

Abstract
Nitroimidazoles such as PA-824 and OPC-67683 are currently in clinical development as members of a promising new class of therapeutics for tuberculosis. While the antitubercular activity of these compounds is high, they both suffer from poor water solubility thus complicating development. We determined the single-crystal X-ray structure of PA-824 and found a close packing of the nitroimidazoles facilitated by a pseudoaxial conformation of the p-trifluoromethoxybenzyl ether. To attempt to disrupt this tight packing by destabilizing the axial preference of this side chain, we prepared the two diastereomers of the 7-methyl-nitroimidazo-oxazine. Determination of the crystal structure of the 7-(S)-methyl derivative (5, cis) revealed that the benzylic side chain remained pseudoaxial while the 7-(R)-methyl derivative (6, trans) adopted the desired pseudoequatorial conformation. Both derivatives displayed similar activities against Mycobacterium tuberculosis, but neither showed improved aqueous solubility, suggesting that inherent lattice stability is not likely to be a major factor in limiting solubility. Conformational analysis revealed that all three compounds have similar energetically accessible conformations in solution. Additionally, these results suggest that the nitroreductase that initially recognizes PA-824 is somewhat insensitive to substitutions at the 7-position.
Keywords: Mycobacterium tuberculosis, PA-824, solubility
Despite decades of research, tuberculosis continues to be a major public health threat. The WHO estimates that, in 2005, 1.6 million people died from tuberculosis (TB) which is caused by the microorganism Mycobacterium tuberculosis (Mtb).1 Current therapy for TB lasts for several months and is called directly-observed therapy short-course (DOTS). This regimen calls for an intensive phase of chemotherapy using four drugs for 2 months, followed by a continuation phase using two drugs for 4–6 months.2 Rifampin is inarguably the most important drug in the DOTS regimen as it allowed shortening the course of therapy to 6 months from the standard 12–18 months of previous regimens. The potency of rifampin is thought to be due in part to its action against both aerobically-growing and anaerobically-adapted, non-replicating mycobacteria.3
Nitroimidazoles are a class of compounds widely used clinically for the management of anaerobic bacterial infections.4 Metronidazole (1) has activity only against anaerobically adapted Mtb5 and is the subject of an on-going clinical trial.6 Bicyclic nitroimidazoles have also been developed that have both aerobic and anaerobic activity against Mtb (Figure 1). CGI-17341 (2) and OPC-67683 (3), both nitroimidazo-oxazoles, and PA-824 (4), a nitroimidazo-oxazine, have activity against aerobic and anaerobic populations of Mtb.7-9 PA-824 has an aerobic MIC (minimum inhibitory concentration) of 0.4μM and substantial anaerobic activity at 8–16μM. Several groups have also confirmed the efficacy of PA-824 in the mouse model of TB infection.10–13 PA-824, and more recently OPC-67683, have entered clinical trials for the treatment of tuberculosis.14,15 Prior work has shown that PA-824 acts as a prodrug requiring bioreductive activation before exerting its biological effect.16 One serious issue with the clinical use of both PA-824 and OPC-67683 lies in their low water solubility. PA-824 is only soluble at 10.2μg/mL17 (Table 1) and the in vivo activity of OPC-67683 requires formulation in 5% gum arabic.15 Similarly, the low water solubility of PA-824 necessitates the use of a complex formulation in animals (CM-2) that may not be suitable for human use and may require development of an expensive alternate.
Figure 1.

Nitroimidazoles with antitubercular activity.
Table 1.
Biological activity and solubility of PA-824 and 7-methyl analogues. The phenotype and genetic changes in mutants H37Rv-T3, H37Rv-5A1 and H37Rv-T2 have been reported.16
| Compound | H37Rv MIC (μM) | H37Rv MAC (μM) | H37Rv-T3MIC (μM) | H37Rv-5A1MIC (μM) | H37Rv-T2MIC (μM) | Solubility (μg/mL) |
|---|---|---|---|---|---|---|
| PA-824 (4) | 0.4 | 8–16 | >100 | >100 | >100 | 10.2 ± 1.6 |
| 7(S)-Methyl-824 (5, cis) | 0.2–0.4 | 16 | >100 | >100 | >100 | 9.89 ± 1.9 |
| 7(R)-Methyl-824 (6, trans) | 0.2 | 8–16 | >100 | >100 | >100 | 10.3 ± 1.6 |
Low solubility can often be related to thermodynamic stability of a particular crystalline polymorph.18 Therefore, in an effort to understand the reasons behind the low water solubility of PA-824, we determined its crystal structure (Figure 2).19, 31 This structure showed PA-824 arranged in antiparallel layers with stacked nitroimidazoles alternating with stacked p-trifluoromethoxybenzyl ethers. The oxazine ring is bent so that C-6, bearing the trifluoromethoxybenzyl side chain, is out of the plane of the imidazole ring. This forces the side chain into a pseudoaxial orientation. We hypothesized that the low water solubility of PA-824 could be due to tight packing in the crystal lattice and that disruption of this arrangement might result in improved water solubility. We postulated that adding a substituent on the oxazine ring adjacent to the trifluoromethoxybenzyl ether would force this side chain into the pseudoequatorial position disrupting the antiparallel stacking observed in the crystal structure.
