

Abstract
Amyloid plaques are a characteristic feature in Alzheimer’s disease (AD). A novel non-toxic contrast agent is presented, Gd-DTPA-K6Aβ1–30, which is homologous to Aβ, and allows plaque detection in vivo. μMRI was performed on AD model mice and controls prior to and following intra-carotid injection with Gd-DTPA-K6Aβ1–30 in mannitol solution, to transiently open the blood brain barrier. A gradient echo T2*-weighted sequence was used to provide 100 μm isotropic resolution with imaging times of 115 min. The scans were examined with voxel-based analysis (VBA) using statistical parametric mapping, for un-biased quantitative comparison of ligand-injected mice and controls. The results indicate that: 1) Gd-DTPA-K6Aβ1–30 is an effective, non-toxic, ligand for plaque detection when combined with VBA (p<0.01–0.001), comparing pre and post-ligand injection scans. 2) Large plaques can be detected without the use of a contrast agent and this detection co-localizes with iron deposition. 3) Smaller, earlier plaques require contrast ligand for MRI visualization. Our ligand when combined with VBA may be useful for following therapeutic approaches targeting amyloid in transgenic mouse models.
Keywords: Alzheimer’s disease, Magnetic resonance imaging, Voxel-based analysis, Transgenic mice, Imaging, Amyloid, Iron
1. INTRODUCTION
One of the defining lesions of Alzheimer’s disease is the amyloid β (Aβ) plaque. With advances in imaging technology there has been extensive interest in developing techniques to detect Aβ plaques in live animals using approaches that may eventually be applied to humans. Positron emission tomography (PET) using a C11 labeled amyloid binding ligand has recently been shown to provide quantitative information on the amyloid burden in Alzheimer’s disease (AD) patients [20]. However, the low spatial resolution of PET does not allow the visualization of individual plaques and it is unclear whether it will be able to detect the earliest stages of amyloid deposition. It is particularly in these early phases of disease, prior to irreversible damage, that a diagnosis is most difficult to make while having potentially greatest value. Treatment initiated at this early stage will likely have the most dramatic effects on the subsequent amyloid burden, as suggested by therapeutic interventions in AD model animals [33;45] Amyloid plaque deposition tends to follow a stereotypic pattern with AD pathology progression in terms of regional distribution and density of plaque burden, while at the same time it is common for elderly individuals to have some degree of plaque burden, but to remain clinically asymptomatic [4;28;40]. Magnetic resonance imaging (MRI) based plaque detection methods coupled with more accurate amyloid quantitation, may be superior to PET based methods at differentiating degrees of amyloid burden that are associated with early AD versus clinically normal aging. Furthermore, MRI is more widely available and does not require the use of radioligands. Aβ plaques have been detected after a 20 hour imaging time in postmortem AD brain by T2*-weighted MR microimaging (μMRI) [3], but this long imaging time is not feasible for in vivo plaque detection. Also, subsequent studies by other investigators failed to detect Aβ plaques in AD brain tissue by T2*-weighted μMRI although both field strength and resolution were substantially higher than in the prior study [5]. We have previously shown that Aβ1–40 tagged with two paramagnetic MRI contrast agents, gadolinium-diethylenetriaminepentaacetic acid (Gd-DTPA) and monocrystalline iron oxide nanoparticles (MION), can be used to detect Aβ plaques ex vivo and in vivo in the brains of both APP [15] and APP/PS1 [14] transgenic AD model mice after intracarotid co-injection with mannitol to transiently open the blood brain barrier (BBB) [37;42]. We and others have also shown that intravenous administration of putrescine modified, Gd-DTPA-Aβ1–40 allows plaque detection ex vivo suggesting that this approach has a potential for in vivo detection of plaques [27]. More recently, we and other investigators have reported on plaque detection ex vivo [22;49] and in vivo [16;36], without a contrast agent in old transgenic mice with mature plaques. It is likely that this is achieved because of enhanced iron content in mature plaques and may also be related to their compact nature. For early detection of plaques, which is more clinically relevant, it is likely that contrast agents will be needed. The use of full-length Aβ for this purpose may have toxic side effects and can actually promote plaque buildup, since Aβ is known to seed amyloid deposition [18]. Hence, we developed a compound, Gd-DTPA-K6Aβ1–30, that does not form amyloid aggregates and is non-toxic while maintaining high affinity for Aβ. We have previously used K6Aβ1–30 and related derivatives as experimental vaccines against AD [34;35]. In this report we have evaluated the utility of this Aβ homologous peptide, which has therapeutic potential as a vaccine, for a dual use as a MRI contrast agent, when coupled with gadolinium. To analyze our data we used voxel-based analysis techniques [8;9], comparing pre- and post-ligand injection images in both control and AD model mice.
2. METHODS
Contrast agents
Peptide Synthesis
K6Aβ1–30 covalently linked on the N-terminus to the chelator diethylenetriaminepentaacetic acid (DTPA) was synthesized on an ABI 430A peptide synthesizer (AME Bioscience, Chicago, IL) at the Keck peptide synthesis facility at Yale University, CT, using a Vydac C18 preparative column, 2.5 x 30 cm (Vydac Separations, Hesperia, CA). Standard protocols for tBOC (tert-butyloxycarbonyl) chemistry were used and the chelator was linked to the amino-terminus as the final step of synthesis. The peptide was subsequently cleaved from the resins using hydrofluoric acid and purified by high-pressure liquid chromatography (HPLC) on a Vydac C18 preparative column using linear gradients from 0–70% of acetonitrile in 0.1% trifluoroacetic acid.
Gadolinium Chelation
Gadolinium (Gd) was chelated to DTPA-K6Aβ1–30 by incubating the peptide in double distilled water, at pH 7.0 for 24 h with threefold molar excess of Gd, derived from Gd (III) chloride hexahydrate (Sigma, St. Louis, MO). The pH of the solution was adjusted to pH 7 with 1 N NaOH and monitored with pH test strips. Mass spectroscopy of the lyophilized end-product was used to verify the expected molecular weight.
