the BRC repeats can disrupt the RAD51 filament, block its assembly and impair recombination, and infers that BRCA2 can maintain RAD51 as a monomer via the BRC repeats (Chen et al., 1999; Davies et al., 2001; Pellegrini et al., 2002; Shin et al., 2003). Indeed, studies of GFP-labelled RAD51 have suggested that some RAD51 exists in the mammalian nucleus in relatively immobile pools, one of which is BRCA2-bound, as well as in another pool of relatively greater mobility (Essers et al., 2002; Yu et al., 2003). Alternatively, the number of BRC repeats may vary to ensure that sufficient RAD51 is available at the site of DNA double strand breaks, the abundance of which might be dependent on genome size and/or sequence complexity (Pellegrini et al., 2002). Such an explanation implies that BRCA2–RAD51 interaction involves more than just sequestration, for which there is accumulating evidence. It is now clear that RAD51 binding to BRCA2 through non-BRC sequences in both mammals (Davies and Pellegrini, 2007; Esashi et al., 2007) and C. elegans (Petalcorin et al., 2007) is specific for, and stabilizes, RAD51 filaments. In vitro studies have also shown that isolated BRC motifs can, in some conditions, bind RAD51 filaments without causing disruption (Galkin et al., 2005), and that a polypeptide from Homo sapiens BRCA2 spanning all eight BRC repeats can promote RAD51 strand exchange (Shivji et al., 2006). Furthermore, a fusion of the BRC repeats from either H. sapiens (Saeki et al., 2006) or U. maydis (Kojic et al., 2005) BRCA2 with RPA, thereby excluding non-BRC-RAD51 binding motifs, also functions in DNA repair and recombination. These data indicate that BRC-RAD51 binding can play an active role in RAD51 recombination, perhaps during the delivery of RAD51 to sites of damage through BRCA2 binding to single-strand DNA tails (Yang et al., 2005).
The evolutionary pressures that have dictated the expansion of BRC repeat numbers in T. brucei are unknown, but could either reflect specific aspects of the biology of this parasite or reveal general aspects of BRCA2 function. The hypothesis we sought to test is that the BRC repeat expansion is a consequence of antigenic variation. VSG switching is, at least in part, reliant on RAD51-dependent homologous recombination, as mutation of RAD51 impairs the process (McCulloch and Barry, 1999), as does mutation of the RAD51 paralogue RAD51-3 (Proudfoot and McCulloch, 2005). The work here adds to that picture by showing that BRCA2 is also critical for efficient VSG switching, as brca2−/− mutants are significantly impaired in this reaction. It should be noted that the impairment of VSG switching in all these DNA repair mutants appears to affect the activation of new VSGs both by gene conversion and by transcriptional switching between VSG ESs, questioning whether these reactions are enzymatically and mechanistically distinct (Proudfoot and McCulloch, 2005). Irrespective of this, antigenic variation can occur at rates measured to be substantially greater than background mutation (Turner and Barry, 1989), are primarily driven by gene conversion (Robinson et al., 1999) and involve the activation of VSGs from an archive of silent genes numbering > 1000 and found dispersed throughout the subtelomeres of most, if not all, T. brucei chromosomes (Barry et al., 2005; Marcello and Barry, 2007). Any, and perhaps all, of these factors might explain why BRCA2 in T. brucei has evolved such a specialized structure. However, we find that BRCA2 with only one BRC repeat, either retaining only the divergent C-terminal repeat or being BRCA2 from T. vivax, is capable of supporting efficient VSG switching. This suggests that the expanded BRC repeat number of T. brucei BRCA2 is not a consequence of antigenic variation, with two important caveats. First, we have examined VSG switching in Lister 427, a monomorphic strain that undergoes VSG switching at rates of only around 1 × 10−6 switches per cell per generation (see Figs 4 and 8), less than the pleomorphic cells in which high switching rates (up to 1 × 10−2) were measured. However, the BRC repeat number of BRCA2 in Lister 427 is among the highest we characterized, and BRC numbers vary considerably in T. brucei, which appears to suggest that the selective pressures that expanded the BRC array are retained in low-switching strains. Second, our assay measures VSG switching only very early in an infection, when telomere-proximal, intact VSG genes are predominantly activated (Pays, 1989; Morrison et al., 2005; Marcello and Barry, 2007). Later in infections VSG pseudogenes and gene fragments become the preferred substrates for switching (Thon et al., 1990; Marcello and Barry, 2007) and it is therefore possible that T. brucei BRCA2 structure reflects distinctive features of these reactions.