Figure 2.
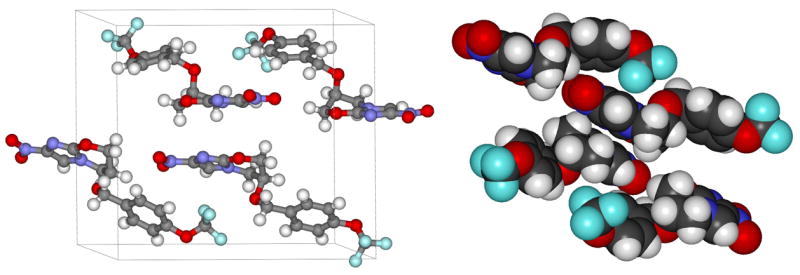
Crystal structure of PA-824. Asymmetric unit of PA-824 crystal shows staggered configuration of individual molecules and four molecules per unit cell (left). Space-filling diagram highlights tight packing arrangement (right).
To this end, we targeted the 7-methyl analogues of PA-824, 5 and 6 (Figure 1). The synthesis of these compounds is shown in Scheme 1. Briefly, crotonyl chloride (7) underwent asymmetric dihydroxylation using AD-mix αto give diol 8.20 Ring-closure under basic conditions yielded epoxy alcohol 10.21 To prepare the opposite diastereomer, racemic allylic alcohol 9 underwent Sharpless epoxidation to give epoxy alcohol 11.22 Compounds 10 and 11 were protected to give 12 and 13,23 respectively. Alkylation of 2,4-dinitroimidazole24 yielded the corresponding secondary alcohols 14 and 15,25 which were converted to the THP-protected derivatives 16 and 17.26 Desilylation and ring cyclization proceeded in one step to give the oxazines 18 and 19.27 Deprotection, giving free alcohols 20 and 21,28 followed by benzylation with p-trifluoromethoxyl bromide delivered desired compounds 7-(S)-methyl-PA-824 (5) and 7-(R)-methyl-PA-824 (6), respectively.29
Scheme 1.

Synthesis of 7-Methyl Analogues of PA-824a
aReagents and conditions: (a) AD-mix, CH3SO2NH2, NaHCO3, tBuOH, H2O, 0°C; (b) KOH, Et2O, 0°C; (c) D-(−)-DIPT, TBHP, Ti(OiPr)4, CH2Cl2, −20°C; (d) TBSCl, imidazole, CH2Cl2, 0°C→rt; (e) 2,4-dinitroimidazole, EtOH, 70°C; (f) 3,4-dihydro-2H-pyran, PPTS, CH2Cl2, rt; (g) TBAF, THF, rt; (h) AcOH, THF, H2O, 45°C; (i) NaH, 4-rifluoromethoxybenzyl bromide, DMF, -45°C→rt.
