

Abstract
An enantiospecific synthesis was developed to generate both enantiomers of 7-(4-methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one. A biological assay utilizing the HCT-116 colon cancer cell line to determine the cytotoxicity of these analogs revealed that only the (R)-enantiomer exhibited appreciable cytotoxicity with an IC50 value of 0.2 µM.
The emergence of resistance to common anti-tumor agents by cancer cells makes it imperative that new compounds, operating through novel modes of action, be developed. Successful endeavors in this arena could allow for the identification of new cancer targets that may not have inherent resistance mechanisms. To this end, research in our laboratories has focused on the synthesis and biological evaluation of cytotoxic compounds that act through unknown mechanisms of action. These pursuits led us to our research into the tyloindicine family, originally disclosed by Ali et al. in 1991.1,2
In a recent communication, the racemic synthesis of a tyloindicine I (1) based diaryl-2,3,8,8-a-tetrahydroindolizin-5(1H)-one analog library was described (Figure 1).3 This library evaluated substitution on both the northern and southern aromatic rings attached to the tetrahydroindolizin-5(1H)-one core.4 Our studies revealed that the lead compound (+/−)-2 (NSC 707904) was the most cytotoxic in the library and that it most likely operates through an unknown, novel mechanism of action.3 Furthermore, (+/−)-2 was selectively active towards colon cancer and leukemia cell lines, and was effective in vivo in the mouse hollow fiber assay.3 Driven by this intriguing biological data, we set out to determine if the stereochemistry of 2 influenced the biological activity.
Figure 1.
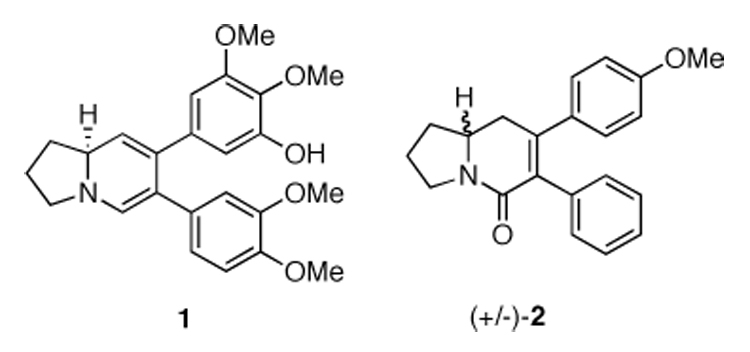
Tyloindicine I (1) and tetrahydroindolizin-5(1H)-one (+/−)-2.
Employing preparative HPLC,5 the racemate (+/−)-2 was separated to provide enantiopure (+)-2 and (−)-2.6 The pure enantiomers were assayed for cytotoxicity against the HCT-116 colon cancer cell line, using racemic tylophorine as a standard. This assay revealed that the (−)-2 enantiomer exhibited submicromolar cytotoxicity whereas (+)-2 was less than 1% as active (Table 1). These results reinforced the necessity to develop an enantiospecific synthesis of (−)-2 so the absolute stereochemistry of the active enantiomer could be determined.
Table 1.
Cytotoxicity of stereoisomers of 2 and tylophorine against HCT-116 cells
| Analog | IC50, µM |
|---|---|
| (+/−)-2 | 0.39 |
| (+)-2 | 46 |
| (−)-2 | 0.20 |
| (+/−)- tylophorine | 0.67 |
The selection of enantiomer (S)-2 as the target for synthesis (Scheme 1) was influenced by Ali’s proposal of the (S)-stereochemistry of tyloindicine I (Figure 1).2
Scheme 1.
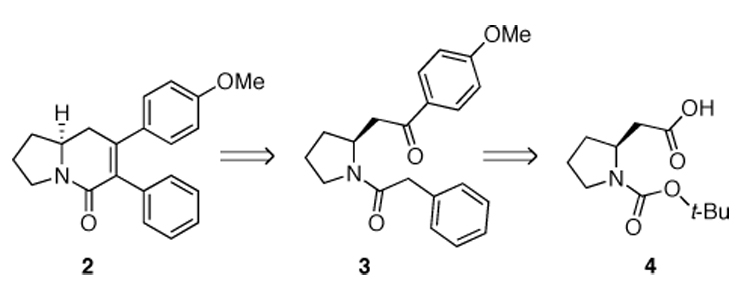
Retrosynthesis of (S)-2.3
Retrosynthetically, title compound (S)-2 can be envisioned as coming from amide 3 via an intramolecular aldol condensation. We reasoned that the desired asymmetry could be effectively derived from a chiral pool approach using N-BOC-L-homoproline (4) as the starting substrate. Thus, in turn, amide 3 may be derived from 4 by Weinreb amide formation, Grignard addition, and BOC-deprotection/amide bond formation sequence (Scheme 1).
In a forward sense, our synthesis commenced with the Weinreb amidation of (S)-2-(1- (tert-butoxycarbonyl) pyrrolidin-2-yl)acetic acid (4) (Scheme 2). A subsequent Grignard addition into the Weinreb amide furnished aryl ketone 5. An acidic BOC-deprotection of ketone 5 formed a stable hydrochloride amine salt, which was subjected to an EDCI coupling protocol to supply the aldol precursor 3.
Scheme 2.
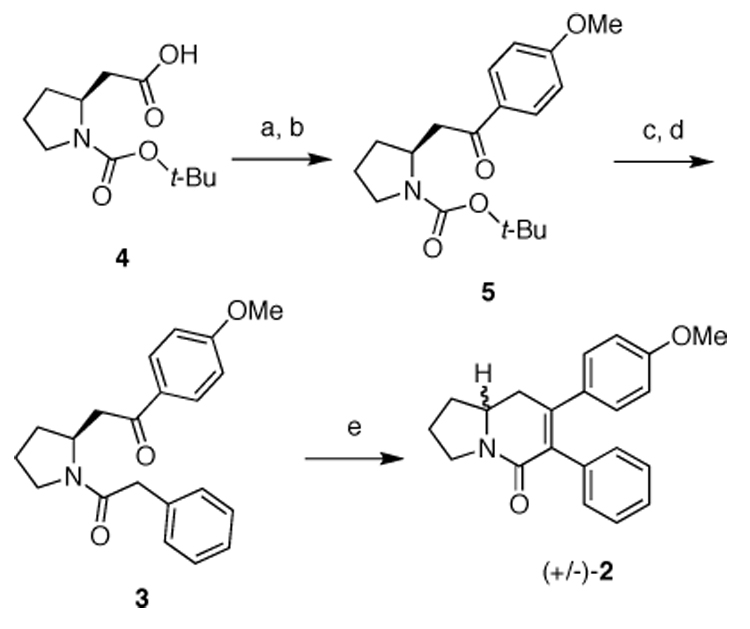
Initial synthetic attempt towards (S)-2. Reagents and conditions: a) N,O-dimethylhydroxylammonium chloride, EDCI, NMM, dry CH2Cl2, −15 °C, 12 h (91%); b) i) Mg0 turnings, cat. I2 crystals, 4-bromoanisol, dry THF, reflux, 1h (87%); ii) add cooled Grignard reagent to Weinreb amide intermediate, −5 °C, 1 h; c) 4 M HCl/dioxane, 24 °C, 15 min; d) phenylacetic acid, EDCI, NMM, dry CH2Cl2, −15 °C, 90 min (74% over two steps); e) 5% KOH(EtOH), reflux, 5h (80%).
It was crucial to handle and store the amine precursor to intermediate 3 as the HCl salt, as the free amine rapidly decomposed. The final step called for a basic intramolecular aldol condensation to deliver title compound 2. To our disappointment, we obtained the racemate (+/−)-2,7 rather than the desired (S)-2 enantiomer as suggested by the lack of optical rotation and confirmed by chiral HPLC. This result led us to hypothesize that at some point in the synthesis, the reaction conditions triggered a retro-Michael-Michael ring opening/closing sequence that racemized the stereogenic center.
Retro-Michael-Michael reactions are known to be catalyzed by either acid or base. Efforts to identify at what stage the racemization occurred were hampered by the inability to resolve the intermediates. Consequently, attempts to revise the synthesis to suppress the racemization had to take the mechanism for the retro-Michael-Michael process into account. The acid-catalyzed racemization mechanism is shown in Scheme 3, although the base-catalyzed racemization would proceed through a similar mechanism.
Scheme 3.
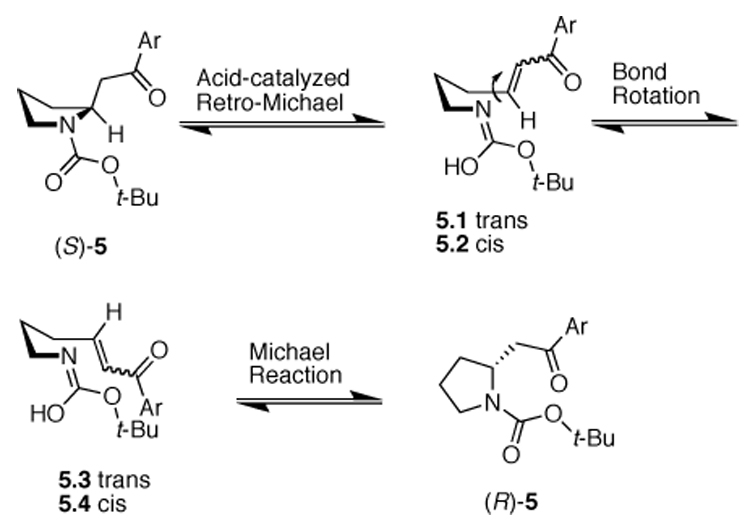
Retro-Michael-Michael racemization mechanism for the conversion of (S)-5 to (R)-5.