The finding that VSG switching may not be the basis for the unusual structure of T. brucei BRCA2 has further support. T. congolense and T. vivax are closely related to T. brucei and also survive in mammals through antigenic variation of a VSG surface coat, which in T. congolense has been shown to involve gene duplication (Majiwa et al., 1985). Despite this, the BRCA2 orthologues of these species possess two to three and one BRC repeats, respectively, which is more typical of single-celled eukaryotes. It is worth noting, however, that the genome annotation of T. congolense BRCA2 predicts three highly related BRC repeats present in an array, perhaps indicating an abridged version of the BRC expansion in T. brucei. It will be interesting to see how the size and organization of the VSG system in these parasites relate to T. brucei. Nevertheless, BRC repeat expansion in protists is not limited to T. brucei. Previous work showed that BRCA2 homologues in Plasmodium have six BRC repeats (Lo et al., 2003), and we now show that this may be a common feature of Apicomplexans. Furthermore, T. vaginalis BRCA2 appears to possess 14 BRC repeats. Whether or not the BRCA2 organization in these parasites shares a functional basis with T. brucei is unknown.
Our data illustrate that, whatever the basis for the selection of BRC repeat expansion, this structural organization of T. brucei BRCA2 is critical for general repair and recombination. BRCA2 with a single repeat does not support efficient homologous recombination and impairs the ability of the cell to repair both MMS and phleomycin-induced DNA damage (C. Hartley and R. McCulloch, submitted). Most likely, this reflects impaired BRCA2 interaction with RAD51, as visible re-localization of RAD51 to subnuclear foci following phleomyin damage was only seen in parasites expressing full-length TbBRCA2 or BRC–RPA. Why, then, can BRCA2 with only one BRC support efficient VSG switching? A number of possibilities might be considered. The first is that general repair/recombination and antigenic variation have differing requirements, the former involving extensive interaction between BRCA2 and RAD51 through a large number of BRC repeats and the latter needing less extensive interaction. A second possibility is that the BRC repeat organization of T. brucei BRCA2 represents a dichotomy in function between the majority, conserved repeats and the downstream divergent repeat: for instance, the former class may provide interactions with RAD51 interaction that directs general repair/recombination, while the latter have evolved for a specific form of RAD51 interaction during antigenic variation. For either of these scenarios to be true, antigenic variation in T. vivax and T. congolense must differ from that of T. brucei and such a distinction from general repair/recombination must be absent. A final possibility therefore is that the T. vivax BRCA2 protein and the T. brucei variant retaining only the divergent C-terminal BRC repeat do not interact with T. brucei RAD51 sufficiently to mediate repair/recombination. If correct, the ability of these proteins to support VSG switching is perplexing, but may suggest that the reaction involved (which is mediated by RAD51, RAD51-3 and BRCA2) has significant mechanistic distinctions from general recombination pathways in the parasite. In this light, it is worth noting that two further proteins that act in general repair and recombination in T. brucei, RAD51-5 and MRE11 (Robinson et al., 2002; Proudfoot and McCulloch, 2005), appear not to influence VSG switching.
Further work will be required to evaluate the above hypotheses. Examining the unusual BRC repeat expansion of T. brucei BRCA2 could shed light on both the biochemistry of recombination during antigenic variation and the evolution of BRCA2 for recombination in general. For instance, it is intriguing that the GCRs found in the T. brucei brca2−/− mutants were very similar to those described previously in mutants of MRE11 (Robinson et al., 2002). In each case the chromosome size changes were primarily due to sequences loss, telomeric elements appeared to be relatively unaffected and rearrangements were only clearly seen in the megabase chromosomes. It is tempting to speculate that this indicates a shared function of the proteins in maintenance or use of the subtelomeric VSG arrays, but it is also possible that this reflects broadly conserved roles in genome stability.