Compounds 5 and 6 displayed comparable antitubercular activities to PA-824 under aerobic conditions (Table 1). The MIC for 7-(S)-methyl-PA-824 (5) is 0.2–0.4 μM and for 7-(R)-methyl-PA-824 (6) is 0.2 μM. Like PA-824, the two methyl-oxazines both require reduced coenzyme F420 as mutants that either fail to reduce this cofactor (H37Rv-T3) or mutants deficient in its biosynthesis (H37Rv-5A1) are cross-resistant to these nitroimidazoles. The reduction of these compounds is probably mediated by the nitroreductase Rv3547 as mutants lacking this enzyme (H37Rv-T2) are also cross-resistant.16 The MAC (minimum anaerobicidal concentration) values for compounds 4–6 were equivalent.30
Unfortunately, when we determined the thermodynamic solubility of compounds 5 and 6, they were not more soluble than PA-824, each having a solubility limit of approximately 10 μg/mL (Table 1).17 To understand the consequence of the methyl substituent on the conformation of these compounds in the crystalline form, we determined the crystal structures of compounds 5 and 6 (Figure 3).31 The crystal structure of compound 5 (cis) shows the oxazine ring in the same conformation as in that of PA-824. Thus, carbon-6 in 5 is out of the plane of the imidazole ring, just as in the crystal structure of PA-824. The side chain in 5 is similarly pseudoaxial. As hoped, in compound 6, the trans isomer, the opposite effect occurred. Carbon-6 in this compound is out of the plane of the imidazole ring, but on the side opposite to that found in PA-824 and compound 5. This conformation places the benzylic side chain in the desired pseudoequatorial conformation. The conformation of compound 6 depicted in the crystal structure places the substituents of both carbon-6 and carbon-7 in the more favorable pseudoequatorial positions. Interestingly, the packing diagrams for compounds 5 and 6 do not show the same arrangement of antiparallel molecules as in the case of PA-824 (Figure 3). Instead, the compounds are arranged linearly in a back-to-back conformation with imidazole rings in a nearly perpendicular arrangement between rows. The similarity between packing diagrams for compounds 5 and 6 indicates that the differences in single-molecule structure do not impact the overall packing between molecules.
Figure 3.
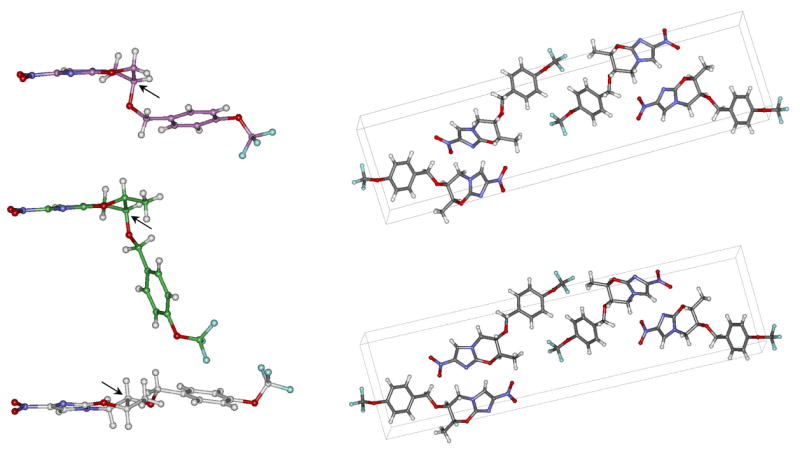
Crystal structures of PA-824 (purple), 7-(S)-methyl-PA-824 (5, cis, green), and 7-(R)-methyl-PA-824 (6, trans, white). View on the left is shown along the plane of the nitroimidazole to highlight the differing conformations of the oxazine rings in each structure. Arrows indicate the C-6 carbon atoms. Crystal packing diagrams of compounds 5 (right, top) and 6 (right, bottom) are distinct from that of PA-824.
Despite changing the angle of the oxazine-trifluoromethoxybenzyl side chain to either more pseudoaxial (in compound 5) or more pseudoequatorial (in compound 6) compared with PA-824, there was a negligible effect on solubility. This result suggests that stability of this crystalline form is not a major contributor to the overall insolubility of this molecule. Further, the melting points and crystal packing densities of these compounds are very similar. The melting points are 149–150°C (PA-824)32, 146–148 °C (compound 5), and 126.5–128.5 °C (compound 6). The crystal packing densities are 1.55–1.59 g/cm3 for all three compounds. The similarities in these numbers support the conclusion that the compounds’ poor solubility is not related to the crystal packing.
Nonetheless, the variation in angle between the imidazo-oxazine and the trifluoromethoxybenzyl side chain seemed incongruent with the minor variations in biological activity, particularly since the R isomer of PA-824 is nearly 50-fold less active than the S.10 To understand this discrepancy, we performed a conformational analysis on these compounds to identify the lowest energy conformations in solution.33 Compounds 4–6 were minimized to yield the 100 lowest energy structures. For PA-824 (4), all 100 structures agreed well with the crystal structure (within 9 kJ/mol), the primary differences arising from rotation of the benzyl side chain about the ether bond. For compound 5 (cis), 73 of the 100 lowest energy conformations agreed with the PA-824 set. The remaining conformations agreed well with the compound 5 crystal structure, although these had minimized energies of at least 12.2 kJ/mol above the global minimum. For compound 6 (trans), 91 of 100 conformations were nearly the same as PA-824 (within 6 kJ/mol). The remaining 9 solutions agreed with the crystal structure of 6, being at least 3.9 kJ/mol above the global minimum. These data indicate that the major conformation in solution of both 7-methyl isomers will most likely be very close to that of PA-824. The slightly higher activity seen with compound 6 is intriguing in that it suggests that the active conformer may, in fact, not be the minimum energy conformer in solution. This result suggests that the active conformer may be more abundant with the trans substituent forcing an even more pronounced pseudoequatorial orientation of the side chain.