In this process, an acid-catalyzed carbamate activation allows for the initial retro-Michael ring opening. Depending on which α-proton is removed, either 5.1 or 5.2 may result. A simple bond rotation can give rise to either 5.3 or 5.4. Once π-orbital overlap is re-established the intramolecular Michael reaction can proceed. In that each step is reversible and there are no stereocontrolling influences elsewhere in the molecule, discrimination of the Re and Si faces of the enone is nullified during the intramolecular Michael reaction and racemization occurs to provide the racemic mixture. It should be noted that this is only one possible scenario. It seemed feasible that epimerization could have occurred at any, or all, of the steps along the way.
With this in mind, we turned to the use of a diastereomeric derivative of starting material 4, the hydroxyproline derivative 6 (Scheme 4). It was reasoned that this would enable us in several respects. First, this additional “chemical reporter” group would be situated in a locale in which racemization is unlikely and, as such, would allow us to readily track the integrity of our core stereogenic center while consequently permitting us to determine at which step the epimerization occurred. Second, if properly chosen, it may provide us a tunable, conformational bias during this retro-Michael-Michael process. Lastly, it would allow for practical separation of epimerized side products. A trait we deemed necessary in this auxiliary group: it must be readily removed and therefore a traceless reporter/controller.
Scheme 4.
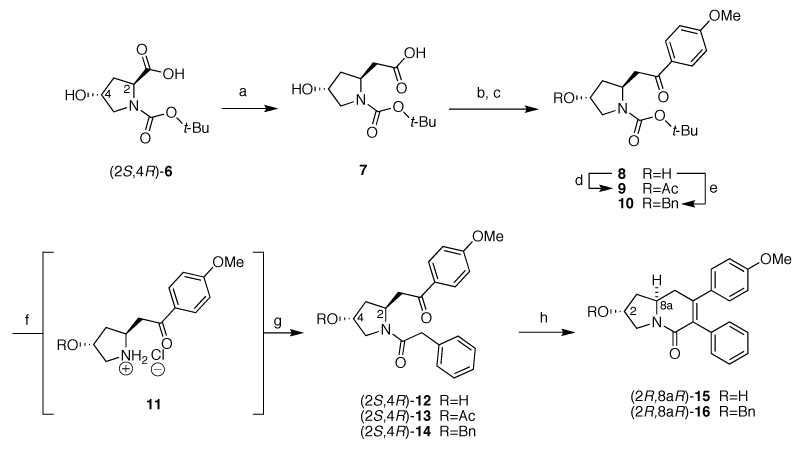
Synthesis of tetrahydroindolizin-5(1H)-one intermediates (R,R)-15 and (R,R)-16. Reagents and conditions: a) i) TEA, ClCO2Et, diazomethane, THF, 0 °C-RT, 12 h; ii) PhCO2Ag, TEA, −25 °C-RT, 12h, (27%, unoptimized); b) N,O-dimethylhydroxylammonium chloride, EDCI, NMM, dry CH2Cl2, 0 °C-RT, 2h (98%); c) Mg0 turnings, cat. I2 crystals, 4-bromoanisol, dry THF, (85%); d) Ac2O, DMAP, pyridine, 0 °C-RT, 1h (99%); e) Ag2O, BnBr, toluene, 50 °C, 3h (74%); f) 4 M HCl/dioxane, (for R=H a 77:23 dr was noted; for R=Ac, Bn the dr was not determined); g) phenylacetic acid, EDCI, NMM, dry CH2Cl2, (R=H 38%, 74:26 dr), (R=Ac 88%, 80:20 dr), (R=Bn, 73%, 60:40 dr); h) KOHMeOH, reflux (see Table 2).
The revised synthesis towards (S)-2 began with the Arndt-Eistert homologation of (2S,4R)-1-(tert-butoxycarbonyl)-4-hydroxyproline (6, Scheme 4). The resultant homolog 7 was then converted to the Weinreb amide to allow for Grignard addition of (4-methoxyphenyl)magnesium bromide into the amide to furnish arylketone 8. The acylated intermediate 9 and benzylated intermediate 10 were then prepared from 8 to probe the significance of protecting groups of increasing size during the retro-Michael-Michael racemization.8 Up to this point, no epimerization was detected. Next, the BOC-protected analogs were deprotected and acylated to give the corresponding amides (2S,4R)-12, (2S,4R)-13, and (2S,4R)-14. After deprotection, ketone 8 afforded the hydrochloride salt 11 (R=H) in a 77:23 diastereomeric ratio, revealing that the retro-Michael-Michael process does occur under the acidic BOC-deprotection conditions. The free hydroxy analog (2S,4R)-12 exhibited the lowest yield for this transformation (38%) while both the acylated and benzylated versions exhibited satisfactory yields (88% and 73%, respectively). The benzylated analog 14 showed the poorest dr at 60:40 and acylated (2S,4R)-13 the best dr at 80:20. All of the diastereomeric mixtures could be resolved through silica chromatography to supply pure diastereomers in preparation for the basic intramolecular aldol condensation (Table 2). This condensation proceeded to furnish (2R,8aR)-15 in 89% yield (70:30 dr) when (2S,4R)-12 was employed as the starting material. (2S,4R)-13 underwent the intramolecular aldol condensation with concomitant acetate-deprotection/to provide the same (2R,8aR)-15 in a slightly lower yield (75%) and dr (64:36 dr). The benzylated (2S,4R)-14 provided the condensed product (2R,8aR)-16 in 44% and 61:39 dr. Analogous to the acidic BOC-deprotection conditions used earlier in the synthesis, the basic aldol conditions also triggered racemization. The purified single diastereomer (2R,8aR)-15 was re-subjected to the basic aldol conditions to ascertain whether the racemization occurs before or after the cyclization takes place. A 92:8 dr was observed following exposure to these basic conditions indicating that the majority of the racemization occurs prior to the cyclization.
Table 2.
Intramolecular aldol condensation for anti-analogs.
| Starting Material | Conditions | Product | Yield | dra |
|---|---|---|---|---|
| (2S,4R)-12 | KOHMeOH | (2R,8aR)-15 | 89% | 70:30 |
| (2S,4R)-13 | KOHMeOH | (2R,8aR)-15 | 75% | 64:36 |
| (2S,4R)-14 | KOHEtOH | (2R,8aR)-16 | 44% | 60:40 |
Diastereomeric ratios determined by 1H NMR.
We also examined the purified syn-diastereomers in the aldol condensation (Table 3). Compounds (2R,4R)-13 and (2R,4R)-14 were subjected to the basic intramolecular aldol condensation conditions. The yields and the dr observed for this transformation were indeed lower than those for the corresponding anti-substrates (Table 2). As expected, the anti-substrates incorporated the more energetically favorable stereochemistry for the aldol condensation whereas the syn-substrates were more prone to racemization. Despite the incorporation of the larger O-benzyl group, analogs (2S,4R)-14 and (2R,4R)-14 surprisingly showed the lowest dr for both the anti and syn stereochemical situations respectively.
Table 3.
Intramolecular aldol condensation for syn-analogs.
| Starting Material | Conditions | Product | Yield | dra |
|---|---|---|---|---|
| (2R,4R)-13 | KOHEtOH | (2R,8aS)-15 | 64% | 56:44 |
| (2R,4R)-13 | KOHMeOH | (2R,8aS)-15 | 73% | 56:44 |
| (2R,4R)-14 | KOHEtOH | (2R,8aS)-16 | 28% | 50:50 |
Diastereomeric ratios determined by 1H NMR.
Overall, from the commercially available homolog 7, the synthesis of 15 was achieved in four steps and in 28% overall yield (via intermediate 12) for the diastereomeric mix and in 15% overall yield for the pure diastereomer (2R,8aR)-15. When protected as the acetate, the synthesis of 15 can be accomplished over five steps (via intermediate 13) in 54% overall yield as a mix of diastereomers of 15 and in 28% overall yield for the pure diastereomer (2R,8aR)-15. The results for the O-benzyl analogs led us not to pursue them further.
With a route to the pure (2R,8aR)-15 diastereomer realized, a Barton-McCombie deoxygenation protocol was followed to provide the target molecule (S)-2.9 As such, (2R,8aR)-15 was converted into the S-methyl xanthate (2R,8aR)-17 (Scheme 5). Completion of the synthesis of (S)-2 was accomplished under radical conditions to supply enantiopure (S)-2 in 61% yield. The optical rotation indicated that (S)-2 was the biologically inactive antipode (+)-2 allowing us to identify the stereochemistry of the biologically active (−)-2 stereoisomer as (R)-2.
Scheme 5.