Experimental procedures
Assaying recombination and VSG switching
Trypanosoma brucei Lister 427 bloodstream cells of strains MITat1.2a and 3174 (McCulloch et al., 1997) were grown at 37°C in HMI-9 medium (Hirumi and Hirumi, 1989) and transformed by electroporation as described previously (Conway et al., 2002; Bell and McCulloch, 2003). Details of the generation of BRCA2 mutants are described elsewhere (C. Hartley and R. McCulloch, submitted). To assay recombination, 5 × 107cells of each cell line, grown to a maximum of 2 × 106 cells ml−1, were transformed with 5 μg of XhoI- and XbaI-digested tub-HYG-tub, recovered in non-selective media for three generations (taking account of growth rate) and transformants were selected with 5 μg ml−1 hygromycin. For WT and +/− cells, 5 × 106 electroporated cells were plated over 24 wells in 1.5 ml well−1, whereas 2 × 107−/− cells were plated over 48 wells. VSG-switching frequencies in 3174 cell lines were determined as described previously (McCulloch and Barry, 1999; Proudfoot and McCulloch, 2005; 2006). For the Lister 427 MITat1.2a cell lines, essentially the same procedure was used, but with some modifications. At least three mice were infected with 2 × 105cells of each BRCA2 variant cell line, and the infections cured by cymelarsan treatment after the parasitaemia reached > 5 × 107 cells ml−1 of blood, generating mice immune to the VSGs expressed. To select for switched variants, 4 or 8 × 107 cells of each BRCA2 variant cell line (growing in vitro) were inoculated into a mouse previously infected with the same variant, and recovered 24 h later by exsanguination. The number of switched variants in the blood was measured as described previously (McCulloch and Barry, 1999; Proudfoot and McCulloch, 2005; 2006). Details of the BRCA2 variant constructs and targeting are described elsewhere (C. Hartley and R. McCulloch, submitted).
MVR mapping
For each T. brucei strain or subspecies, the BRCA2 ORF was PCR-amplified from genomic DNA with Taq DNA Polymerase (ABgene) and the primers TbBRCA2for (5′-ATGAGCCACAAAAAAGGAAGACAAGGC) and TbBRCA2rev (5′-TTCTCGCATAAGATCAGCG) to provide a substrate for MVR PCR reactions, which contained 5 μM primers TbBRCrepfor (5′-GCGGTACAAGGAAATTCCAC) and TbBRCreprev (5′-AGGAGGCACCTGCTCCCGAA) and 5 U of Taq DNA polymerase (ABgene). MVR PCR was performed in 75 mM Tris-HCl (pH 8.8), 20 mM (NH4)2SO4, 0.01% (v/v) Tween-20 and 1.5 mM MgCl2 for 18, 21 or 28 cycles of 95°C for 1 min, 55°C for 1 min and 72°C for 4 min, and the products were separated by 1.5% agarose gel electrophoresis.
RAD51 immunofluoresence
Trypanosoma brucei in culture were harvested by centrifugation at 583 g for 10 min at room temperature, washed in PBS and re-suspended in 100 μl of PBS. One millilitre of 1% (v/v) formaldehyde in PBS was added and the sample incubated at 4°C for 1 h. The cells were then centrifuged at 6000 g for 1 min, washed twice in chilled PBS, washed once in 500 μl of chilled 1% BSA in H2O, re-suspended in 30 μl of the same solution and smeared across a slide and allowed to air dry for 3 h. After re-hydration in PBS for 5 min, the slides were blocked by adding 50% fetal bovine serum in PBS (FBS/PBS) for 15 min. Blocking solution was then removed and the slides were transferred to a dark, humid chamber before adding rabbit polyclonal anti-TbRAD51 antibody (Proudfoot and McCulloch, 2005) diluted 1:500 in 3% FBS/PBS for 45 min. The slides were then washed twice in PBS for 5 min, returned to the humid chamber and Alexa 488-conjugated goat-derived anti-rabbit IgG, diluted 1:1000 in 3% FBS/PBS, added and incubated for 45 min at room temperature. Finally, the slides were washed twice in PBS for 5 min, allowed to air dry, then mounted in Vectashield with DAPI (4,6-diamidino-2-phenylindole) (Vector Laboratories). Fluorescence microscopic analysis was performed using an Axioskop 2 microscope (Zeiss) and images were obtained with Openlab software (Improvision).