Thus, these results suggest that the low solubility of PA-824 in aqueous media may not be the result of an intrinsically stable crystalline form. Future efforts will focus on the introduction of solubilizing groups while preserving activity. Such studies would be greatly facilitated by an in vitro nitroreductase assay from which detailed SAR could be obtained. The activity of compounds 5 and 6 against wild-type H37Rv and all three classes of mutants resistant to PA-824 suggests that these compounds are acting through the same mechanism as PA-824 and are substrates of Rv3547. The activity of 5 and 6 demonstrates that the nitroreductase accepts at least small substituents at the 7-position. These results suggest that further derivatives at the 7-position of the oxazine may be fruitfully explored to improve activity and solubility of these important compounds.
Acknowledgments
We thank the Division of Intramural Research (NIAID, NIH) for support of this work. Crystal structures were determined by Lee Daniels at Rigaku/MSC, Inc. (PA-824) and Kenneth Hardcastle of Emory University (compounds 5 and 6).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.WHO International Fact sheet N°104. 2007. http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 2.Treatment of Tuberculosis: Guidelines for National Programmes WHO/CDS/TB 2003.313. WHO; Geneva: 2003. [Google Scholar]
- 3.Duncan K, Barry CE., 3rd Curr Opin Microbiol. 2004;7:460. doi: 10.1016/j.mib.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Barry CE, 3rd, Boshoff HI, Dowd CS. Curr Pharm Des. 2004;10:3239. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 5.Wayne LG, Sramek HA. Antimicrob Agents Chemother. 1994;38:2054. doi: 10.1128/aac.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.http://www.clinicaltrials.gov/ct2/show/NCT00425113?term=metronidazole&cond=tuberculosis&rank=1
- 7.Ashtekar DR, Costa-Perira R, Nagrajan K, Vishvanathan N, Bhatt AD, Rittel W. Antimicrob Agents Chemother. 1993;37:183. doi: 10.1128/aac.37.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, Sasaki H, Shimokawa Y, Komatsu M. PLoS Med. 2006;3:2131. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. Nature. 2000;405:962. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 10.Baker WR, Shaopei C, Keeler EL. 5,668,127. US Patent. 1997
- 11.Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM. Antimicrob Agents Chemother. 2005;49:2294. doi: 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manjunatha UH, Dowd CS, Zhang L, Barry CE., III Unpublished work. [Google Scholar]
- 13.Tyagi S, Nuermberger E, Yoshimatsu T, Williams K, Rosenthal I, Lounis N, Bishai W, Grosset J. Antimicrob Agents Chemother. 2005;49:2289. doi: 10.1128/AAC.49.6.2289-2293.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Global Alliance for TB Drug Development (http://www.tballiance.org/home/home.php)
- 15.Sasaki H, Haraguchi Y, Itotani M, Kuroda H, Hashizume H, Tomishige T, Kawasaki M, Matsumoto M, Komatsu M, Tsubouchi H. J Med Chem. 2006;49:7854. doi: 10.1021/jm060957y. [DOI] [PubMed] [Google Scholar]
- 16.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE., 3rd Proc Natl Acad Sci USA. 2006;103:431. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The thermodynamic solubility determination was based on the method of Avdeef and Testa Avdeef A, Testa B. Cell Mol Life Sci. 2002;59:1681. doi: 10.1007/PL00012496. Briefly, 3–5mg of compound was exposed to 0.5mL water and the suspension is shaken for 3 days. The sample is centrifuged at 10,000g for 5 minutes. The supernatant is diluted in water and are injected onto an LC/MS where the UV peak areas are recorded at 310nm. The concentration of the sample solution is calculated using a previously determined calibration curve, corrected for the dilution factor of the sample.
- 18.Singhal D, Curatolo W. Adv Drug Delivery Rev. 2004;56:335. doi: 10.1016/j.addr.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 19.Crystal data for compound 4: C14H12N3O5F3, MW=359.26, monoclinic, P21; Z=4, a=14.7024(17), b=5.7699(8), c=17.702(3) Å; β=94.913(8) °; V=1496.2(3) Å3, F(000)= 736.00; Dx=1.595 g/cm3; 2θmax=136.7° (Rigaku RAXIS RAPID detector with graphite monochromated Cu-Kα radiation.), R=0.0430 (4733 data with I>2σI, 451 parameters).