Binding and Toxicity Studies
Experiments were performed to assess the ability of the magnetically labeled K6Aβ1–30 peptide to bind to Aβ1–42 peptide which is a major constituent of amyloid plaques both in AD and in its mouse models. Therefore, the calculated affinity constants approximate the binding of the MR ligand to Aβ plaques in the AD-transgenic mice. The interaction between Gd-DTPA-K6Aβ1–30 and Aβ1–42 was evaluated by enzyme-linked immunosorbent solid phase assays, similar to previously described protocols [42]. Here, Gd-DTPA-K6Aβ1–30 was coated onto the plate and binding of Aβ1–42 was detected with an Aβ1–42 specific antibody, whereas in our previous study, the plate was coated with Aβ1–42 and binding of Gd-DTPA-Aβ1–40 was detected with an Aβ1–40 specific antibody. This modification was necessary because an antibody that specifically recognizes K6Aβ1–30 is not available. The specificity of Gd-DTPA-K6Aβ1–30 binding to Aβ1–42 could also be assessed by displacing it by DTPA-K6Aβ1–30. All binding studies were performed in triplicate. Briefly, polystyrene microtiter plates (Immulon 2HB, Thermo Electron Corp., Milford, MA) were coated overnight at 4°C with freshly dissolved Gd-DTPA-K6Aβ1–30 in 50 mM NaHCO3 (0.5 μg/well) and then blocked for 1 h (Superblock, Pierce, Rockford, IL). The plates were washed 3 times before blocking and 3–4 times between all subsequent steps. The Gd-DTPA-K6Aβ1–30 coated wells were then exposed to increasing concentrations (0–11,000 nM) of Aβ1–42 in Tris-buffered saline for 3 h. Bound Aβ1–42 was detected using an anti-Aβ1–42 antibody (Biosource International, Hopkinton, MA) that does not cross-react with K6Aβ1–30. The plates were incubated for 1 h with an anti-rabbit horseradish peroxidase-linked antibody (Amersham Biosciences, UK), developed for 15 min with a TMB peroxidase kit (Bio-Rad, Hercules, CA), and quantified at 450 nm on a 7520 microplate reader (Cambridge Technology, Watertown, MA). The data were analyzed by a nonlinear regression fit algorithm in Prism (v3.0, Graph-Pad Software, San Diego, CA). Controls for nonspecific binding included wells without coating with Gd-DTPA-K6Aβ1–30 and omission of the anti-Aβ1–42 or secondary antibodies.
The potential neurotoxicity of the MRI ligand, Gd-DTPA-K6Aβ1–30, was evaluated at 6 days in a human neuroblastoma cell line (SK-N-SH) using the MTT assay as described previously [35]. Aβ1–42, K6Aβ1–30 and DTPA-K6Aβ1–30 were used as controls. The dose of each peptide was 10 μmol/l, at which dose Aβ1–42 is toxic in this culture model. Briefly, cells were plated at 10,000 cells per 100 μl of culture medium per well in flat-bottom, 96-well microtiter plates. After cell attachment to the plate overnight in an incubator (37°C; 5.0% CO2), 10 μl of freshly prepared peptide solution (in sterile H2O) was added. Subsequent steps were as described previously [35].
Mannitol Co-Injection
As with Gd-DTPA-Aβ1–40 used in our prior report [42], Gd-DTPA-K6Aβ1–30 does not cross the BBB in sufficient quantities for μMRI plaque labeling, and must be co-injected into the carotid artery with mannitol. For mannitol co-injection, 400 μg of Gd-DTPA-K6Aβ1–30 or DTPA-K6Aβ1–30 (controls) was dissolved in 100 μl of water, and then diluted in 600 μl of 15% mannitol in PBS immediately before infusion.
Mouse Preparation and Injections
The peptides were infused via the intracarotid artery using a PHD2000 computer-controlled syringe pump (Harvard Apparatus, Hollison, MA), as we have previously described [42]. The infusion cannula was constructed from polyethylene tubing PE-10 (Intramedic, Becton Dickinson, Parsippany, NJ; ID=0.28 mm and OD=0.61 mm) by heating and stretching it to reduce its OD to approximately 0.25 mm. Anesthesia was induced with 5% isoflurane in air for 3–5 min, followed by 1 to 1.5% isoflurane in air to maintain anesthesia. The skin was subsequently shaved and cleaned with 70% ethanol around the injection site. Fine scissors were then used on the neck to expose the common carotid artery (CCA). For injection into the CCA, a 5-0 silk suture was tied loosely at the cephalic end of the right common artery and an identical suture was ligated at its central portion. Between the ligations, a puncture was made with a 30-gauge needle. Then, the modified PE-10 tubing, attached to a 1-cc syringe filled with labeled peptide, was introduced into the right CCA through the small puncture. The suture at the cephalic CCA was then tightened around the intraluminal catheter to prevent bleeding. During the injection, the left CCA was temporarily clamped with a microvascular clip. Clamping briefly increases intra-arterial pressure that promotes the osmotic BBB passage of the contrast agent. The clamping interval needs to be determined empirically and may depend on the animal model, age, contrast agent, its dose and solvent, as well as the volume and rate of injection. Under our conditions, periodic clamping for a few seconds at a time throughout the injection interval, while maintaining a normal breathing pattern in the mouse, enhances consistency in plaque labeling with contrast agent. Prolonged clamping, however, may lead to non-specific vascular labeling which can be identified on MRI images by its streak-like vascular pattern. The Gd-DTPA-K6Aβ1–30/mannitol mixture (600 μl) was injected into the carotid artery at a rate of 60 μl per minute. Following the injection into the right CCA, the microvascular clip was removed, the catheter withdrawn from the right CCA, and the cephalic CCA was ligated. The puncture was subsequently sealed with cyanoacrylate glue. The CCA was then unligated and with the blood flow restored the wound was closed with suture, and the mice were kept warm on a heating pad until they regained full mobility.
In Vivo MR Brain Imaging
MR Micro-Imaging System (μMRI)
The scans were obtained with a SMIS console (Guildford, UK) interfaced to a 7-T horizontal bore magnet equipped with 250-mT/m actively shielded gradients with 200-μs rise time (Magnex Scientific, Abingdon UK). A quadrature litz coil (Doty Scientific, Columbia, SC USA ) designed to fit the mouse head (inner diameter, ID = 25 mm; length along the magnet bore axis, L = 22 mm) and tuned to 301-MHz was used for imaging the brain. The coil was incorporated into an in-house custom made holder containing a tooth bar that stabilized the head of the mouse during MRI and was fitted with a nosecone for isoflurane (Aerane, Baxter, Deerfield, IL) anesthesia delivery and with various physiological monitoring devices. The mice were imaged 4–6 hours following injection of the contrast agent to ensure its clearance from the blood stream and to reduce the background signal. For induction of anesthesia, the mice received 5% isoflurane in air for 3 to 5 minutes and subsequently 1 to 1.5% isoflurane in air to maintain anesthesia during the MRI scans. Gd-DTPA-K6Aβ1–30 was administered by an intracarotid injection with mannitol to osmotically open the BBB to enhance its uptake into the brain (n=9; see Table 1). Each mouse was scanned 3 times. A 3-D gradient echo T2*-weighted sequence was used for imaging (TE = 15 ms; TR = 50ms; flip angle = 20º), providing a 100 μm isotropic resolution with an imaging time of 115 min. The advantage of the 3D-imaging approach is that the image set can be reprocessed in any desired slice orientation, facilitating image comparison during longitudinal studies and co-registration with histology. A pre-ligand injection scan was performed on an intact mouse to determine if plaques could be detected without a contrast agent and to assess the specificity of plaque detection. An injection scan was performed two weeks later, 4–6 hours following injection of a contrast agent. Finally, a post-ligand injection scan was performed two weeks later to determine if the contrast agent was cleared from the labeled plaques. These studies were performed in 18–20 months old Tg2576 mice and 6–8 months old APP/PS1 mice that already have substantial Aβ plaque burden. Control mice included APP/PS1 mice injected with DTPA-K6Aβ1–30, without Gd (n=3) and wild-type mice injected with Gd-DTPA-K6Aβ1–30 (n=5) or Gd-DTPA (Magnevist (Berlex, Montville, NJ), n=5). Additional controls were repeated pre-ligand scans of the same transgenic mice (11 sets of two pre-ligand scans).