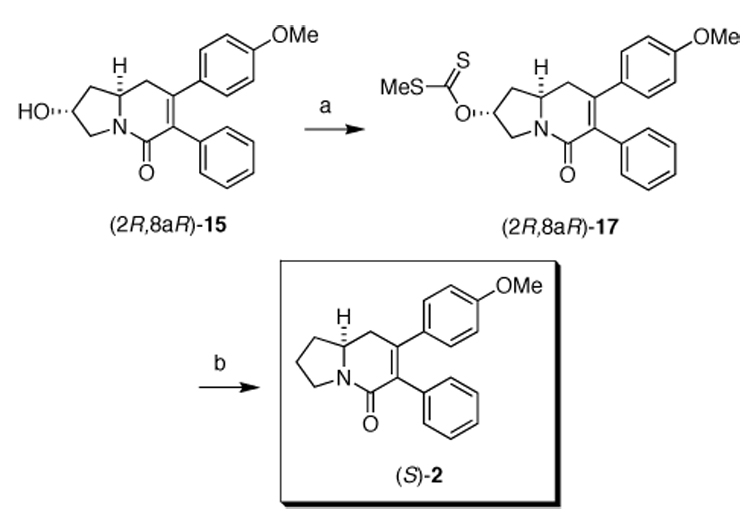
Synthesis of (S)-2. Reagents and conditions: a) 95% NaH, carbon disulfide, MeI, dry THF, 24 °C, 1h (76%, 100:0 dr); b) Bu3SnH, AIBN, dry toluene, reflux, 3h (61%, 100:0 er).
The synthesis of (R)-2 was achieved by subjecting the previously isolated minor diastereomer (2R,8aS)-15 to the same Barton-McCombie deoxygenation protocol. Thus, (2R,8aS)-15 was treated with NaH, carbon disulfide, and iodomethane in dry THF to furnish the S-methyl xanthate (2R,8aS)-17 (Scheme 6). The synthesis was successfully completed under radical conditions to deliver the biologically active enantiomer (−)-(R)-2.
Scheme 6.
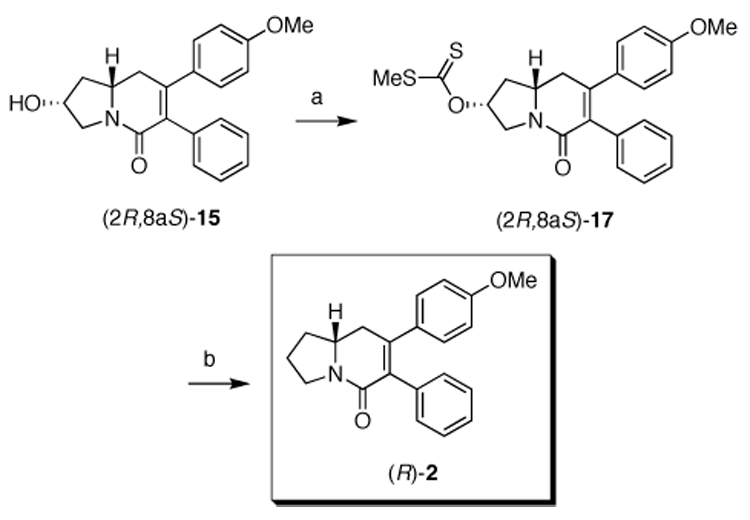
Synthesis of (R)-2. Reagents and conditions: a) 95% NaH. carbon disulfide, MeI, dry THF, 24 °C, 1h 45min (85%, 99:1 dr)a; b) Bu3SnH, AIBN, dry toluene, reflux, 3h (85%, 99:1 er)a. aDid not exhibit the anticipated 100:0 dr and er presumably due to incomplete resolution of the hydroxy recursor.
In summary, it was determined that stereochemistry indeed plays a significant role in the biological activity for the lead indolizidinone (+/−)-2. A stereospecific synthesis was developed for (S)-2 to determine if this indolizidinone, which shares the same stereochemistry with the natural product tyloindicine I, is the cytotoxic enantiomer. This endeavor identified enantiomer (S)-2 as the biologically inactive (−)-2 isomer. As a consequence of this synthetic study, it was determined that the R-stereochemistry was needed for biological activity. As such, antipode (R)-2 was also generated using this route although conceptually (2R,4S)-6 could be employed as the starting substrate to more efficiently prepare (−)-(R)-2. Our results suggest that the potentially novel cancer target involved may have a very ligand-specific binding pocket and thus be susceptible to potent and directed inhibition by these specific substrates.
Experimental section for Scheme 2
(S)-Tert-butyl 2-(2-(4-Methoxyphenyl)-2-oxoethyl)-pyrrolidine-1-carboxylate (5) Step 1
N-BOC-L-homoproline 4 (10.92 g, 47.62 mmol) was dissolved in CH2Cl2 and cooled to −15 °C. To this mixture was added N,O-dimethylhydroxylamine HCl (9.848 g, 101.0 mmol), NMM (11.66 mL, 101.0 mmol) and EDCI (19.35 g, 101.0 mmol). The reaction solution was allowed to come to room temperature over 2 h after which it was again cooled to 0 °C. The reaction was quenched by the addition of an ice cold 10% HCl solution and allowed to stir at this temperature for 5 min. The reaction was diluted with water and extracted with CH2Cl2. The combined organic layers were washed with saturated sodium bicarbonate and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (33% EtOAc/hexanes) provided (S)-tert-butyl 2-(2-(methoxy(methyl)amino)-2-oxoethyl)pyrrolidine-1-carboxylate in 91% yield (11.78 g) as a colorless oil. Rotamers were evident using NMR spectroscopy: 1H NMR (400 MHz, CDCl3) δ 1.38 (s, 9H), 1.62–1.82 (m, 3H), 1.93–2.00 (m, 1H), 2.32–2.38 (m, 1H), 2.87 (bs, 1H), 3.15 (s, 3H), 3.38 (bs, 2H), 3.64 (s, 3H), 4.15 (bs, 1H); 13C NMR (100 MHz, CDCl3) δ 23.6, 28.9, 31.5, 36.7, 46.8, 54.2, 56.8, 61.5, 79.5, 154.5, 172.7; IR 3564, 2973, 2879, 1694, 1394, 1170, 1110 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C13H25N2O4 : 273.1814, found 273.1819; [α]22 D −58 (c = 0.76, CHCl3). Step 2: In a 25 mL two-necked round-bottom flask equipped with a reflux condenser was placed activated magnesium(0) turnings (0.089 g, 3.67 mmol). THF (3 mL) was added and stirring was commenced. Through the side neck port was injected p-bromoanisol (0.23 mL, 1.8 mmol) and the resultant solution was heated towards reflux. Before appreciable heating had taken place, a catalytic amount of iodine (2 crystals) was added and the solution immediately turned brown but quickly dissipated back to the originally clear solution. As the solution was heated near 60 °C, the radical initiation process catalyzed by iodine commenced and the reaction solution bubbled violently for 2–5 min. The Grignard reagent preparation was allowed to proceed over 1 h and the reaction mixture was then cooled to −5 °C. In a separate flask was dissolved (S)-tert-butyl 2-(2-(methoxy(methyl)amino)-2-oxoethyl)pyrrolidine-1-carboxylate (0.100 g, 0.367 mmol) in THF (1 mL) at −5 °C. The Grignard reagent solution was added to the Weinreb amide solution and the reaction mixture was stirred for 2 h. Ice cold 10% HCl (1 mL) was used to quench the reaction to pH 2–4. EtOAc was used to extract the crude product and the combined organic fractions were dried over Na2SO4 and condensed under vacuum. Column chromatography with silica gel (10% EtOAc/hexanes) was employed to isolate the desired product 5 in 87% yield (0.102 g) as a colorless oil. Rotamers were evident by NMR spectroscopy. 1H NMR (400 MHz, CDCl3) δ 1.48 (s, 9H), 1.86 (m, 3H), 2.05 (m, 1H), 2.78 (m, 1H), 3.42 (m, 2H), 3.73–3.91 (m, 1H), 3.73 (s, 3H), 4.32 (t, 1H), 6.95 (d, J = 8.68 Hz, 2H), 7.99 (q, 2H); 13C NMR (100 MHz, CDCl3) δ 23.17, 23.93, 28.95, 30.55, 31.65, 43.24, 46.63, 47.04, 54.75, 55.03, 55.85, 79.56, 80.06, 114.13, 130.38, 130.88, 131.11, 154.82, 163.84, 163.97, 197.63, 198.12; IR 1168, 1259, 1396, 1600, 1689, 2877, 2935, 2972 cm−1; HRMS (ES+) m/z calc’d for [M+Na] C18H25NO4Na : 341.1681, found 342.1672; [α]22D-18 (c = 1.0, CHCl3).