- 20.Richardson TI, Rychnovsky SD. J Org Chem. 1996;61:4219. doi: 10.1021/jo960218x. [DOI] [PubMed] [Google Scholar]
- 21.Vanhessche KPM, Wang ZM, Sharpless KB. Tetrahedron Lett. 1994;35:3469. [Google Scholar]
- 22.White JD, Kang MC, Sheldon BG. Tetrahedron Lett. 1983;24:4539. [Google Scholar]
- 23.Hindupur RM, Panicker B, Valluri M, Avery M. Tetrahedron Lett. 2001;42:7341. [Google Scholar]
- 24.Sudarsanam VNK, George T, Shenoy SJ, Iyer VV, Kaulgud AP. Indian J Chem, Sect B. 1982;21B:1022. [Google Scholar]
- 25.Preparation of 14: (2S, 3S)-3-(TBDMS)-1-(2,4-dinitro-imidazol-1-yl)-butan-2-ol. A mixture of 2,4-dinitroimidazole (0.25 g, 1.58 mmol) and epoxide 12 (0.48 g, 2.37 mmol) in ethanol (0.53 mL) was heated at 70 ºC for 18 h in a sealed tube. After removal of the solvent, the residue was separated by silica gel column chromatography (10–20% EA in hexanes) to give 14 as an oil (0.22 g, 38%). 1H NMR (300 MHz, CDCl3) δ 7.99 (s, 1 H), 4.77 (dd, J = 13.8, 2.1 Hz, 1 H), 4.29 (dd, J = 13.8, 9.9 Hz, 1 H), 3.89 (m, 1 H), 3.74 (m, 1 H), 2.58 (d, J = 6.9 Hz, 1 H), 1.31 (d, J = 6.3 Hz, 3 H), 0.92 (s, 9 H), 0.15 (s, 3 H), 0.14 (s 3 H). 13C NMR (75 MHz, CDCl3) δ 143.14, 125.11, 74.10, 69.21, 60.05, 54.63, 25.86, 19.94, 18.10, −4.07, −4.81. [α]D = +16.9 (c = 1.0 in CH2Cl2). HRMS calcd for C13H23N3O4Si [M – NO2]+: 314.1536; found 314.1418. Compound 15 (2S, 3R)-3-(TBDMS)-1-(2,4-dinitro-imidazol-1-yl)-butan-2-ol was prepared in the same way from epoxide 13 giving (0.193g, 34%). 1H NMR (300 MHz, CDCl3) δ 8.03 (s, 1 H), 4.92 (dd, J = 1.5, 13.6 Hz, 1 H), 4.24 (dd, J = 9.9, 13.6 Hz, 1 H), 4.00 (m, 1 H), 3.86 (m, 1 H), 2.62 (d, J = 6.0 Hz, 1 H), 1.27 (d, J = 6.3 Hz, 3 H), 0.92 (s, 9 H), 0.13 (s, 3 H), 0.12 (s, 3 H). 13C NMR (75 MHz, CDCl3) δ 143.20, 131.30, 125.15, 74.20, 70.19, 53.26, 25.92, 19.12, 18.16, −4.11, −4.72. [α]D = +8.9 (c = 1.0 in CHCl3). HRMS calcd for C13H24N3O4Si [M – NO2]+: 314.1536; found 314.1548.