Table 1.
Summary of the Sets of Scans from AD-Transgenic- and Wild-type Mice Imaged In Vivo with μMRI*
| Mice |
Set 1 (2 scans compared without contrast agent) |
Set 2 (Gd-K6Aβ1–30 injection scan compared to pre-ligand injection scan) |
Set 3 (Magnevist control injection scan compared to pre-ligand injection scan) |
|---|---|---|---|
| Transgenic | 11 APP/PS1 Tg (In the first scan no injection was given. In the second scan 8 mice received no injection and 3 mice received DTPA-K6Aβ1–30 with no Gd.) | 9 (3 APP/PS1 Tg and 6 APP Tg) | |
| Wild-type | 10 | 5 | 5 |
Quantitative analysis of these sets is presented in Figure 5, in which the wild-type scans were combined.
Group Assignments
A total of 30 mice were examined (20 transgenic AD model mice and 10 wild-type controls), and 3 groups created:
Transgenic Control AD model mice (Tg2576 and APP/PS1; n=11) that were not injected (n=8) or received control peptide (n=3; Gd free DTPA-K6Aβ1–30).
Transgenic AD model mice (n=9) that were injected with Gd-DTPA-K6Aβ1–30.
Wild-type (Wt) mice (n=10) injected with Gd-DTPA (Magnevist; n=5) or Gd-DTPA-K6Aβ1–30 (n=5).
Data Analysis and Statistical Parametric Mapping
The MRI images were analyzed on a Sun Ultra 60 workstation (Sun Microsystems, Mountain View, CA) using MATLAB 5.3 (the MathWorks, Natick, MA) and Statistical Parametric Mapping [SPM 2, Wellcome Department of Cognitive Neurology, London, UK [8;9]]. The steps involved in processing data for this modified voxel-based analysis (VBA) are realignment, spatial normalization, brain extraction, smoothing and modulation. After being rescaled to an isotropic voxel size of 100 × 100 × 100 μm, the MR images were realigned to correct for positioning errors. This is done by estimating mispositioning relative to the first scan using a least-squares analysis, and realigning the scans post hoc using these estimates with rigid-body transformations and SINC interpolation. To implement the VBA of imaging data, data from different subjects must derive from homologous parts of the brain. Spatial transformations are therefore applied that deform and “warp” the images such that they all conform to a standard brain image (i.e., an anatomical template image). The transformation of an image into a standardized anatomical space as that described in the atlas of Paxinos and Watson (1998) corresponds to spatial normalization. The normalizing transformations are computed on the basis of the MRI data themselves.
The template image was created from all the MRI scans from the entire study group, including the baseline and the follow-up scans, for a total of 30 scans acquired on the same MRI scanner with the same scanning parameters, in order to reduce any scanner-specific bias and provide a template appropriate to the study sample. In order to create such a template, we first chose a brain with a high degree of symmetry, aligned according to the anterior commissure - posterior commissure (AC-PC) line, and free of artifacts. Using SPM, this scan was smoothed with a convolution Gaussian kernel characterized by a full-width at half-maximum (FWHM) = 3 times the voxel dimensions so that the normalization step would not be biased by any peculiar features of the brain. Next, all the MRIs from the entire study cohort were spatially transformed to match this image. This involves registering each scan to the reference image by using the residual sum of squared differences as the matching criterion. The first step in spatial normalization involves estimating the optimum 12-parameter affine transformation to match images [8;9]. SPM utilizes a Bayesian framework, whereby the maximum a posteriori estimate of the spatial transformation is made using prior knowledge of the normal variability in brain size. The second step accounts for global non-linear shape differences, which are modeled by a linear combination of 7x8x7 smooth spatial basis functions. A masking procedure is used to weight the normalization to brain rather than non-brain tissue. The normalized images were averaged to create a mean image that was then smoothed with a convolution Gaussian kernel to create the template brain image representing the prototypical mouse brain.
After creating the template image (as described above), each MRI scan in native space was spatially normalized to this image. The spatially normalized MRI images (now in stereotactic space) were resliced with SINC interpolation. We used a semi-automated procedure to remove scalp tissue, skull and dural venous sinus voxels, as well as CSF, from all the structural MRI volumes. With this procedure, voxels outside the brain were automatically isolated from a brain mask. The brain mask was constructed as the largest contiguous structure that is only weakly connected to adjacent structures (e.g., optic nerve connection to the eyes). The normalized images were smoothed with a Gaussian kernel. Smoothing is done to: 1) Increase signal relative to noise, 2) Condition the data to conform more closely to a Gaussian field model, by rendering the data more normally distributed, and 3) Account for anatomical variability among the brains and reducing the impact of possible small misregistrations [8;9]. In order to preserve the volumes of a particular tissue compartment [grey or white matter or cerebrospinal fluid (CSF)] within each voxel, we modulated the images by the Jacobian determinants derived from the spatial normalization to examine differences in the absolute amounts (volumes) of tissue in each voxel [1;11].
For the analysis of MRI scans the baseline scan (naive mice) was compared to the post-contrast injection scan or control scan (naïve mice or Gd free peptide ligand) in all transgenic and control mice. After injection of the Gd-DTPA-K6Aβ1–30 tracer, plaques are visualized on MRI as spots characterized by signal intensity different from gray- and white matter, resulting in a loss of signal, as we have observed following Gd-DTPA-Aβ1–40 injections [42;43]. The MRI scans were compared using the General Linear Model (GLM) as implemented in SPM 2 [9]. In cross-section, regional specific differences were assessed statistically by comparing the two scans using the GLM/univariate analysis with a two-tailed contrast, namely testing for an increased or decreased probability of particular voxel containing plaques. Longitudinal effects were examined by comparing the two scans within each group using the GLM/repeated measures. For all analyses, we controlled for global differences in voxel intensity across scans by including the global mean voxel intensity value as a confounding covariate in all analyses. Because SPM uses a whole-brain analysis, a correction for multiple comparisons is applied according to the random field theory [47]. In addition, we performed a separate analysis using the mean voxel intensity in the cerebellum as a confounding covariate. The cerebellum was chosen since this is an area of the brain not subject to amyloid deposition. Corrections for the search volume (and implicit multiple comparisons) in terms of the P values were made using Gaussian random field theory, which accommodates spatial correlations inherent in the data and is now established as the conventional approach to inference in smooth spatially extended data. Significance levels for one-tailed T statistics were set at p<0.05, cluster-level corrected for multiple comparisons. Non-parametric Mann-Whitney U tests were used to test for differences between the subgroups of wild-type mice injected with different ligands at p<0.05. The clusters were primarily located in: 1) the dorsal- and dorsolateral cortex (retrosplenial- and parietal association cortex); 2) the ventral- and ventrolateral cortex (entorhinal-, perirhinal and pyriform cortex) as well as the amygdala; and 3) the hippocampus.