1-(4-Methoxyphenyl)-2-(1-(2-phenylacetyl)-pyrrolidin-2-yl)ethanone (3)
Carbamate 5 (0.100 g, 0.313 mmol) was taken into 4 M HCl/dioxane (2 mL) and stirred at room temperature for 20 min. The solvent was removed under reduced pressure and the crude amine salt was allowed to sit under high vacuum for 12 h. The amine salt was then dissolved in CH2Cl2 (4 mL) with stirring and cooled to −15 °C. Solid phenylacetic acid (0.0512 g, 0.376 mmol), NMM (0.037 mL, 0.34 mmol) and EDCI (0.0720 g, 0.376 mmol) were added to the reaction vessel and stirring was continued for 2 h. The reaction mixture was diluted with water and the organic product was extracted with CH2Cl2 and dried over Na2SO4. The solvent was removed in vacuo and the product purified via flash column chromatography on silica gel (25% EtOAc/hexanes) to provide the pure amide 3 in 74% yield (0.078 g) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.93 (m, 4H), 2.60 (q, 1H), 3.45 (m, 1H), 3.47 (m, 1H), 3.69 (s, 2H), 3.87 (s, 3H), 3.89 (m, 1H), 4.56 (m, 1H), 6.94 (d, J = 8.80 Hz, 2H), 7.34 (m, 5H), 8.11 (d, J = 8.80 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 24.32, 29.64, 42.49, 42.92, 47.79, 55.64, 55.86, 114.20, 114.26, 127.24, 129.09, 129.33, 129.43, 130.10, 130.67, 131.37, 135.14, 163.98, 170.22, 197.96, 203.30; MS (EI) [M+H]+ C21H24NO3 : m/z 338; [α]22 D +7.4 (c = 1.0, CHCl3).
7-(4-Methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one (+/−)-2)
Compound 3 (0.500g, 1.5 mmol) was added to 5% (w/v) potassium hydroxide in 200 proof EtOH (125 mL, 111 mmol) and the reaction mixture was heated to 78 °C for 5 h. After cooling the solvent was removed in vacuo, and the crude residue was dissolved in CH2Cl2, washed twice with water and once with brine. The organic layer was dried over Na2SO4, filtered, and concentrated. Silica gel column chromatography (25% EtOAc/hexanes to 50% EtOAc/hexanes) afforded the desired product (+/−)-2 (0.383 g, 80%) as an off-white solid. mp 184.8–186.4 °C (acetone); 1H NMR (400 MHz, CDCl3) δ 1.73 (m, 1H), 1.88 (m, 1H), 2.08 (m, 1H), 2.29 (m, 1H), 2.77 (m, 2H), 3.59–3.73 (m, 2H), 3.73 (s, 3H), 3.94 (m, 1H), 6.46 (d, J = 7.24 Hz, 2H), 6.66 (d, J = 8.72 Hz, 2H), 7.14 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 23.59, 34.23, 37.94, 45.20, 55.55, 56.00, 113.68, 127.05, 127.98, 130.31, 131.55, 132.70, 132.75, 136.79, 145.11, 159.21, 164.54; IR 1033, 1247, 1434, 1604, 1643, 2925, 2962 cm−1; HRMS (ES+) m/z calc’d for [M+H]+ C21H22NO2 : 320.1651, found 320.1635.
Experimental section for Scheme 4
N-BOC-L-(4-Hydroxy)homoproline (7)
Warning: Large amounts of diazomethane were used for this transformation. Proper care should be taken when handling this highly explosive reagent. All glassware used was free of cracks, scratches or ground-glass joints and a blast shield was used. N-BOC-L-(4-Hydroxy)proline (20.00 g, 86.49 mmol) was taken into THF (250 mL) with stirring and cooled to 0 °C with an ice bath. The reaction solution was treated with triethylamine (12.04 mL, 86.42 mmol) and allowed to react for 15 min to fully deprotonate the carboxylic acid. With the addition of ethyl chloroformate (8.23 mL, 126 mmol), the resultant anhydride product precipitated out of solution as a thick white solid. Stirring was continued for 15 min and then stopped. In a separate flask, an ice-cold solution of diazomethane was prepared (see procedure below) and, without stirring, was carefully decanted into the freshly prepared anhydride reaction flask using a glass funnel. The reaction solution was lightly stirred for 4 seconds then stirring was stopped. The mixture was allowed to warm to room temperature and react overnight. Any unreacted diazomethane was carefully quenched with acetic acid (0.5 N, 69 mL). The drop-wise addition of saturated sodium bicarbonate regulated the solution back to a basic pH 8–9 with gentle stirring. The organic and aqueous layers were separated. The organic phase was washed twice each with saturated sodium bicarbonate and brine then dried over Na2SO4. The solvent was evaporated under reduced pressure and placed under high vacuum overnight. The crude diazo intermediate was taken into water (150 mL) and THF (1500 mL) and cooled to −25 °C and stirred for 30 min. The reaction solution turned into a thick slush. Foil was used to cover the reaction flask so as to exclude light from the reaction solution and silver benzoate (2.56 g, 11.2 mmol), dissolved in triethylamine (40.0 mL, 287 mmol), was then added. The reaction temperature was allowed to slowly warm to room temperature and the solution was stirred overnight. The reaction mixture was evaporated to dryness and the residue stirred for 1 h with a saturated sodium bicarbonate solution (100 mL). The resultant black solution was partitioned with water and EtOAc (100 mL). The organic layer with black suspension was collected and washed with brine once then twice with saturate sodium bicarbonate. The organic phase was set aside and not used again. All aqueous layers were combined in a large Erlenmeyer flask with a large stir bar and cooled to 0 °C. EtOAc (100 mL) was added and stirring was commenced. The drop-wise addition of 5 N HCl to pH 2 resulted in the emission of carbon dioxide gas. The organic layer was separated and the aqueous phase was extracted an additional three times with EtOAc. The combined organic layers were dried over Na2SO4 and the solvent was removed in vacuo. Flash column chromatography with silica gel (33% EtOAc/hexanes) was used to purify homolog 7 from its progenitor N-BOC-L-(4-hydroxy)proline in 27% yield (10.92 g) as a colorless oil. At least two purifications were performed with iodine stain utilized to monitor the product on TLC. Diazomethane preparation: Warning Glassware was free of cracks, scratches and ground glass joints. An ice cold solution of potassium hydroxide (80.00 g, 1425 mmol) in water (200 mL) was prepared and cold ether (1000 mL) followed by 1-methyl-3-nitro-1-nitrosoguanidine (MNNG, 50.89 g, 346.0 mmol) were added. With the addition of MNNG the basic solution turned clear yellow and bubbled moderately as the reagent dissolved and volatile diazomethane was generated. The solution was reacted for 2 min without stirring. Using a glass funnel, the mixture was taken into a separatory funnel and the bottom aqueous layer was collected and set in a negative pressure hood overnight. The yellow organic layer was collected in an ice cold Erlenmeyer flask which was layered on the bottom with potassium hydroxide pellets to absorb any remaining moisture. The diazomethane solution was used immediately. The remaining aqueous layer was carefully quenched the following day with dilute acid. All other glassware was allowed to sit in a negative pressure hood and was carefully washed with dilute acid. For product 7, rotameric peaks were evident for NMR spectroscopy methods: 1H NMR (500 MHz, CDCl3) δ 1.41 (s, 9H), 1.79 (m, 3H), 2.03 (m, 1H), 2.28 (q, 1H), 2.76 (bd, J = 13.65 Hz, 0.5H), 2.93 (bd, J = 15 Hz, 0.5H), 3.31 (bs, 2H), 4.08 (m, 1H), 7.61 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 22.61, 23.31, 28.13, 28.30, 28.45, 30.67, 31.23, 38.52, 39.02, 46.08, 46.48, 53.79, 79.65, 79.88, 154.36, 176.24.