- 26.Preparation of compound 16: (2S, 3S)1-[3-(TBDMS)-2-(tetrahydro-pyran-2-yloxy)-butyl]-2,4-dinitro-1H-imidazole. A mixture of the 14 (115 mg, 0.319 mmol), PPTS (16.7 mg, 0.0665 mmol), 3,4-dihydro-2H-pyran (60.6 μL, 0.664 mmol) in methylene chloride (1.8 mL) was stirred at rt for 20 h. The reaction was quenched with water (1.8 mL). The organic layer was separated and the aqueous layer was extracted with methylene chloride (3 × 1.8 mL). The combined organic portions were washed with brine and dried over Na2SO4. Solvent removal yielded a pair of diastereomers (133 mg, 94%, ~1:1) as a white solid. The solid was used in the next reaction without further purification. 1H NMR (300 MHz, CDCl3) δ 7.94 (s, 1 H), 7.88 (s, 1 H), 4.99 (dd, J = 2.6, 13.6 HZ, 1 H), 4.86 (dd, J = 2.4, 13.8 Hz, 1H), 4.36 (dd, J = 10.2, 13.8 Hz, 1 H), 4.26−4.08 (m, 4 H), 3.91 (m, 2 H), 3.75 (m, 2 H), 3.36 (m, 2 H), 3.09 (m, 1 H), 1.65−1.09 (m, 18 H), 0.85 (s, 18 H), 0.034 (m, 12 H). 13C NMR (75 MHz, CDCl3) δ 143.01, 142.58, 141.97, 141.60, 125.69, 124.90, 101.77, 100.25, 80.54, 76.93, 68.59, 68.22, 65.29, 63.83, 51.70, 50.56, 30.99, 30.81, 25.84, 25.80, 25.05, 24.88, 21.34, 20.07, 18.24, 18.09, 18.04, 17.01, −4.54, −4.84, −4.85, −4.95. HRMS calcd for C18H33N4O7Si [M + H]+: 445.2119; found 445.2118. Compound 17, (2S, 3R)-3-1-[3-(TBDMS)-2-(tetrahydro-pyran-2-yloxy)-butyl]-2,4-dinitro-1H-imidazole, was prepared in the same way from 15 to give 0.174g (94%). 1H NMR (300 MHz, CDCl3) δ 7.91 (s, 1 H), 7.86 (s, 1 H), 4.93 (d, J = 14.1 Hz, 2 H), 4.42 (dd, J = 9.0, 14.1 Hz, 1 H), 4.30−3.72 (m, 8 H), 3.37−3.08 (m, 3 H), 1.69−1.10 (m, 12 H), 1.24 (d, J = 6.3 Hz, 3 H), 1.23 (s, J = 6.3 Hz, 3 H), 0.90 (s, 9 H), 0.89 (s, 9 H), 0.11 (s, 3 H), 0.86 (s, 3 H), 0.075 (s, 6 H). 13C NMR (75 MHz, CDCl3) δ 143.07, 142.69, 142.02, 141.60, 125.77, 125.16, 101.55, 100.03, 80.62, 78.19, 70.55, 69.35, 65.48, 64.55, 52.26, 51.18, 31.12, 31.05, 25.95, 25.87, 25.16, 24.95, 21.53, 20.75, 20.63, 20.37, 18.23, 18.15, −4.32, −4.36, −4.48, −4.67. HRMS calcd for C18H33N4O7Si [M + H]+: 445.2119; found 445.2123.
- 27.Preparation of 18: (6S, 7S)-7-Methyl-2-nitro-6-(THP)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine. To a solution of 16 (133 mg, 0.299 mmol) in THF (1.2 mL) was added TBAF (0.90 mL, 0.9 mmol, 1 M in THF) at rt. After stirring 0.5h, the solvent was removed and the residue was dissolved in chloroform (2.0 mL). The mixture was washed with saturated NaHCO3 (1.5 mL), water (1.5 mL), and was dried over MgSO4. After removing the solvent, the residue was chromatographed (2.5–5% MeOH in EA) to give the desired product (67.6 mg, 80%). 1H NMR (300 MHz, CDCl3) δ 7.42 (s, 2 H), 4.77 (m, 2 H), 4.64 (m, 1 H), 4.54 (m, 1 H), 4.39 (dd, J = 2.7, 12.9 Hz, 1 H), 4.28 (m, 1 H), 4.21−4.04 ( m, 4 H), 3.86 (m, 1 H), 3.68 (m, 1 H), 3.53 (m, 2 H), 1.59 (d, J = 6.6 Hz, 3 H), 1.51 (d, J = 6.3 Hz, 3 H), 1.82−1.46 (m, 12). 13C NMR (75 MHz, CDCl3) δ 147.98, 147.92, 143.47, 115.70, 115.57, 102.05, 95.67, 76.40, 76.05, 70.26, 65.02, 63.37, 62.97, 48.93, 45.52, 30.59, 30.24, 25.11, 25.02, 19.59, 19.25, 16.80, 16.47. HRMS calcd for C12H18N3O5 [M + H]+: 284.1246; found 284.1249. Compound 19, (6S, 7R)-7-Methyl-2-nitro-6-(THP)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine, was prepared in the same way from 17 (59.6mg, 96%). 1H NMR (300 MHz, CDCl3) δ 7.46 (s, 1 H), 7.43 (s, 1 H), 4.76 (m, 3 H), 4.56 (dq, J = 6.4, 6.4 Hz, 1 H), 4.32 (dd, J = 4.6, 12.8 Hz, 1 H), 4.26 (dd, J = 3.4, 12.4 Hz, 1 H), 4.14−3.99 (m, 4 H), 3.78 (m, 2 H), 3.52 (m, 2 H), 1.74−1.49 (m, 12 H), 1.44 (d, J = 6.6 Hz, 3 H), 1.44 (d, J = 7.2 Hz, 3 H). 13C NMR (75 MHz, CDCl3) δ 147.16, 147.07, 143.87, 115.40, 115.23, 99.92, 97.46, 77.07, 75.73, 70.22, 67.91, 63.42, 62.79, 46.83, 44.74, 30.62, 30.50, 25.20, 25.14, 19.54, 19.02, 17.87, 17.55. HRMS calcd for C12H18N3O5 [M + H]+: 284.1246; found 284.1249.