Histology
These procedures were performed as we have described elsewhere [31;32]. Briefly, following μMRI, the animals were anesthetized with sodium pentobarbital (150 mg/kg, i.p.), perfused transaortically with phosphate buffer and subsequently with 2% periodate-lysine-paraformaldehyde (PLP). The brains were immersion-fixed further overnight in PLP and then placed in 2% DMSO/20% glycerol in PBS overnight or until sectioned. Serial coronal sections (40 μm) were cut and every fifth section was stained with a combination of 6E10 and 4G8, both monoclonal anti-Aβ antibodies (Signet, Dedham, MA). Free-floating sections were incubated in a mixture of the antibodies at a 1:1000 dilution. An immunodetection kit was employed (MOM, Vector Laboratories, Burlingame, CA), with the anti-mouse IgG secondary antibody used at a 1:1000 dilution. Antibody staining was revealed with 3,3′-diaminobenzidine tetrahydrochloride (Sigma) with nickel ammonium sulfate (Mallinckrodt, Paris, KY) intensification.
Perls iron stain was performed on every fifth section by placing defatted and hydrated slide-mounted sections for 30 min in a solution containing 5% potassium ferrocyanide and 10% hydrochloric acid. The slides were then rinsed in distilled water, and counterstained for 5 min with nuclear fast red solution. Following washes in distilled water, the sections were subsequently dehydrated, cleared in CitriSolv (Fisher Scientific), and cover slipped.
3. RESULTS
Binding Affinity and Toxicity Studies
Prior to determining the utility of Gd-DTPA-K6Aβ1–30, its binding affinity towards Aβ was assessed by ELISA and its predicted lack of toxicity was verified in cell culture (Figure 1). Its affinity for Aβ1–42 (3.2 nM ± 1.2) was higher than our previously reported binding of Gd-DTPA-Aβ1–40 [42]; however, the same protocol could not be used as described in Methods, and we have observed some variability in this type of affinity studies which may be explained by different conformational states of Aβ1–42, as has been previously reported [10;12]. We found that Gd-DTPA-K6Aβ1–30 was not toxic in human neuronal culture as compared to Aβ1–42 and measured by the MTT assay at the concentration tested (Figure 1).
Figure 1.
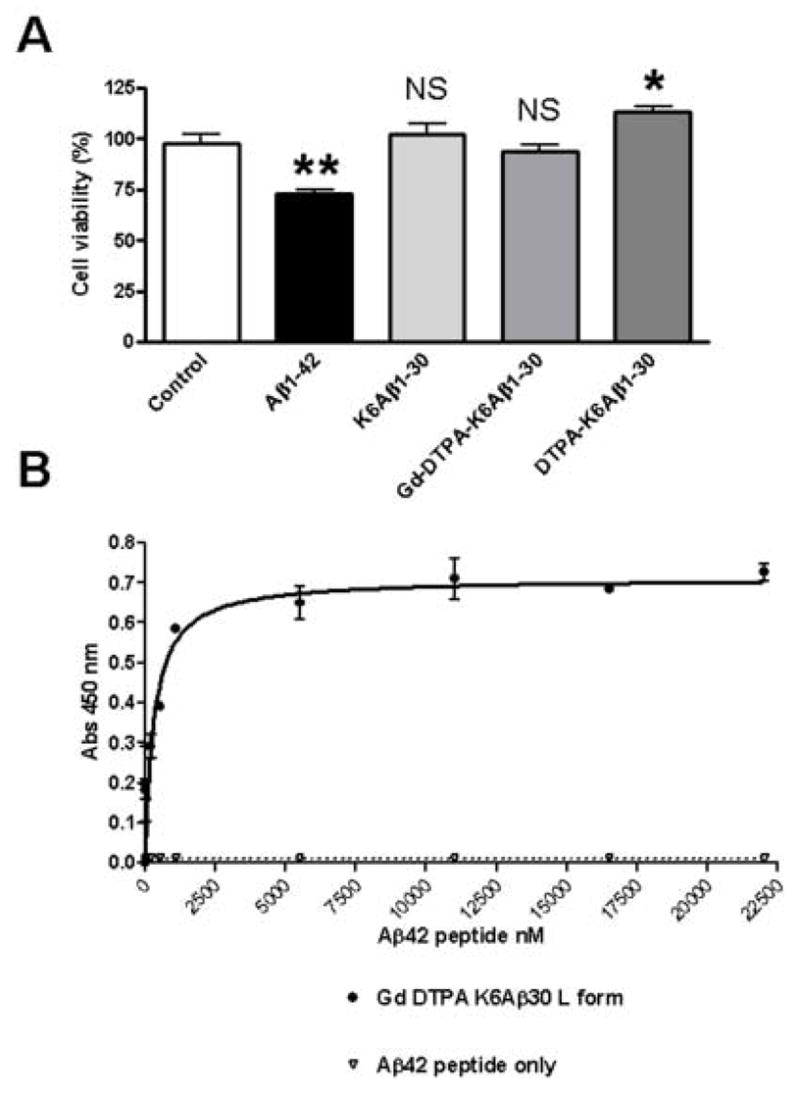
(A) Gd-DTPA-K6Aβ1–30 is not toxic in cell culture
As determined by the MTT assay, Gd-DTPA-K6Aβ1–30 and K6Aβ1–30 had no effect on viability of human neuroblastoma cells (SK-N-SH). DTPA-K6Aβ1–30 was slightly neurotrophic, whereas Aβ1–42 was toxic as is well established. *, p<0.05 **, p<0.01; (One-Way Analysis of Variance, Neuman Keuls post hoc test versus control).
(B) Gd-DTPA-K6Aβ1–30 binds to Aβ1–42 with high affinity (KD=3.2 nM ± 1.2).
μMRI and Voxel-Based Analysis (VBA)
In the pre-ligand injection scans of the old Tg2576 animals some dark spots were observed on T2* gradient echo images that matched large plaques on histological sections. This phenomenon was observed both in mice with high (Figure 2A and C) and low amyloid burden (Figure 3A and C). Iron staining of the brain sections showed a good spatial correlation between Aβ plaques with high iron content (Figure 3D) and plaques detected by MRI (Figure 3A). However, more spots that matched a greater number of plaques were observed following the ligand injection (Figures 2B and 3B), as indicated by VBA. The 2-week post-ligand injection scans were always similar to the pre-ligand injection scans, indicating that the contrast agent was cleared from the brain (data not shown).
Figure 2.