PFG electrophoresis
Agarose plugs containing genomic DNA from 5 × 107 cells were prepared as described previously (Barnes and McCulloch, 2007). A CHEF-DR III (Bio-Rad) apparatus was used for electrophoretic separation. 1× TB1/10E (90 mM Tris base, 90 mM boric acid, 2 mM EDTA) and 1.2% agarose (Seakem LE, BioWhittaker Molecular Applications) was used for the separation of megabase chromosomes, whereas 0.5× TBE (45 mM Tris base, 45 mM boric acid, 10 mM EDTA) and 1.0% agarose was used for the separation of smaller chromosomes. Agarose plugs were prepared by three rounds of dialysis in the appropriate electrophoresis buffer. Gels were electrophoresed at 15°C, either at 2.5 V cm−1 for 144 h with an initial switch time of 1400 s and final switch time of 700 s for separation of megabase chromosomes, or 5.8 V cm−1 for 24 h with initial and final switch times of 20 s for separation of smaller chromosomes. DNA was visualized by UV illumination following staining of the gels in 0.2 μg ml−1 ethidium bromide. Southern blots were prepared by capillary transfer onto hybond-XL membrane (Amersham), and were probed with [α-32P]-labelled DNA generated by random priming and washed to 0.2× SSC, 0.1% SDS at 65°C.
Acknowledgments
We thank L. Marcello, R. Dobson and C. Stockdale for comments, and L. Pellegrini, J. Nickoloff, C.R. Machado and colleagues in WCMP for discussions. The Medical Research Council, Royal Society and Wellcome Trust are thanked for funding.
Supplementary material
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1365-2958.2008.06230.x (This link will take you to the article abstract).
Please note: Blackwell Publishing is not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Barbet AF, Kamper SM. The importance of mosaic genes to trypanosome survival. Parasitol Today. 1993;9:63–66. doi: 10.1016/0169-4758(93)90039-i. [DOI] [PubMed] [Google Scholar]
- Barnes RL, McCulloch R. Trypanosoma brucei homologous recombination is dependent on substrate length and homology, though displays a differential dependence on mismatch repair as substrate length decreases. Nucleic Acids Res. 2007;35:3478–3493. doi: 10.1093/nar/gkm249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry JD, McCulloch R. Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite. Adv Parasitol. 2001;49:1–70. doi: 10.1016/s0065-308x(01)49037-3. [DOI] [PubMed] [Google Scholar]
- Barry JD, Marcello L, Morrison LJ, Read AF, Lythgoe K, Jones N, et al. What the genome sequence is revealing about trypanosome antigenic variation. Biochem Soc Trans. 2005;33:986–989. doi: 10.1042/BST20050986. [DOI] [PubMed] [Google Scholar]
- Becker M, Aitcheson N, Byles E, Wickstead B, Louis E, Rudenko G. Isolation of the repertoire of VSG expression site containing telomeres of Trypanosoma brucei 427 using transformation-associated recombination in yeast. Genome Res. 2004;14:2319–2329. doi: 10.1101/gr.2955304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JS, McCulloch R. Mismatch repair regulates homologous recombination, but has little influence on antigenic variation, in Trypanosoma brucei. J Biol Chem. 2003;278:45182–45188. doi: 10.1074/jbc.M308123200. [DOI] [PubMed] [Google Scholar]
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, Bartholomeu DC, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- Bignell G, Micklem G, Stratton MR, Ashworth A, Wooster R. The BRC repeats are conserved in mammalian BRCA2 proteins. Hum Mol Genet. 1997;6:53–58. doi: 10.1093/hmg/6.1.53. [DOI] [PubMed] [Google Scholar]
- Bork P, Blomberg N, Nilges M. Internal repeats in the BRCA2 protein sequence. Nat Genet. 1996;13:22–23. doi: 10.1038/ng0596-22. [DOI] [PubMed] [Google Scholar]
- Carlton JM, Hirt RP, Silva JC, Delcher AL, Schatz M, Zhao Q, et al. Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science. 2007;315:207–212. doi: 10.1126/science.1132894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CF, Chen PL, Zhong Q, Sharp ZD, Lee WH. Expression of BRC repeats in breast cancer cells disrupts the BRCA2–Rad51 complex and leads to radiation hypersensitivity and loss of G(2)/M checkpoint control. J Biol Chem. 1999;274:32931–32935. doi: 10.1074/jbc.274.46.32931. [DOI] [PubMed] [Google Scholar]
- Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA. 1998;95:5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway C, Proudfoot C, Burton P, Barry JD, McCulloch R. Two pathways of homologous recombination in Trypanosoma brucei. Mol Microbiol. 2002;45:1687–1700. doi: 10.1046/j.1365-2958.2002.03122.x. [DOI] [PubMed] [Google Scholar]
- Cortez AP, Ventura RM, Rodrigues AC, Batista JS, Paiva F, Anez N, et al. The taxonomic and phylogenetic relationships of Trypanosoma vivax from South America and Africa. Parasitology. 2006;133:159–169. doi: 10.1017/S0031182006000254. [DOI] [PubMed] [Google Scholar]
- Criss AK, Kline KA, Seifert HS. The frequency and rate of pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol. 2005;58:510–519. doi: 10.1111/j.1365-2958.2005.04838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, West SC. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell. 2001;7:273–282. doi: 10.1016/s1097-2765(01)00175-7. [DOI] [PubMed] [Google Scholar]
- Davies OR, Pellegrini L. Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol. 2007;14:475–483. doi: 10.1038/nsmb1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitsch KW, Moxon ER, Wellems TE. Shared themes of antigenic variation and virulence in bacterial, protozoal, and fungal infections. Microbiol Mol Biol Rev. 1997;61:281–293. doi: 10.1128/mmbr.61.3.281-293.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray E, Siaud N, Dubois E, Doutriaux MP. Interaction between Arabidopsis Brca2 and its partners Rad51, Dmc1, and Dss1. Plant Physiol. 2006;140:1059–1069. doi: 10.1104/pp.105.075838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, West SC. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol. 2007;14:468–474. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, et al. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 2002;21:2030–2037. doi: 10.1093/emboj/21.8.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci USA. 2005;102:8537–8542. doi: 10.1073/pnas.0407266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover L, McCulloch R, Horn D. Sequence homology and microhomology dominate chromosomal double-strand break repair in African trypanosomes. Nucleic Acids Res. 2008. in press. [DOI] [PMC free article] [PubMed]
- Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- Horn D, Barry JD. The central roles of telomeres and subtelomeres in antigenic variation in African trypanosomes. Chromosome Res. 2005;13:525–533. doi: 10.1007/s10577-005-0991-8. [DOI] [PubMed] [Google Scholar]
- Hughes K, Wand M, Foulston L, Young R, Harley K, Terry S, et al. A novel ISWI is involved in VSG expression site downregulation in African trypanosomes. EMBO J. 2007;26:2400–2410. doi: 10.1038/sj.emboj.7601678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamper SM, Barbet AF. Surface epitope variation via mosaic gene formation is potential key to long-term survival of Trypanosoma brucei. Mol Biochem Parasitol. 1992;53:33–44. doi: 10.1016/0166-6851(92)90004-4. [DOI] [PubMed] [Google Scholar]
- Kojic M, Kostrub CF, Buchman AR, Holloman WK. BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2002;10:683–691. doi: 10.1016/s1097-2765(02)00632-9. [DOI] [PubMed] [Google Scholar]
- Kojic M, Zhou Q, Lisby M, Holloman WK. Brh2-Dss1 interplay enables properly controlled recombination in Ustilago maydis. Mol Cell Biol. 2005;25:2547–2557. doi: 10.1128/MCB.25.7.2547-2557.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic M, Zhou Q, Lisby M, Holloman WK. Rec2 interplay with both Brh2 and Rad51 balances recombinational repair in Ustilago maydis. Mol Cell Biol. 2006;26:678–688. doi: 10.1128/MCB.26.2.678-688.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R. DNA damage checkpoint and repair centers. Curr Opin Cell Biol. 2004;16:328–334. doi: 10.1016/j.ceb.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Lo T, Pellegrini L, Venkitaraman AR, Blundell TL. Sequence fingerprints in BRCA2 and RAD51: implications for DNA repair and cancer. DNA Repair (Amst) 2003;2:1015–1028. doi: 10.1016/s1568-7864(03)00097-1. [DOI] [PubMed] [Google Scholar]