- 28.Preparation of 20: (6S, 7S)-7-Methyl-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ol. A mixture of 18 (67.6 mg, 0.239 mmol) in acetic acid (1.0 mL), THF (0.52 mL) and water (0.26 mL) was stirred for 11 h at 45 ºC. Prep TLC (5% MeOH in EA) of the reaction mixture gave the desired compound (39.7 mg, 83%) as a white solid, mp (dec.) >200 ºC. 1H NMR (300 MHz, DMSO-d6) δ 8.06 (s, 1 H), 5.71 (bs, 1 H), 4.59 (q, J = 6.3 Hz, 1 H), 4.18 (d, J = 13.0 Hz, 1 H), 4.02 (s, 1 H), 4.00 (d, J = 13.0 Hz, 1 H), 1.36 (d, J = 6.3 Hz, 3 H). 13C NMR (75 MHz, DMSO-d6) δ 147.81, 142.26, 118.04, 76.31, 61.69, 49.97, 16.52. [α]D = −26.4 (c = 1.0 in DMSO-d6). HRMS calcd for C7H10N3O4 [M + H]+: 200.0671; found 200.0690. Compound 21, (6S, 7R)-7-Methyl-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazin-6-ol, was prepared in the same way from 19 to give 33.1 mg (86%). Mp: 182.1–182.6 ºC. 1H NMR (300 MHz, CD3OD) δ 7.77 (s, 1 H), 4.55 (ddq, J = 0.9, 5.3, 6.6 Hz, 1 H), 4.31 (dd, J = 4.2, 12.7 Hz, 1 H), 4.07 (ddd, J = 4.2, 5.3, 5.3 Hz, 1 H), 3.97 (ddd, J = 0.9, 5.3, 12.7, 1 H), 1.98 (bs, exchanged, 1 H), 1.45 (d, J = 6.6 Hz, 3 H). 13C NMR (75 MHz, CD3OD) δ 149.05, 144.25, 117.76, 80.02, 65.37, 48.37, 17.74. [α]D = 22.4 (c = 0.5 in CH3OH). HRMS calcd for C7H10N3O4 [M + H]+: 200.0671; found 200.0683.
- 29.Preparation of 5: (6S, 7S)-7-Methyl-2-nitro-6-(4-trifluoromethoxy-benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine. To a solution of 20 (27 mg, 0.136 mmol) and 4-(trifluoromethoxy)-benzylbromide (26.3 μL, 0.163 mmol) in anhydrous DMF (0.17 mL) was added NaH (3.9 mg, 0.163 mmol) at −45 ºC. After stirring for 1 h, the reaction mixture was allowed to warm to rt where stirring continued for 30 min. The mixture was separated by prep TLC (2% MeOH in EA) to give the target molecule (41.9 mg, 83%) as white needles. Mp: 146–148 ºC. 1H NMR (300 MHz, CD3COCD3) δ 7.70 (s, 1 H), 7.50 (d, J = 8.2 Hz, 2 H), 7.30 (d, J = 8.2 Hz, 2 H), 4.88 (d, J = 12.2 Hz), 4.78 (m, 1 H), 4.73 (d, J = 12.2), 4.61 (dd, J = 13.8, 1.8 Hz, 1 H), 4.34 (dd, J = 10.5, 3.2 Hz, 2 H), 4.23 (m, 1 H) 1.50 (d, J = 6.3 Hz, 3 H). 13C NMR (75 MHz, CD3COCD3) δ 149.00, 149.50, 138.32, 130.43, 123.30, 121.91, 119.80, 117.30, 76.88, 71.32, 71.11, 46.84, 17.08. [α]D = −11.6 (c = 0.5 in acetone). HRMS calcd for C15H15F3N3O5 [M + H]+: 374.0964; found 374.0970. Compound 6, (6S, 7R)-7-Methyl-2-nitro-6-(4-trifluoromethoxy-benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine, was prepared in the same way from 21 to give 37.1 mg (62%) of the product. Mp: 126.5–128.5 ºC. 1H NMR (300 MHz, CD3COCD3) δ 7.77 (s, 1 H), 7.50 (d, J = 8.1 Hz, 2 H), 7.30 (d, J = 8.1 Hz, 2 H), 4.89 (dq, J = 3.6, 6.9 Hz, 1 H), 4.81 (s, 2 H), 4.49 (dd, J = 3.8, 13.4 Hz, 1 H), 4.39 (dd, J = 3.5, 13.4 Hz, 1 H), 4.19 (ddd, 3.5, 3.6, 3.8, 1 H), 1.47 (d, J = 6.9 Hz, 3 H). 13C NMR (75 MHz, CD3COCD3) δ 149.72, 148.02, 138.66, 130.61, 123.49, 122.21, 120.10, 117.66, 76.83, 72.48, 71.13, 45.58, 18.08. [α]D = 6.4 (c = 0.5 in acetone). HRMS calcd for C15H15F3N3O5 [M + H]+: 374.0964; found 374.0966.