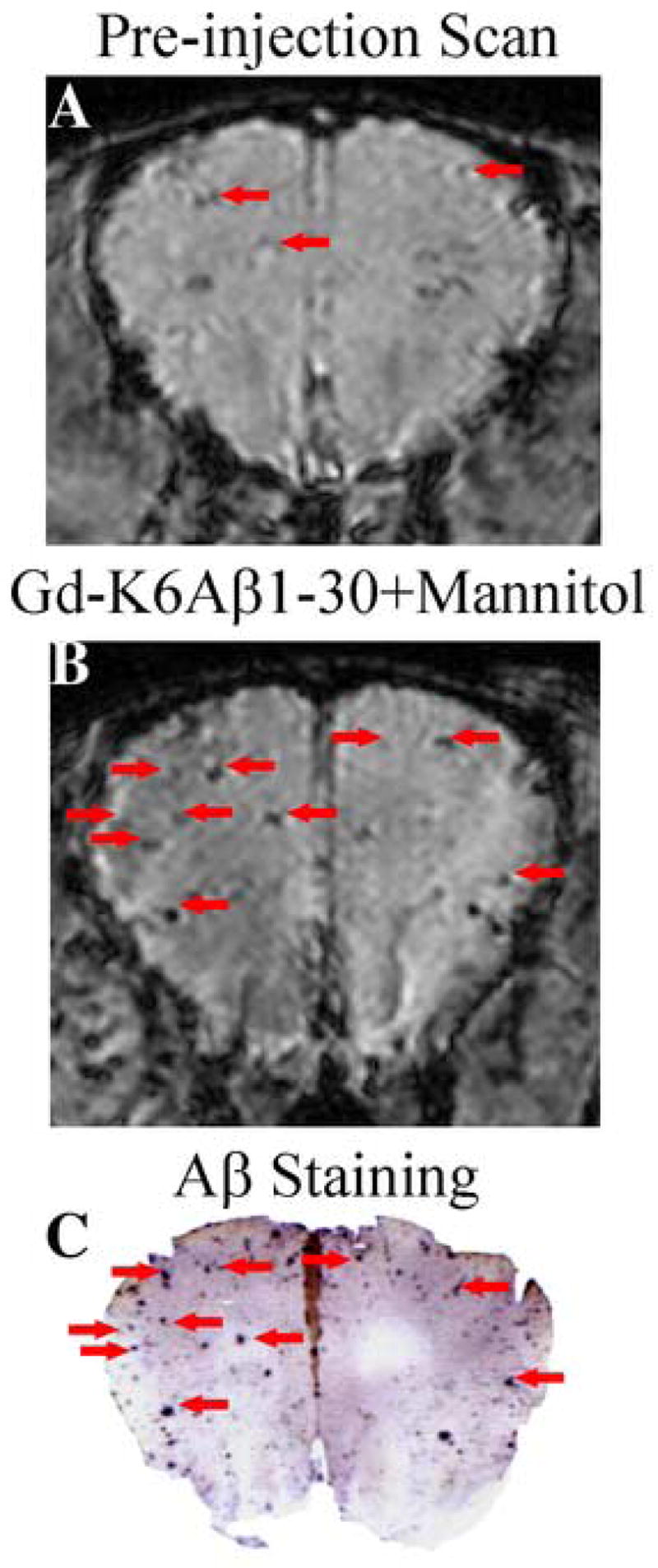
Enhancement of in vivo plaque detection following intracarotid injection of Gd-DTPA-K6Aβ1–30 with mannitol in 20 month old Tg2576 mouse. (A) A few large plaques can be visualized in the pre-ligand scan and their detection is likely related to high iron content in Aβ plaques in older mice (see Figure 3). (B) Following intracarotid injection of Gd-DTPA-K6Aβ1–30, the number of visualized plaques is substantially increased, and these plaques co-register with plaques detected with 6E10 antibody on histological sections as depicted in C.
Figure 3.
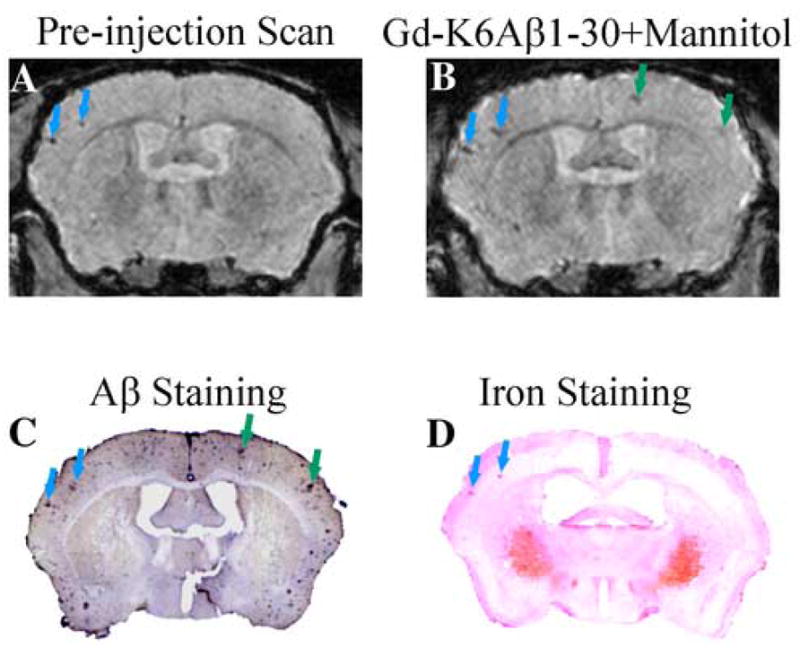
Detection of Aβ plaques without contrast agent correlates with plaques detected by Perl’s iron stain. (A) A few large plaques are detected in the parietal cortex in the pre-ligand injection scan of a 20 month old Tg2576 mouse (blue arrows). Following the intracarotid injection of a contrast agent (Gd-DTPA-K6Aβ1–30), the intensity of these plaques is enhanced and additional plaques are detected (green arrows). These plaques co-register with Aβ deposits on tissue section (C), and the plaques detected without a contrast agent stain for iron as depicted in D. At this magnification, the Perls’ stained iron deposits (blue) appear pink because of the nuclear fast red counterstain used for nuclei and cytoplasm.
Comparison of the pre-ligand injection MRI scans between transgenic and control wild type mice showed no significant differences (data not shown). This was despite the identification of a few amyloid lesions in transgenic mice without the use of the contrast agent. This small number of lesions, which can be detected by visual inspection of the scans, is not detected by voxel-based analysis. Also, on non-parametric analysis, no differences were found between wild-type mice injected with different tracers so those were combined in Figure 5.
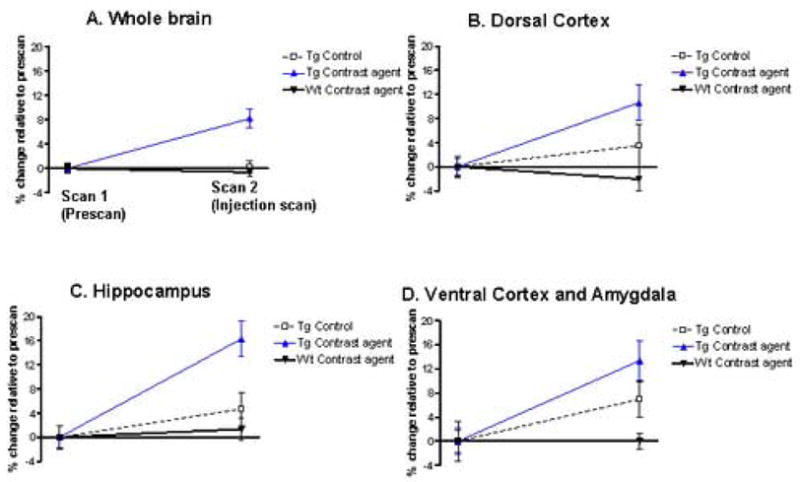
Figure 5. Quantitative voxel based analysis of the in vivo MRI scans of AD model mice.