(2S,4R)-Tert-butyl 4-Hydroxy-2-(2-(4-methoxyphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (8). Step 1
The N-BOC-L-(4-hydroxy)homoproline 7 (3.115 g, 12.70 mmol) was dissolved in CH2Cl2 (180 mL) and cooled to −15 °C. To this mixture was added N,O-dimethylhydroxylamine. HCl (2.617 g, 26.93 mmol), NMM (3.006 mL, 27.31 mmol) and EDCI (5.158 g, 26.93 mmol). The reaction solution was allowed to come to room temperature over 2 h after which it was again cooled to 0 °C. The reaction was quenched by the addition of an ice cold 10% HCl solution and allowed to stir at this temperature for 5 min. The reaction was diluted with water and extracted with CH2Cl2. The combined organic layers were washed with saturated sodium bicarbonate and then dried over Na2SO4, filtered and concentrated. Silica gel column chromatography (33% EtOAc/hexanes) was used to purify the desired intermediate (2S,4R)-tert-butyl 4-hydroxy-2-(2-(methoxy(methyl)amino)-2-oxoethyl)pyrrolidine-1-carboxylate in 98% yield (3.556 g) as a colorless oil. Rotamers were evident using NMR spectroscopy. The alcohol proton was found to exhibit variable chemical shifts depending on product concentration: 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 9H), 1.86 (bs, 1H), 2.16 (bs, 1H), 2.40 (bs, 1H), 3.01–3.09 (m, 4H), 3.37–3.51 (bs, 3H), 3.62 (s, 3H), 4.23 (bs, 1H), 4.31 (t, 1H); 13C NMR (100 MHz, CDCl3, δ 13.53, 18.94, 20.82, 28.37, 29.49, 30.45, 31.86, 36.31, 37.31, 40.13, 40.54, 52.70, 54.20, 54.50, 61.08, 64.19, 68.67, 69.15, 79.17, 79.61, 95.94, 154.54, 171.13, 172.07; IR 1120, 1159, 1365, 1400, 1668, 1691, 2935, 2974, 3434; HRMS (ES+) m/e calc’d for [M+H]+ C13H25N2O5 : 289.1764, found 289.1759; [α]22 D –77 (c = 1.0, CHCl3). Step 2: In a 100 mL two-necked round-bottom flask equipped with a reflux condenser were placed activated Mg(0) turnings (5.000 g, 205.8 mmol). THF (50 mL) was added and stirring was commenced. Through the side neck port was injected 4-bromoanisol (10.6 mL, 193 mmol) and the resultant solution was heated towards reflux. Before appreciable heating had taken place, a catalytic amount of iodine (4 crystals) was added and the solution immediately turned brown but quickly dissipated back to the originally clear solution. As the solution was heated near 60 °C, the radical initiation process catalyzed by iodine commenced and the reaction solution bubbled violently for 2–5 min. The Grignard reagent preparation was allowed to proceed over 1 h and was then cooled to −5 °C. In a separate flask was dissolved the intermediate (2S,4R)- tert-butyl 4-hydroxy-2-(2-(methoxy(methyl)amino)-2-oxoethyl)pyrrolidine-1-carboxylate (4.898 g, 16.99 mmol) in THF (200 mL) at −5 °C. The Grignard reagent solution was added to the Weinreb amide solution and the reaction mixture was stirred for 2 h. Ice cold 10% HCl was used to quench the reaction to pH 2–4. EtOAc was used to extract the crude product and the combined organic fractions were dried over Na2SO4 and condensed under vacuum. Column chromatography with silica gel (66% EtOAc/hexanes) was employed to isolate the desired product 8 in 85% yield (100:0 dr) (4.846 g) as a colorless oil. Rotamers were evident by NMR spectroscopy: mp 105–109 °C; 1H NMR (500 MHz, CDCl3) δ 1.44 (s, 9H), 1.89 (bs, 1H), 2.19 (bs, 1H), 2.80 (bs, 1H), 3.04 (bs, 1H), 3.42 (s, 1.5H), 3.58 (bs, 1H), 3.84 (s, 3.5H), 4.38 (s, 2H), 6.89 (d, J = 8.45 Hz, 2H), 7.93 (bd, J = 10.85 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 28.42, 39.90, 40.60, 43.02, 44.06, 52.69, 53.27, 54.33, 54.58, 55.32, 68.88, 69.33, 79.43, 79.97, 96.00, 111.44, 113.66, 129.79, 130.01, 130.37, 130.59, 154.67, 163.49, 196.89, 197.605; IR 1170, 1259, 1366, 1401, 1600, 1673, 2359, 2934, 2974, 3437 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C18H26NO5 : 336.1811, found 336.1783; [α]22 D –33 (c = 2.6, CHCl3).
(2S,4R)-Tert-butyl 4-Acetoxy-2-(2-(4-methoxyphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (9)
Pyridine (2 mL, xs) was used to dissolve substrate 8 (0.091 g, 0.27 mmol, 100:0 dr). With stirring, the reaction temperature was lowered to 0 °C and DMAP (0.033 g, 0.27 mmol) and acetic anhydride (0.13 mL, 1.4 mmol) were added. The reaction mixture was allowed to come to room temperature over 1 h and the reaction was then quenched with water. EtOAc was used to extract the organic product. The organic layer was dried over Na2SO4 and the solvent removed under reduced pressure. Silica gel column chromatography (20% EtOAc/hexanes) was used to isolate the desired product 9 in 99% yield (100:0 dr) (0.101 g) as a colorless oil. Rotamers were evident using NMR spectroscopy: 1H NMR (400 MHz, CDCl3) δ 1.46 (s, 9H), 1.97– 2.04 (m, 1H), 2.03 (s, 3H), 2.35 (bs, 1H), 2.28 (q, 1H), 3.54–3.89 (m, 3H), 3.86 (s, 3H), 4.38 (m, 1H), 5.20 (bs, 1H), 6.92 (d, J = 8.52 Hz, 2H), 7.96 (bs, 2H); 13C NMR (100 MHz, CDCl3) δ 14.57, 21.40, 21.48, 28.86, 30.06, 37.57, 38.78, 43.16, 44.28, 52.16, 52.72, 53.15, 53.55, 55.84, 60.74, 67.44, 72.29, 72.82, 80.17, 114.14, 130.32, 130.51, 130.88, 154.84, 163.96, 170.97, 171.47, 196.96, 197.47; IR 1170, 1246, 1367, 1398, 1600, 1692, 1739, 2930, 2974 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C20H28NO6 : 378.1917, found 378.1905; [α]22 D –25 (c = 1.0, CHCl3).
(2S,4R)-Tert-butyl 4-(Benzyloxy)-2-(2-(4-methoxyphenyl)-2-oxoethyl)pyrrolidine-1-carboxylate (10)
Toluene (8 mL) was used to dissolve the carbamate 8 (0.100 g, 0.298 mmol, 100:0 dr) with stirring. Foil was wrapped around the reaction vessel so as to exclude light. The addition of benzyl bromide (0.284 mL, 2.39 mmol) and Ag2O (0.691 g, 2.98 mmol) followed and the mixture was allowed to react at room temperature for 3 h and 20 min. The reaction was quenched with water and the organic product was extracted with EtOAc. The organic phase was dried over Na2SO4 and filtered to remove any remaining silver reagent/salts and was then evaporated under vacuum. The desired product 10 was isolated using flash column chromatography with 10% EtOAc/hexanes as mobile phase in 74% yield (100:0 dr) (0.094 g) as a colorless oil. Rotamers could be visualized using NMR spectroscopy: mp 61–62 °C; 1H NMR (400 MHz, CDCl3) δ 1.48 (s, 9H), 2.00 (bs, 1H), 2.30 (bs, 1H), 2.80 (q, 1H), 3.40–3.89 (m, 4H), 3.87 (s, 3H), 4.41–4.51 (m, 3H), 6.93 (d, J = 8.88 Hz, 2H), 7.32 (m, 5H), 7.97 (bd, 2H); 13C NMR (100 MHz, CDCl3, trace impurities) δ 23.06, 25.68, 28.93, 32.00, 35.06, 37.10, 38.55, 43.63, 44.65, 51.14, 51.51, 52.36, 53.92, 55.87, 71.30, 76.16, 76.70, 79.94, 80.42, 114.17, 128.09, 128.83, 130.33, 131.00, 131.06, 138.41, 155.03, 163.96, 197.32, 197.93; IR 1112, 1157, 1170, 1259, 1365, 1396, 1600, 1689, 2933, 2974 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C25H32NO5 : 426.2280, found 426.2290; [α]22 D –16 (c = 1.0, CHCl3).
(2S,4R)-2-(4-Hydroxy-1-(2-phenylacetyl)pyrrolidin-2-yl)-1-(4-methoxyphenyl)ethanone ((2S,4R)-12) and (2R,4R)-2-(4-Hydroxy-1-(2-phenylacetyl)pyrrolidin-2-yl)-1-(4-methoxyphenyl)ethanone ((2R,4R)-12)
The carbamate 8 (100:0 dr) (1.904 g, 5.677 mmol) was taken into 4 M HCl/dioxane (15 mL) and stirred at room temperature for 30 min. With the addition of the acid, the reaction solution changed colors from a cloudy orange to a cloudy scarlet within 5 min. The solvent was removed under reduced pressure and the crude amine salt was allowed to sit under high vacuum overnight and appeared as a magenta solid. The amine salt was then dissolved in CH2Cl2 (35 mL) with stirring and cooled to −15 °C. Solid phenylacetic acid (0.927 g, 6.81 mmol), NMM (0.686 mL, 6.25 mmol) and EDCI (1.306 g, 6.812 mmol) were added to the reaction vessel and stirring was continued for 2 h. The orange reaction mixture was quenched with 10% HCl (2 mL) and diluted with water. The organic product was extracted with CH2Cl2 three times. As the organic layer was washed twice with saturated sodium bicarbonate the cloudy orange organic layer color was observed to change to a clear yellow color. The combined saturated sodium bicarbonate washes were back extracted twice with CH2Cl2. The combined organic fractions were dried over Na2SO4. The solvent was removed in vacuo and the two diastereomeric products were purified from a number of impurities via precipitations in 80% EtOAc/hexanes. The precipitate was collected by filtration to provide the diastereomeric product 12 in 38% yield (74:26 dr favoring the anti diastereomer) (0.759 g). The diastereomers were resolved using flash column chromatography (1.5% methanol/CH2Cl2). The anti-diastereomer proved to be slightly more polar than the syn-counterpart. Rotamers could be visualized using NMR spectroscopy: For (2S,4R)-12 (major diastereomer): colorless crystals, mp 125 °C; 1H NMR (500 MHz, CDCl3) δ 2.03 (m, 1H), 2.14 (m, 1H), 2.76 (q, 0.92H, rotameric), 3.06 (d, J = 6.80 Hz, 0.08H, rotameric), 3.49 (d, J = 10.90 Hz, 1H), 3.58 (m, 1H), 3.65 (d, J = 5.2 Hz, 2H), 3.85 (s, 3H), 3.93 (q, 1H), 4.46 (bs, 1H), 4.62 (m, 1H), 6.91 (d, J = 8.80 Hz, 2H), 7.24–7.33 (m, 5H), 7.77 (d, J = 8.65 Hz, 0.16H, rotameric), 8.01 (d, J = 8.80 Hz, 1.84H, rotameric); 13C NMR (125 MHz, CDCl3) δ 39.27, 42.23, 42.67, 53.78, 55.47, 69.94, 113.81, 126.88, 128.70, 128.98, 129.41, 130.79, 134.61, 163.66, 170.37, 197.34; IR 1178, 1257, 1311, 1438, 1602, 1618, 1662, 2850, 2923, 3438 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C21H24NO4 : 354.1705, found 354.1714. For (2R,4R)-12 (minor diastereomer), colorless oil: 1H NMR (500 MHz, CDCl3) δ 1.99 (m, 1H), 2.18 (m, 1H), 3.01 (q, 0.17H, rotameric), 3.20 (q, 0.83H, rotameric), 3.58–3.69 (m, 4H), 3.85 (s, 2.49H, rotameric), 3.87 (s, 0.51H, rotameric), 3.92 (q, 1H), 4.50 (m, 1H), 4.59 (t, 0.83H rotameric), 4.70 (t, 0.17H, rotameric), 6.90 (d, J = 8.85 Hz, 2H), 7.28 (m, 5H), 7.85 (d, J = 8.80 Hz, 0.4H, rotameric), 8.03 (d, J = 8.80 Hz, 1.6H, rotameric); 13C NMR (125 MHz, CDCl3) δ 38.08, 42.27, 42.50, 54.57, 55.46, 55.80, 71.27, 113.76, 126.85, 128.97, 129.40, 129.82, 130.83, 134.42, 163.65, 169.99, 198.32; IR 1170, 1259, 1311, 1419, 1598, 1668, 2852, 2923, 3388 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C21H24NO4 : 354.1705, found 354.1710.