- McCulloch R, Barry JD. A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev. 1999;13:2875–2888. doi: 10.1101/gad.13.21.2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch R, Rudenko G, Borst P. Gene conversions mediating antigenic variation in Trypanosoma brucei can occur in variant surface glycoprotein expression sites lacking 70-base-pair repeat sequences. Mol Cell Biol. 1997;17:833–843. doi: 10.1128/mcb.17.2.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majiwa PA, Matthyssens G, Williams RO, Hamers R. Cloning and analysis of Trypanosoma (Nannomonas) congolense ILNat 2.1 VSG gene. Mol Biochem Parasitol. 1985;16:97–108. doi: 10.1016/0166-6851(85)90052-0. [DOI] [PubMed] [Google Scholar]
- Marcello L, Barry JD. Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res. 2007;17:1344–1352. doi: 10.1101/gr.6421207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, Ouchi T, Aaronson SA. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc Natl Acad Sci USA. 1998;95:13869–13874. doi: 10.1073/pnas.95.23.13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ. RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair. Mol Cell Biol. 2005;25:3127–3139. doi: 10.1128/MCB.25.8.3127-3139.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville SE, Leech V, Navarro M, Cross GA. The molecular karyotype of the megabase chromosomes of Trypanosoma brucei stock 427. Mol Biochem Parasitol. 2000;111:261–273. doi: 10.1016/s0166-6851(00)00316-9. [DOI] [PubMed] [Google Scholar]
- Mizuta R, LaSalle JM, Cheng HL, Shinohara A, Ogawa H, Copeland N, et al. RAB22 and RAB163/mouse BRCA2: proteins that specifically interact with the RAD51 protein. Proc Natl Acad Sci USA. 1997;94:6927–6932. doi: 10.1073/pnas.94.13.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison LJ, Majiwa P, Read AF, Barry JD. Probabilistic order in antigenic variation of Trypanosoma brucei. Int J Parasitol. 2005;35:961–972. doi: 10.1016/j.ijpara.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Narod SA, Foulkes WD. BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer. 2004;4:665–676. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- Neto JL, Lira CB, Giardini MA, Khater L, Perez AM, Peroni LA, et al. Leishmania replication protein A-1 binds in vivo single-stranded telomeric DNA. Biochem Biophys Res Commun. 2007;358:417–423. doi: 10.1016/j.bbrc.2007.04.144. [DOI] [PubMed] [Google Scholar]
- Overath P, Haag J, Lischke A, O'hUigin C. The surface structure of trypanosomes in relation to their molecular phylogeny. Int J Parasitol. 2001;31:468–471. doi: 10.1016/s0020-7519(01)00152-7. [DOI] [PubMed] [Google Scholar]
- Palmer GH, Brayton KA. Gene conversion is a convergent strategy for pathogen antigenic variation. Trends Parasitol. 2007;23:408–413. doi: 10.1016/j.pt.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Patel KJ, Yu VP, Lee H, Corcoran A, Thistlethwaite FC, Evans MJ, et al. Involvement of Brca2 in DNA repair. Mol Cell. 1998;1:347–357. doi: 10.1016/s1097-2765(00)80035-0. [DOI] [PubMed] [Google Scholar]
- Pays E. Pseudogenes, chimaeric genes and the timing of antigen variation in African trypanosomes. Trends Genet. 1989;5:389–391. doi: 10.1016/0168-9525(89)90181-9. [DOI] [PubMed] [Google Scholar]
- Pays E. Regulation of antigen gene expression in Trypanosoma brucei. Trends Parasitol. 2005;21:517–520. doi: 10.1016/j.pt.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Venkitaraman A. Emerging functions of BRCA2 in DNA recombination. Trends Biochem Sci. 2004;29:310–316. doi: 10.1016/j.tibs.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR. Insights into DNA recombination from the structure of a RAD51–BRCA2 complex. Nature. 2002;420:287–293. doi: 10.1038/nature01230. [DOI] [PubMed] [Google Scholar]
- Petalcorin MI, Galkin VE, Yu X, Egelman EH, Boulton SJ. Stabilization of RAD-51-DNA filaments via an interaction domain in Caenorhabditis elegans BRCA2. Proc Natl Acad Sci USA. 2007;104:8299–8304. doi: 10.1073/pnas.0702805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot C, McCulloch R. Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucleic Acids Res. 2005;33:6906–6919. doi: 10.1093/nar/gki996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot C, McCulloch R. Trypanosoma brucei DMC1 does not act in DNA recombination, repair or antigenic variation in bloodstream stage cells. Mol Biochem Parasitol. 2006;145:245–253. doi: 10.1016/j.molbiopara.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Robinson NP, Burman N, Melville SE, Barry JD. Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol Cell Biol. 1999;19:5839–5846. doi: 10.1128/mcb.19.9.5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson NP, McCulloch R, Conway C, Browitt A, Barry JD. Inactivation of Mre11 does not affect VSG gene duplication mediated by homologous recombination in Trypanosoma brucei. J Biol Chem. 2002;277:26185–26193. doi: 10.1074/jbc.M203205200. [DOI] [PubMed] [Google Scholar]