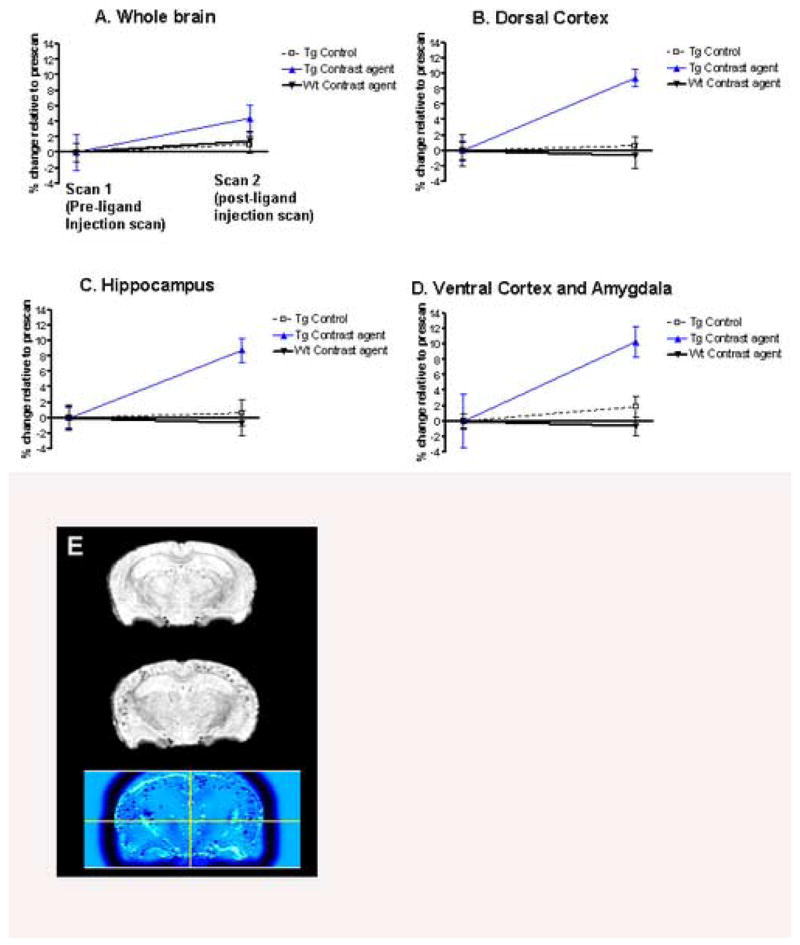
(A-D) The first scan (pre-ligand injection scan) was always in naïve animals (no injection), transgenic (Tg) (n=11) or wild-type (n=10). The second scan (Scan 2) was performed: 1) In Tg controls that were imaged two weeks earlier with no ligand injection (n=8); 2) in Tg controls injected with Gd free DTPA-K6Aβ1–30 (n=3); 3) In Tg mice injected with Gd-DTPA-K6Aβ1–30 (n=9), and; 4) in wild-type controls injected with Gd-DTPA-K6Aβ1–30 (n=5) or Gd-DTPA (Magnevist; n=5). Significant differences were observed in the AD transgenic group injected with Gd-DTPA-K6Aβ1–30 (Tg Contrast ligand) compared to Tg controls. This effect was observed within the whole brain (A; p<0.01), dorsal cortex (B; p<0.001), hippocampus (C; p<0.001), and the ventral cortex and amygdala (D; p=0.002). When the animals were injected, Scan 2 was performed 4–6 hours later. On non-parametric analysis, no differences were found between wild-type mice injected with different tracers so those were combined. The dorsal cortex encompasses the dorsal- and dorsolateral cortex (retrosplenial- and parietal association cortex). The ventral cortex contains the ventral- and ventrolateral cortex (entorhinal-, perirhinal- and pyriform cortex). In the graphs, the data is presented as percentage change (± SEM) in signal intensity relative to the prescans within the same group.
(E) Shows the co-registered and spatially normalized pre-ligand (top) and Gd-DTPA-K6Aβ1–30 post-ligand injection (middle) scans of one Tg mouse. The resulting difference image is shown on the bottom and was generated with SPM (see methods) as the mathematical subtraction of the post-ligand from the pre-ligand MRI scans. Non-brain voxels were excluded from the analysis by applying a brain mask (see methods). The difference image is displayed using a blue-black color-coded scale to highlight the amyloid-plaques load in the Gd-DTPA-K6Aβ1–30 injection scan as compared to the pre-injection scan. The cross-bars in the bottom image are set at the origin of the normalized MRI scan.
As compared to the pre-ligand injection MRI, significant loss of signal intensity, reflecting amyloid plaques, was observed on the post-ligand injection scans only for the group of transgenic mice injected with Gd-DTPA-K6Aβ1–30 (Figure 4A, Figure 5). Clusters of voxels representing statistically significant amyloid plaque burden were observed with whole brain analysis (Figure 5A; t(8)=3.09, p<0.01). Greater significance was observed when the analysis focused on brain regions with high amyloid burden which included the dorsal- and dorsolateral cortex (Figure 5B; t(8)=7.67, p<0.001), hippocampus (Figure 5C; t(8)=7.69, p<0.001), the amygdala, and ventral- and ventrolateral cortex (Figure 5D; t(8)=4.41, p=0.002).
Figure 4. 3D rendering of cortical Aβ plaques.
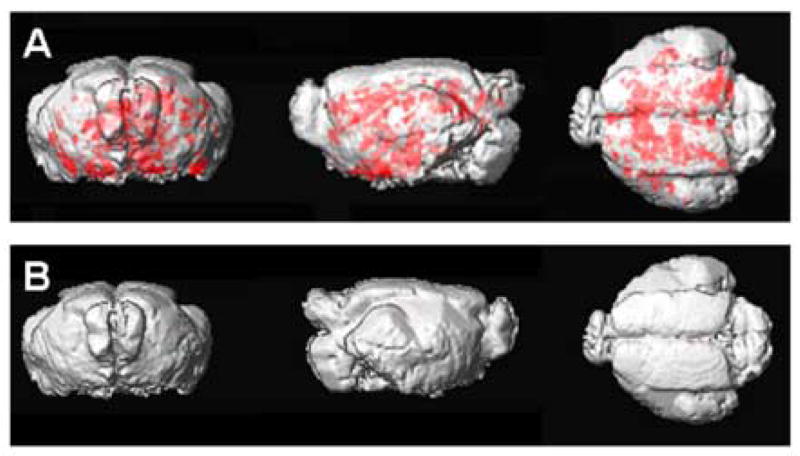
(A) A three-dimensional rendering of the transgenic mouse template showing the distribution of amyloid deposits as depicted in red, in the MRI images following Gd-DTPA-K6Aβ1–30 injection.
(B) The same three-dimensional rendering of the wild-type mice template shows no amyloid deposits in the MRI images following Gd-DTPA-K6Aβ1–30 injection in the group of wild-type mice.