(3R,5S)-5-(2-(4-Methoxyphenyl)-2-oxoethyl)-1-(2-phenylacetyl)pyrrolidin-3-yl Acetate ((2S,4R)-13) and (3R,5R)-5-(2-(4-Methoxyphenyl)-2-oxoethyl)-1-(2-phenylacetyl)pyrrolidin-3-yl Acetate ((2R,4R)-13)
The carbamate 9 (0.424 g, 1.13 mmol) was taken into 4 M HCl/dioxane (2 mL) and stirred at room temperature for 20 min. The solvent was removed under reduced pressure and the crude amine salt was allowed to sit under high vacuum overnight. The amine salt was then dissolved in CH2Cl2 with stirring and cooled to −15 °C. Solid phenylacetic acid (0.1823 g, 1.337 mmol), NMM (0.1356 mL, 1.260 mmol) and EDCI (0.2584 g, 1.348 mmol) were added to the reaction vessel and stirring was continued for 2 h. The reaction mixture was diluted with water and the organic product was extracted with CH2Cl2 and dried over Na2SO4. The solvent was removed in vacuo and the product purified via preparative TLC (1.5% methanol/CH2Cl2; 4 runs) to provide the amide 13 in 88% yield (80:20 dr favoring the anti diastereomer) (0.392 g). The anti product could also be recrystallized in minimal isopropanol. Although the purification successfully resolved the slightly less polar anti-product from the syn-product, the yield reflects the diastereomeric mix. Rotamers could be visualized using NMR spectroscopy: For (2S,4R)-13 (major diastereomer): colorless crystals, mp 108 °C; 1H NMR (500 MHz, CDCl3) δ 1.89 (s, 3H), 2.05 (m, 1H), 2.31 (m, 1H), 2.92 (q, 1H), 3.63 (s, 2H), 3.63–3.69 (m, 2H), 3.83 (s, 3H), 3.88 (q, 1H), 4.59 (m, 1H), 5.19 (s, 1H), 6.90 (d, J = 8.55 Hz, 2H), 7.28 (m, 5H), 7.98 (d, J = 8.60 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 20.84, 36.24, 41.82, 42.68, 52.74, 53.46, 55.36, 72.53, 113.69, 126.78, 128.61, 128.74, 129.69, 130.51, 134.34, 163.54, 169.91, 170.38, 196.79; IR 1026, 1172, 1226, 1247, 1259, 1311, 1371, 1421, 1600, 1643, 1668, 1737, 2839, 2906, 2935, 3029 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C23H26NO5 : 396.1811, found 396.1810; [α]22 D + 10.0 (c = 1.80, CHCl3). For (2R,4R)-13 (minor diastereomer), colorless oil: 1H NMR (500 MHz, CDCl3) δ 2.07 (m, 4H), 2.25 (m, 0.8H, rotameric), 2.35 (m, 0.2H, rotameric), 2.95 (q, 1H), 3.66 (m, 3H), 3.79 (q, 1H), 3.81 (s, 2.4H, rotameric), 3.87 (s, 0.6H, rotameric), 3.97 (q, 1H), 4.67 (t, 0.8H), 4.70 (t, 0.2H), 5.30 (s, 0.8H, rotameric), 5.35 (s, 0.2H, rotameric), 6.94 (d, J = 8.80 Hz, 2H), 7.28 (m, 5H), 7.85 (d, J = 8.80, 0.4H, rotameric), 8.05 (d, J = 8.75 Hz, 1.6 Hz, rotameric); 13C NMR (125 MHz, CDCl3) δ 21.12, 34.63, 41.88, 42.39, 53.38, 54.37, 55.37, 73.56, 113.73, 126.89, 128.64, 128.96, 129.69, 130.60, 134.10, 163.53, 169.69, 170.26, 197.20; IR 1170, 1230, 1247, 1375, 1417, 1598, 1643, 1668, 1737, 2840, 2937, 3028, 3060 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C23H26NO5 : 396.1811, found 396.1812.
2-((2S,4R)-4-(Benzyloxy)-1-(2-phenylacetyl)pyrrolidin-2-yl)-1-(4-methoxyphenyl) ethanone ((2S,4R)-14) and 2-((2R,4R)-4-(Benzyloxy)-1-(2-phenylacetyl)pyrrolidin-2-yl)-1-(4-methoxyphenyl)ethanone ((2R,4R)-14)
The carbamate 10 (0.654 g, 1.54 mmol, 100:0 dr) was taken into 4 M HCl/dioxane (10 mL) and stirred at room temperature for 30 min. The solvent was removed under reduced pressure and the crude amine salt was allowed to sit under high vacuum overnight. The amine salt was then dissolved in CH2Cl2 (30 mL) with stirring and cooled to −15 °C. Solid phenylacetic acid (0.252 g, 1.85 mmol), NMM (0.170 mL, 1.58 mmol) and EDCI (0.351 g, 1.83 mmol) were added to the reaction vessel and stirring was continued for 2 h. The reaction mixture was diluted with water and the organic product was extracted with CH2Cl2 and dried over Na2SO4. The solvent was removed in vacuo and the product purified via preparative TLC (50% EtOAc/hexanes; multiple runs) to provide the pure amide 14 in 73% yield (60:40 dr favoring the anti diastereomer) (0.496 g). The anti product could also be obtained via recrystallization from EtOAc or isopropanol. The yield reflects the diastereomeric mix although the slightly less polar anti diastereomer and more polar syn counterpart were successfully resolved. Rotamers could be visualized using NMR spectroscopy. For (2S,4R)-14 (major diastereomer): colorless crystals, mp 119 °C; 1H NMR (400 MHz, CDCl3) δ 2.07 (m, 1H), 2.29 (m, 1H), 2.78 (q, 1H), 3.58–3.64 (m, 4H), 3.87 (s, 3H), 3.95 (q, 1H), 4.19 (t, 1H), 4.33 (d, J = 11.88 Hz, 1H), 4.45 (d, J = 11.96 Hz, 1H), 4.66 (bd, J = 6.44 Hz, 1H), 6.94 (d, J = 8.80 Hz, 2H), 7.21–7.37 (m, 10H), 8.06 (d, J = 8.76 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 36.44, 42.80, 43.09, 52.91, 54.36, 55.88, 71.39, 77.06, 114.23, 127.25, 127.98, 128.21, 128.89, 129.12, 129.37, 130.12, 131.25, 135.03, 138.23, 164.08, 170.55, 197.76; IR 1026, 1099, 1174, 1261, 1602, 1627, 1668, 2837, 2925, 3031, 3060 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C28H30NO4 : 444.2175, found 444.2173; [α]22 D +8 (c = 1, CHCl3). For (2R,4R)-14 (minor diastereomer), colorless oil: 1H NMR (400 MHz, CDCl3) δ 2.06 (m, 1H), 2.25 (d, J = 14.08 Hz, 1H), 2.90 (q, 0.2H, rotameric), 3.14 (q, 0.8H, rotameric), 3.63–3.76 (m, 4H), 3.87 (s, 3H), 3.94 (q, 1H), 4.19 (bs, 1H), 4.39–4.65 (m, 3H), 6.83 (d, J = 8.84 Hz, 0.4H, rotameric), 6.91 (d, J = 8.88 Hz, 1.6 H, rotameric), 7.26–7.37 (m, 10H), 7.81 (d, J = 8.84 Hz, 0.4H, rotameric), 8.04 (d, J = 8.88 Hz, 1.6H, rotameric); 13C NMR (100 MHz, CDCl3) δ 34.18, 42.52, 42.92, 53.84, 53.92, 54.96, 55.84, 71.20, 114.12, 127.29, 128.05, 128.34, 128.92, 129.11, 129.48, 130.33, 131.17, 134.58, 137.98, 163.89, 170.00, 198.34; IR 1027, 1170, 1259, 1417, 1598, 1643, 1666, 2839, 2869, 2935, 3029, 3060 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C28H30NO4 : 444.2175, found 444.2174.