- Saeki H, Siaud N, Christ N, Wiegant WW, van Buul PP, Han M, et al. Suppression of the DNA repair defects of BRCA2-deficient cells with heterologous protein fusions. Proc Natl Acad Sci USA. 2006;103:8768–8773. doi: 10.1073/pnas.0600298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechman EV, Rohrer MS, Seifert HS. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol. 2005;57:468–483. doi: 10.1111/j.1365-2958.2005.04657.x. [DOI] [PubMed] [Google Scholar]
- Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature. 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- Shin DS, Pellegrini L, Daniels DS, Yelent B, Craig L, Bates D, et al. Full-length archaeal Rad51 structure and mutants: mechanisms for RAD51 assembly and control by BRCA2. EMBO J. 2003;22:4566–4576. doi: 10.1093/emboj/cdg429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji MK, Venkitaraman AR. DNA recombination, chromosomal stability and carcinogenesis: insights into the role of BRCA2. DNA Repair (Amst) 2004;3:835–843. doi: 10.1016/j.dnarep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Shivji MK, Davies OR, Savill JM, Bates DL, Pellegrini L, Venkitaraman AR. A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucleic Acids Res. 2006;34:4000–4011. doi: 10.1093/nar/gkl505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- Tarsounas M, Davies D, West SC. BRCA2-dependent and independent formation of RAD51 nuclear foci. Oncogene. 2003;22:1115–1123. doi: 10.1038/sj.onc.1206263. [DOI] [PubMed] [Google Scholar]
- Tarsounas M, Davies AA, West SC. RAD51 localization and activation following DNA damage. Philos Trans R Soc Lond B Biol Sci. 2004;359:87–93. doi: 10.1098/rstb.2003.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JE, Rudenko G. Switching trypanosome coats: what's in the wardrobe? Trends Genet. 2006;22:614–620. doi: 10.1016/j.tig.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Thon G, Baltz T, Giroud C, Eisen H. Trypanosome variable surface glycoproteins: composite genes and order of expression. Genes Dev. 1990;4:1374–1383. doi: 10.1101/gad.4.8.1374. [DOI] [PubMed] [Google Scholar]
- Turner CM, Barry JD. High frequency of antigenic variation in Trypanosoma brucei rhodesiense infections. Parasitology. 1989;99(Part 1):67–75. doi: 10.1017/s0031182000061035. [DOI] [PubMed] [Google Scholar]
- Warren M, Smith A, Partridge N, Masabanda J, Griffin D, Ashworth A. Structural analysis of the chicken BRCA2 gene facilitates identification of functional domains and disease causing mutations. Hum Mol Genet. 2002;11:841–851. doi: 10.1093/hmg/11.7.841. [DOI] [PubMed] [Google Scholar]
- Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997;272:31941–31944. doi: 10.1074/jbc.272.51.31941. [DOI] [PubMed] [Google Scholar]
- Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, et al. BRCA2 function in DNA binding and recombination from a BRCA2–DSS1–ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
- Yang H, Li Q, Fan J, Holloman WK, Pavletich NP. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA–ssDNA junction. Nature. 2005;433:653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, Venkitaraman AR. Dynamic control of Rad51 recombinase by self association and interaction with BRCA2. Mol Cell. 2003;12:1029–1041. doi: 10.1016/s1097-2765(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Yu VP, Koehler M, Steinlein C, Schmid M, Hanakahi LA, van Gool AJ, et al. Gross chromosomal rearrangements and genetic exchange between nonhomologous chromosomes following BRCA2 inactivation. Genes Dev. 2000;14:1400–1406. [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Kojic M, Cao Z, Lisby M, Mazloum NA, Holloman WK. Dss1 interaction with Brh2 as a regulatory mechanism for recombinational repair. Mol Cell Biol. 2007;27:2512–2526. doi: 10.1128/MCB.01907-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.