The analysis of the images was repeated using the cerebellum as a confounding covariate (Figure 6). This analysis confirmed that significant loss of signal intensity on the post-ligand injection MRI as compared to the pre-ligand injection MRI was found exclusively for the group of transgenic mice injected with Gd-DTPA-K6Aβ1–30, reflecting amyloid plaques. In cross-section, there were no differences in the pre-ligand MRI scans across groups in any regions (p’s>0.1). Post-ligand injection MRI scans showed group effects for the whole brain (Figure 6A; F(2,25)=10.31, p=0.001), that were maximized in the dorsal- and dorsolateral cortex (Figure 6B; F(2,25)=4.92, p=0.016), hippocampus (Figure 6C; F(2,25)=8.35, p=0.002), and ventral- and ventrolateral cortex (Figure 6D; F(2,25)=5.98, p=0.008). In these regions, post-hoc analyses showed significantly greater amyloid burden for the group of transgenic mice injected with Gd-DTPA-K6Aβ1–30 as compared to both other groups (p’s<0.01), and no differences between wild-type mice and transgenic mice imaged without contrast agent. By contrasting the pre-ligand to the post-ligand injection MRI scans of the transgenic mice injected with DTPA-K6Aβ1–30, regional differences were 11% in the dorsal- and dorsolateral cortex (t(8)=3.01, p=0.01), 16% in the hippocampus (t(8)=10.8, p<0.001), 13% in the ventral- and ventrolateral cortex (t(8)=4.31, p=0.002).
Figure 6. Quantitative voxel based analysis on the in vivo MRI scans of AD model mice using the cerebellum as a confounding covariate.
The mice analyzed are the same as those described in Figure 5. In that figure, global intensity was used as a confounding covariate which can lead to underestimation of differences between pre- and post-ligand injection scans. Because the cerebellum is not prone to Aβ deposition, the images were reanalyzed with the cerebellum as the reference region. This was done by sampling values in anterior cerebellar vermis on the coronal plane of each scan by using a standardized two dimensional spherical region of interest with a radius of 300 μm.
By using the cerebellum as a covariate, the analyses confirmed that significant loss of signal intensity on the post-ligand injection MRI as compared to the pre-ligand injection MRI was found exclusively for the group of transgenic mice injected with Gd-DTPA-K6Aβ1–30, reflecting amyloid plaques. In cross-section, there were no differences in the pre-ligand MRI scans across groups in any regions (p’s>0.1). Post-ligand injection MRI scans showed group effects for the whole brain (A; F(2,25)=10.31, p=0.001), that were maximized in the dorsal- and dorsolateral cortex (B; F(2,25)=4.92, p=0.016), hippocampus (C; F(2,25)=8.35, p=0.002), and ventral- and ventrolateral cortex (D; F(2,25)=5.98, p=0.008). In these regions, post-hoc analyses showed significantly greater amyloid burden for the group of transgenic mice injected with Gd-DTPA-K6Aβ1–30 as compared to both other groups (p’s<0.01), and no differences between wild-type mice and transgenic mice imaged without contrast agent. By contrasting the pre-ligand to the post-ligand injection MRI scans of the transgenic mice injected with DTPA-K6Aβ1–30, regional differences were 11% in the dorsal- and dorsolateral cortex (t(8)=3.01, p=0.01), 16% in the hippocampus (t(8)=10.8, p<0.001), 13% in the ventral- and ventrolateral cortex (t(8)=4.31, p=0.002).
No group differences were found on Mann-Whitney non-parametric analysis contrasting wild-type mice injected with different ligands (p’s>0.1).
These findings from the regional analysis support the specificity of our approach and strengthen the feasibility of using gadolinium labeled Aβ homologous peptides for early in vivo plaque detection.
4. DISCUSSION
We and others have previously shown the successful use of gadolinium labeled Aβ peptides to target amyloid plaques in transgenic AD model mice [26;27;37;42;43]. This approach for labeling amyloid lesions takes advantage of the high affinity binding Aβ peptides have to other Aβ peptides [18;42]. However, Aβ peptides are toxic and deposited Aβ can seed further amyloid deposition [18;48]. It has previously been shown that circulating Aβ peptides can cross the BBB in vivo and contribute directly to amyloid lesion growth [23]. Therefore in the present study we sought to develop Aβ homologous peptides as amyloid targeting agents, which are non-toxic and non-fibrillogenic. We designed these homologous peptides so that they still have a high binding affinity to wild-type Aβ peptides and also have a similar BBB permeability. In prior studies we have shown that these Aβ homologous peptides do not self-assemble or promote the fibrillization of endogenous Aβ peptides [2;34;35]. In the current study we used K6-Aβ1–30 chelated to gadolinium via incorporation of DPTA at the amino terminus. In our previously published studies we used K6-Aβ1–30 as a vaccine therapy in AD model mice and have shown that this peptide is both non-toxic and non-fibrillogenic [35]. In the current study we have also demonstrated that this Aβ homologous peptide is non-toxic when labeled with chelated Gd (Figure 1). Importantly, we show that Gd-DTPA-K6Aβ1–30 maintains its high affinity binding to Aβ1–42, allowing its use as an amyloid targeting agent. As we have previously reported using Aβ1–40, the introduction of Gd-DTPA lowers BBB permeability significantly [42]. Hence, in order to use this ligand for amyloid labeling the BBB has to be transiently disrupted with mannitol. In this study we document that Gd-DPTA-K6Aβ1–30 is able to label amyloid lesions in vivo when the BBB is disrupted with mannitol.
Similar to previous ex vivo- [22;49] and in vivo reports [16;17;36;41], we were able to image some amyloid lesions in our AD transgenic mice without the use of a contrast agent. With our relatively short imaging times, only a small proportion of amyloid lesions could be detected without the use of a contrast agent. This direct detection of amyloid lesions most likely reflects iron content within plaques [6;16;17;41;46]. Iron deposition has been previously reported both in AD and in transgenic AD mouse model plaques, in particular in more mature lesions [38;39]. We detected plaques without contrast agent mainly in older animals and in large plaques, most likely related to the increased iron content within some of these more mature lesions. However, when we performed voxel based analysis comparing the pre-ligand MRI scans of controls versus the Tg AD model mice, there were no significant differences, reflecting the small percentage of lesions which can be detected without contrast agent. Detection of AD pathology is most important at the earliest stages of disease, since that is the point at which therapeutic interventions, which are currently under development, will have their greatest effect [45]. For the identification of these earlier lesions contrast agents will likely be needed. Other promising approaches under development include MRI detection of 19F-labelled amyloidophilic compounds as reported in transgenic mice [13]. Significantly, our MRI based methods are able to detect amyloid deposits in transgenic mouse models of AD even when the amyloid burden is relatively low. PET based methods, such as those using PIB are unable to detect plaques in mice even when their amyloid burden is high as in 12 month old PS1/APP mice [21]. Hence, for studies developing amyloid clearing agents where transgenic models are being used to evaluate efficacy, MRI approaches are preferred.