(2R,8aR)-2-Hydroxy-7-(4-methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one ((2R, 8aR)-15) and (2R,8aS)-2-Hydroxy-7-(4-methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one ((2R, 8aS)-15). Via aldol condensation using 2% KOHMeOH (xs)
The diastereomerically pure substrate (2S,4R)-12 (0.385 g, 1.09 mmol) was dissolved in 2% KOHMeOH (xs) and refluxed with stirring for 4 h. The solvent was removed under reduced pressure and the residue partitioned between water and CH2Cl2. A 10% HCl solution was added to the biphasic system until a pH 4 had been attained to break apart an emulsion. The organic product was extracted with CH2Cl2 and dried over Na2SO4. The solvent was removed in vacuo to give the product 15 in 89% yield (70:30 dr favoring the anti diastereomer) (0.324 g) without further purification. The yield reflects the diastereomeric mix although the diastereomers were successfully resolved. Via aldol condensation of acetate protected substrate using 5%KOH (xs) in either ethanol or methanol: The diastereomerically pure acetate protected substrate (2S,4R)-13 (0.145 g, 0.367 mmol) was taken in 5% KOH (24 mL, 22 mmol) dissolved in either ethanol or methanol and refluxed. The ethanolic solution was allowed to react for 90 min whereas the methanolic solution was allowed to stir for 4 h. The solvent was removed in vacuo and the resultant residue was partitioned between water and CH2Cl2. The organic product was extracted with CH2Cl2 and dried over Na2SO4. Regardless of the alcoholic solvent used, the solvent was evaporated and the desired product 15 was isolated via preparative TLC (EtOAc) in 75% yield (64:36 dr favoring the anti diastereomer) (0.092 g). When (2R,4R)-13 was used, the ethanolic solution provided the product 15 in 64% yield (56:44 dr favoring the syn diastereomer) (0.018 g) and the methanolic solution generated the product in 73% yield (56:44 dr favoring the syn diastereomer) (0.021 g). The yield reflects the diastereomeric mix although the diastereomers were successfully resolved. For (2R, 8aR)-15 (anti diastereomer), colorless oil: 1H NMR (500 MHz, CDCl3) δ 1.70 (m, 1H), 2.18 (q, 1H), 2.61 (t, 1H), 2.75 (q, 1H), 3.56 (d, J = 13.4 Hz, 1H), 3.65 (s, 3H), 3.71 (m, 1H), 4.05 (br s, 1H), 4.11 (bt, 1H), 4.47 (s, 1H), 6.58 (d, J = 8.70, 2H), 6.86 (d, J = 8.70 Hz, 2H), 7.07 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 36.28, 41.36, 52.20, 53.15, 54.12, 68.14, 112.29, 125.70, 126.58, 128.74, 130.14, 131.07, 131.18, 135.13, 144.16, 157.87, 163.19; IR 1031, 1178, 1249, 1440, 1510, 1604, 1633, 2929, 3382 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C21H22NO3 : 336.1600, found 336.1606; For (2R, 8aS)-15 (syn diastereomer), colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.85 (m, 1H), 2.51 (m, 1H), 2.75 (q, 1H), 2.94 (t, 1H), 3.67–3.77 (m, 3H), 3.74 (s, 3H), 4.04 (m, 1H), 4.52 (t, 1H), 6.66 (d, J = 8.76 Hz, 2H), 6.94 (d, J = 8.80 Hz, 2H), 7.11 (m, 2H), 7.18 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 37.71, 41.24, 52.53, 53.99, 55.14, 69.43, 113.30, 126.75, 127.63, 129.85, 131.08, 132.08, 132.23, 136.22, 145.43, 158.90, 164.00; IR 1033, 1178, 1249, 1440, 1510, 1604, 1633, 2923, 3382 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C21H22NO3 : 336.1600, found 336.1618.
(2R,8aR)-2-(Benzyloxy)-7-(4-methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one ((2R, 8aR)-16) and (2R,8aS)-2-(Benzyloxy)-7-(4-methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one ((2R, 8aS)-16)
The aldol precursor substrate (2S,4R)-14 (0.168 g, 0.395 mmol, 100:0 dr) was taken into 5% KOHEtOH (26.53 mL, 23.69 mmol) and refluxed for 1 h and 45 min. The solvent was removed under reduced pressure and the residue partitioned between water and CH2Cl2. The organic product was extract with CH2Cl2 and dried over Na2SO4. The solvent was evaporated under vacuum and product 16 was isolated in 44% yield (60:40 dr favoring the anti diastereomer) (0.073 g) using preparative TLC (33% EtOAc/hexanes; 3 runs). When (2R,4R)-14 was used, the product 16 could be isolated in 28% yield (0.022 g) as an equal mix of the syn and anti diastereomers. The yield reflects the diastereomeric mix although the slightly more polar anti diastereomer and less polar syn counterpart were successfully resolved. For (2R,8aR)-16 (anti diastereomer), colorless oil: 1H NMR (500 MHz, CDCl3) δ 1.71 (m, 1H), 2.38 (q, 1H), 2.65 (q, 1H), 2.76 (q, 1H), 3.65 (s, 3H), 3.68 (q, 1H), 3.79 (d, J = 13.55 Hz, 1H), 4.21 (m, 2H), 4.43 (d, J = 11.75 Hz, 1H), 4.52 (d, J = 11.70 Hz, 1H), 6.58 (d, J = 8.75 Hz, 2H), 6.86 (d, J = 8.75 Hz, 2H), 7.02–7.29 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 36.29, 38.90, 49.54, 52.48, 54.10, 69.71, 74.92, 112.22, 125.66, 126.39, 126.58, 126.83, 127.47, 128.82, 130.07, 131.18, 131.23, 135.13, 136.71, 143.66, 157.75, 163.14; HRMS (ES+) m/e calc’d for [M+H]+ C28H28NO3 : 426.2069, found 426.2064. For (2R,8aS)-16 (syn diastereomer), colorless oil: 1H NMR (500 MHz, CDCl3) δ 1.95 (m, 1H), 2.55 (m, 1H), 2.76 (q, 1H), 2.89 (q, 1H), 3.72 (s, 3H), 3.77 (d, J = 6.10 Hz, 2H), 4.02 (m, 1H), 4.26 (m, 1H), 4.53 (d, J = 11.75 Hz, 1H), 4.60 (d, J = 11.70 Hz, 1H), 6.65 (d, J = 8.70 Hz, 2H), 6.93 (d, J = 8.65 Hz, 2H), 7.09–7.37 (m, 10H); 13C NMR (125 MHz, CDCl3) δ 37.59, 39.21, 49.60, 53.73, 55.12, 71.82, 75.77, 113.23, 126.70, 127.61, 127.78, 127.88, 128.51, 129.88, 131.06, 131.84, 132.11, 136.15, 137.70, 145.15, 158.81, 164.03; HRMS (ES+) m/e calc’d for [M+H]+ C28H28NO3 : 426.2069, found 426.2074.
Experimental section for Scheme 5
(2R,8aR)-O-7-(4-Methoxyphenyl)-5-oxo-6-phenyl-1,2,3,5,8,8a-hexahydroindolizin-2-yl S-Methyl Carbonodithioate ((2R,8aR)-17)
(2R,8aR)-15 (0.024 g, 0.072 mmol), dissolved in THF (2 mL), was added to a solution of 95% NaH (0.0034 g, 0.14 mmol) in THF (0.5 mL) at ambient temperature and stirred for 5 min. Carbon disulfide (0.301 mL, 5.01 mmol) and iodomethane (0.312 mL, 5.01 mmol) were added and stirring was continued for an additional 1 h. The reaction solution was condensed under reduced pressure. The remaining residue was dissolved in minimal CH2Cl2 and applied directly to a preparative silica TLC plate for purification. A 33% EtOAc/hexanes mobile phase (2 runs) was used to purify the desired xanthate (2R,8aR)-17 in 76% yield (100:0 dr) (0.023 g) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 2.05 (m, 1H), 2.60 (s, 3H), 2.65 (q, 1H), 2.80 (q, 1H), 2.90 (q, 1H), 3.75 (s, 3H), 3.93 (d, J = 14.64 Hz, 1H), 4.02 (q, 1H), 4.30 (m, 1H), 6.15 (t, 1H), 6.68 (d, J = 8.88 Hz, 2H), 6.96 (d, J = 8.88 Hz, 2H), 7.13 (m, 2H), 7.18 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 18.19, 36.23, 38.71, 50.22, 52.80, 54.13, 79.94, 112.34, 125.81, 126.63, 128.85, 130.08, 131.00, 131.12, 134.90, 143.90, 157.95, 163.35, 214.00; IR 1033, 1051, 1080, 1188, 1213, 1249, 1434, 1510, 1604, 1645, 2837, 2925, 2954 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C23H24NO3S2 : 426.1198, found 426.1202.