- 30.Isoniazid, rifampicin, metronidazole and methylene blue were obtained from Sigma-Aldrich. All stocks were made in 20 mM DMSO. The broth dilution method was used to determine the MIC99 of all compounds against Mtb (H37Rv, ATCC 27294) and all H37Rv mutants as described previously in Domenech et al., Infect. Immun. 2005, 73, 3492–501. For oxygen depletion assays (MAC) early log phase Mtb cultures in Dubos broth was diluted 100-fold and 20-mL was transferred to tubes (Pyrex 16 × 125 mm culture tubes) to maintain a head space ratio of 0.5 as described previously (Wayne in Mycobacterium tuberculosis Protocols, edited by T. Parish and N. G. Stoker, 2001, Humana Press, New Jersey, pp 247–270). The tubes were sealed with paraplast and incubated for 20 days under uniform stirring at 180 rpm using magnetic stirring bars. Methylene blue (1.5 μg/mL) was added to a reference tube to visualize oxygen depletion. Mtb NRP-2 stage cells (100 μL) were exposed to 1.95–500 μM of drug in a 96-well microplate in two-fold drug dilutions. Handling of NRP-2 cells was done in a vinyl anaerobic chamber (Coy Laboratories, Michigan) fitted with a Coy Model 10 gas analyzer and vacuum air lock chamber. The anaerobic chamber was maintained under 90% nitrogen and 10% hydrogen. The 96-well plates were placed in a Type A Bio-bag anaerobic chamber (Bection and Dickinson, Maryland) with an oxygen indicator strip and incubated at 37°C for 7 days. After drug exposure the cells were washed three times with fresh Dubos broth. The MAC against Mtb was estimated by measuring number of viable bacilli spotted on a 7H11 agar-containing 96-well plate. For spotting, 5 μL of a cell suspension from each well is spotted on a 7H11 agar-containing 96-well plate, incubated for 3 weeks at 37°C, and the concentration of the drug at which 90% reduction in visible growth is recorded.
- 31.Crystal data for compound 5: C15H14F3N3O5, MW=373.29, orthorhombic, P212121; Z=4, a=4.6223(2), b=9.3690(4), c=36.8642(14) Å; V=1596.45(11) Å3, F(000)=768; Dx=1.553 Mg/m3; θ range=8.63–50.08° (Bruker CCD area detector with graphite monochromated Cu-Kα radiation.), R=0.0197 (1648 data with I>2σI, 248 parameters). For compound 6: C15H14F3N3O5, MW=373.29, orthorhombic, P212121; Z=4, a=4.42660(10), b=9.6295(3), c=37.3504(10) Å; V=1592.10(7) Å3, F(000)=768; Dx=1.557 Mg/m3; θ range=2.37–66.30° (Bruker CCD area detector with graphite monochromated Cu-Kα radiation.), R=0.0264 (2318 data with I>2σI, 236 parameters). Crystallographic data (excluding structure factors) for these compounds have been deposited at the Cambridge Crystallographic Data Centre [CCDC 669894 (for compound 4), 669895 (for compound 5), and 669896 (for compound 6)]. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44(0)-1223-336033 or email: deposit@ccdc.cam.ac.uk].
- 32.Baker WR, Shaopei C, Keeler EL. 6,087,358. US Patent. 2000
- 33.All calculations performed with Macromodel 9.1 (Schrödinger) using the conformational search module. Automatic setup was used to describe the conformational search parameters. OPLS2005 was the force field used along with an implicit water solvent model. The torsional sampling model (MCMM) was used with up to 10,000 steps saving the 100 lowest energy unique conformations.