(+)-(S)-7-(4-Methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one ((+)-(S)-2)
To a two necked flask equipped with a reflux condenser was added toluene (2 mL) and tributyltinhydride (0.018 mL, 0.067 mmol). Catalytic AIBN (0.0008 g, 0.005 mmol) was added and heating was commenced towards 75 °C. The S-methyl xanthate (2R,8aR)-17 (0.022 g, 0.052 mmol, 100:0 dr) was dissolved in toluene (3 mL) and slowly added to the reaction mixture. The reaction was allowed to progress over 3 h. Although minimal amounts of the starting xanthate could be visualized with TLC under UV light, the reaction solvent was removed in vacuo. The remaining residue was dissolved in CH2Cl2 and applied directly to a preparative silica TLC plate. A mobile phase of 66% EtOAc/hexanes was used to isolate enantiomerically pure indolizidinone (+)-(S)-2 in 61% yield (0.010 g): mp 199.8–201.7 °C [α]22 D +42 (c = 1.0, CHCl3); colorless crystals, mp 199.8–201.7 °C (acetone); 1H NMR (400 MHz, CDCl3) δ 1.73 (m, 1H), 1.88 (m, 1H), 2.08 (m, 1H), 2.29 (m, 1H), 2.77 (m, 2H), 3.59–3.73 (m, 2H), 3.73 (s, 3H), 3.94 (m, 1H), 6.46 (d, J = 7.24 Hz, 2H), 6.66 (d, J = 8.72 Hz, 2H), 7.14 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 23.59, 34.23, 37.94, 45.20, 55.55, 56.00, 113.68, 127.05, 127.98, 130.31, 131.55, 132.70, 132.75, 136.79, 145.11, 159.21, 164.54; IR 1033, 1247, 1434, 1604, 1643, 2925, 2962 cm−1; HRMS (ES+) m/z calc’d for [M+H]+ C21H22NO2 : 320.1651, found 320.1635.
Experimental section for Scheme 6
(2R,8aS)-O-7-(4- Methoxyphenyl)-5-oxo-6- phenyl-1,2,3,5,8,8a-hexahydroindolizin-2-yl S-Methyl Carbonodithioate ((2R,8aS)-17)
(2R,8aS)-15 (0.025 g, 0.075 mmol, 99:1 dr), dissolved in THF (2 mL), was added to a solution of 95% NaH (0.0035 g, 0.15 mmol) in THF (1 mL) at ambient temperature and stirred for 5 min. Carbon disulfide (0.314 mL, 5.22 mmol) and iodomethane (0.325 mL, 5.22 mmol) were added and stirring was continued for 1 h. After monitoring the sluggish reaction progress via TLC, additional NaH (95%, 3 mg) was added and the reaction was stirred for 45 min. After a total reaction time of 1 h and 45 min, the reaction solvent was removed under vacuum to leave a thick moist white solid. The solid was dissolved in minimal CH2Cl2 and applied directly to a preparative silica TLC plate for purification. A 33% EtOAc/hexanes mobile phase (2 runs) was used to purify the desired xanthate (2R,8aS)-17 in 85% yield (99:1 dr) (0.027 g) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 2.13 (m, 1H), 2.58 (s, 3H), 2.80–2.93 (m, 3H), 3.72 (s, 3H), 3.88 (q, 1H), 4.05– 4.16 (m, 2H), 5.99 (t, 1H), 6.66 (d, J = 8.40 Hz, 2H), 6.94 (d, J = 8.40 Hz, 2H), 7.09 (d, J = 6.90 Hz, 2H), 7.17 (t, 3H); 13C NMR (125 MHz, CDCl3) δ 19.32, 37.68, 38.29, 49.89, 53.76, 55.14, 80.45, 113.32, 126.82, 127.65, 129.89, 131.06, 131.82, 131.90, 135.98, 145.25, 158.97, 164.04, 215.28; IR 1033, 1064, 1180, 1207, 1249, 1434, 1510, 1604, 1645, 2837, 2927, 3053 cm−1; HRMS (ES+) m/e calc’d for [M+H]+ C23H24NO3S2 : 426.1198, found 426.1203.
(−)-(R)-7-(4-Methoxyphenyl)-6-phenyl-2,3,8,8a-tetrahydroindolizin-5(1H)-one ((−)-(R)-2)
To a two necked flask equipped with a reflux condenser was added toluene (2 mL) and tributyltinhydride (0.021 mL, 0.076 mmol). Catalytic AIBN (0.0009 g, 0.006 mmol) was added and heating was commenced towards 75 °C. The S-methyl xanthate (2R,8aS)-17 (0.025 g, 0.059 mmol, 99:1 dr) was dissolved in toluene (3 mL) and slowly added to the reaction mixture. The reaction was allowed to progress over 3 h then the reaction solvent was removed in vacuo. The remaining residue was dissolved in CH2Cl2 and applied directly to a preparative silica TLC plate. A mobile phase of 66% EtOAc/hexanes was used to isolate the indolizidinone (R)-2 in 85% yield (99:1 er) (0.016 g): mp 199.5–201.4 °C [α]22 D –42 (c = 1.0, CHCl3); colorless crystals mp 199.5–201.4 °C (acetone); 1H NMR (400 MHz, CDCl3) δ 1.73 (m, 1H), 1.88 (m, 1H), 2.08 (m, 1H), 2.29 (m, 1H), 2.77 (m, 2H), 3.59–3.73 (m, 2H), 3.73 (s, 3H), 3.94 (m, 1H), 6.46 (d, J = 7.24 Hz, 2H), 6.66 (d, J = 8.72 Hz, 2H), 7.14 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 23.59, 34.23, 37.94, 45.20, 55.55, 56.00, 113.68, 127.05, 127.98, 130.31, 131.55, 132.70, 132.75, 136.79, 145.11, 159.21, 164.54; IR 1033, 1247, 1434, 1604, 1643, 2925, 2962 cm−1; HRMS (ES+) m/z calc’d for [M+H]+ C21H22NO2 : 320.1651, found 320.1635.
Acknowledgments
These studies were supported by the National Cancer Institute (CA 90602 and N01-CM-67259). B.J.T. was supported by the Department of Defense Breast Cancer Predoctoral Fellowship (DAMD17-02-1-0435). We thank Deborah A. Bohlander for conducting the cytotoxicity assays and Dr. Dinah Dutta for the synthesis of (+/−)-tylophorine (Table 1).10
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Ali M, Ansari SH, Grever MR. Pharmazie. 2001;56:188. [PubMed] [Google Scholar]
- 2.Ali M, Ansari SH, Qadry JS. J. Nat. Prod. 1991;54:1271. [Google Scholar]
- 3.Kimball FS, Tunoori AR, Victory SF, Dutta D, White JM, Himes RH, Georg GI. Bioorg. Med. Chem. Lett. 2007;17:4703. doi: 10.1016/j.bmcl.2007.05.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.For related investigations of the tetrahydroindolizin-5(1H)-one core see:Sharma VM, Adi Seshu KV, Vamsee Krishna C, Prasanna P, Chandra Sekhar V, Venkateswarlu A, Rajagopal S, Ajaykumar R, Deevi DS, Rao Mamidi NVS, Rajagopalan R. Bioorg. Med. Chem. Lett. 2003;13:1679. doi: 10.1016/s0960-894x(03)00263-4.
- 5.Chiralcel OJ. elution with 20–50% isopropanol in hexanes, flow rate 5 mL / min. Retention time for (+)-2 of 15 min. Retention time of 20.8 min for (−)-2
- 6.(+)-2: Colorless crystals, [α]22 D +42 (c = 1.0 CHCl3), mp 199.8–201.7 °C (acetone); (−)-2: Colorless crystals, [α]22 D −42 (c = 1.0 CHCl3), mp 199.5–201.4 °C (acetone); (+/−)-2: Colorless crystals, mp 184.8 – 186.4 °C (acetone).
- 7.The enantiomeric ratio varied between experiments from 50:50 (racemic) to 60:40 (+)-(S)-2:(−)-(R)-2.
- 8.Some degree of racemization was noticed in step e, with the isolation of 5% yield of the O-benzyl, N-benzyl side-product. This product was isolated in a 60:40 dr suggesting that perhaps the racemization does not require strong acid to occur. Furthermore, this side-product also indicated, that either the BOC-protected amine or the free amine, rather than the amine salt, are prone to racemization.
- 9.Barton DHR, Mc Combie SW. J. Chem. Soc., Perkin Trans. 1. 1975:1574. [Google Scholar]
- 10.Prepared according to reference 4 andCommins DL, Morgan LA. Tetrahedron Lett. 1991;32:5919–5922.