In this study we report the first use of a voxel based analysis (VBA) of AD model μMRI. Statistical Parametric Mapping [SPM’99, Wellcome Department of Cognitive Neurology, London, UK] [8;9] is a collection of tools available in the public domain for basic visualization and analysis of brain images. It is routinely applied for the analysis of structural and functional brain images in humans and it has been recently applied to an autoradiographic study in rats [25]. SPM is predominantly used for its convenience in statistical examination of group differences. VBA, as performed with SPM, relies on semi-automated image registration, spatial normalization and smoothing procedures to standardize all brains to a common space and allow assessment of the brain images on a voxel-wise basis. With registration and normalization, one assumes that all structures occupy the same volume and have the same shape. During VBA facilitates examination of large data sets through the rapid creation of statistical maps that enable to localize significant changes in the whole brain and on a voxel-wise basis. This represents a powerful, unbiased tool to assess the potential effectiveness of therapeutic interventions for amyloid removal, many of which are currently being developed using AD model mice. In the present study VBA comparison of pre and post-ligand injection allowed us to definitively identify ligand binding associated with amyloid. In our studies the pre-ligand and post-ligand injection scans were done 2 weeks apart; over such a short interval it is very unlikely significant changes in the volume of brain structural will have occurred. However, age related volume changes in the brain structures in AD model and wild-type mice do occur and could confound comparisons between scans done too far apart temporally [29]. With our imaging protocol, it was important to have pre-ligand injection scans so that dark spots that are associated with vessels (or other dark structures) rather than plaques can be differentiated. Binding of our ligand to amyloid lesions results in increased darkness on the T2* imaging sequences in areas of plaques. Without comparison to pre-ligand injection scans (or amyloid immunohistochemical staining on tissue sections) it is not possible to definitively identify dark spots as plaques. Hence, application of our ligand and imaging protocols for amyloid detection need to be combined with VBA for making a definitive distinction between mice with amyloid plaques and control non-transgenic mice. As shown in figure 5, when our ligand was injected into control mice, no significant difference was noted between pre and post ligand injection scans, contrasting with our findings when our ligand was injected into AD transgenic mice. Hence, our ligand is associated with little or no non-specific signal change on T2* imaging in mice without amyloid lesions.
In the present study we have not directly compared the sensitivity and specificity of VBA versus a region-of-interest (ROI) approach for quantitation of differences in the amyloid burden. It is possible that an ROI approach may have greater sensitivity; however, we focused on VBA as this method is accurate and much more rapid to apply [24].
A limitation of our imaging protocol is the need to open the BBB with the use of mannitol, along with carotid artery clamping during the injection of the ligand. The use of modifications of gadolinium labeled Aβ1–40, such as putrescine conjugation, to increase the BBB permeability have been reported [19;27;44]; however, when we performed putrescine modification of Gd-K6Aβ1–30 we could not obtain consistent labeling of amyloid deposits with this ligand (data not shown). Several other modifications of ligands to increase their BBB permeability have been reported such as incorporation of poly-cationic domains and coupling with proteins that are actively transported into the brain [7;30]. We are currently investigating such methods to increase the BBB permeability of our ligand. It is only with overcoming this problem of BBB permeability that our ligand would have the possibility of being applied to humans.
Our present findings support the use of a contrast imaging agent for early plaque detection, using non-toxic, non-fibrillogenic Aβ derivatives such as K6Aβ1–30. Such an amyloid contrast probe enhances the sensitivity of plaque detection that allows following the amyloid burden in individual AD mice longitudinally. This will greatly aid the development of therapeutic agents which aim to remove existing amyloid plaques.
Acknowledgments
Supported by NIH grants AG20245, AG20197 and AG08051, and the Alzheimer’s Association.
Footnotes
Disclosure Statement: The authors (EMS, YZW, DHT and TW) have filed a patent (No.: 6,821,504) for MRI based amyloid imaging. The authors have no other conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Ashburner J, Friston KJ. Voxel-based morphometry- the methods. Neuroimage. 2000;11:805–821. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- 2.Asuni A, Boutajangout A, Scholtzova H, Knudsen E, Li Y, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM. Aβ derivative vaccination in alum adjuvant prevents amyloid deposition and does not cause brain microhemorrhages in Alzheimer’s model mice. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benveniste H, Einstein G, Kim KR, Hulette C, Johnson GA. Detection of neuritic plaques in Alzheimer’s disease by magnetic resonance microscopy. Proc Natl Acad Sci (USA) 1999;96:14079–14084. doi: 10.1073/pnas.96.24.14079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18:S85–S88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- 5.Dhenain M, Privat N, Duyckaerts C, Jacobs RE. Senile plaques do not induce susceptibility effects in T2* weighted MR microscopic images. NMR Biomed. 2002;15:197–203. doi: 10.1002/nbm.760. [DOI] [PubMed] [Google Scholar]
- 6.Falangola MF, Lee SP, Nixon RA, Duff K, Helpern JA. Histological co-localization of iron in Abeta plaques of PS/APP transgenic mice. Neurochem Res. 2005;30:201–205. doi: 10.1007/s11064-004-2442-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franc BL, Mandl SJ, Siprashvili Z, Wender P, Contag CH. Breaching biological barriers: protein translocation domains as tools for molecular imaging and therapy. Mol Imaging. 2003;2:313–323. doi: 10.1162/15353500200303148. [DOI] [PubMed] [Google Scholar]
- 8.Friston KJ, Ashburner J, Frith CD, Poline JB, Heather J, Frackowiak RSJ. Spatial registration and normalization of images. Hum Brain Mapping. 1995;3:165–189. [Google Scholar]
- 9.Friston KJ, Frith CD, Liddle PF, Frackowiak RSJ. Comparing functional (PET) images: the assessment of significant change. J Cereb B Flow Met. 1991;11:690–699. doi: 10.1038/jcbfm.1991.122. [DOI] [PubMed] [Google Scholar]
- 10.Golabek AA, Soto C, Vogel T, Wisniewski T. The interaction between apolipoprotein E and Alzheimer’s amyloid β-peptide is dependent on β-peptide conformation. J Biol Chem. 1996;271:10602–10606. doi: 10.1074/jbc.271.18.10602. [DOI] [PubMed] [Google Scholar]
- 11.Good CD, Scahill RI, Fox NC, Ashburner J, Friston KJ, Chan D, Crum WR, Rossor MN, Frackowiak RS. Automatic differentiation of anatomical patterns in the human brain: validation with studies of degenerative dementias. Neuroimage. 2002;17:29–46. doi: 10.1006/nimg.2002.1202. [DOI] [PubMed] [Google Scholar]
- 12.Halverson KJ, Fraser PE, Kirschner DA, Lansbury PT. Molecular determinants of amyloid deposition in Alzheimer’s disease: conformational studies of synthetic β-protein fragments. Biochem. 1990;29:2639–2644. doi: 10.1021/bi00463a003. [DOI] [PubMed] [Google Scholar]
- 13.Higuchi M, Iwata N, Matsuba Y, Sato K, Sasamoto K, Saido TC. 19F and 1H MRI detection of amyloid beta plaques in vivo. Nat Neurosci. 2005;8:527–533. doi: 10.1038/nn1422. [DOI] [PubMed] [Google Scholar]
- 14.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Saad WK, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nature Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 15.Hsiao KK, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 16.Jack CR, Garwood M, Wengenack TM, Borowski B, Curran GL, Lin J, Adriany G, Grohn OHJ, Grimm R, Poduslo JF. In vivo visualization of Alzheimer’s amyloid plaques by magnetic resonance imaging in transgenic mice without a contrast agent. Magn Reson Med. 2004;52:1263–1271. doi: 10.1002/mrm.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CR, Jr, Wengenack TM, Reyes DA, Garwood M, Curran GL, Borowski BJ, Lin J, Preboske GM, Holasek SS, Adriany G, Poduslo JF. In vivo magnetic resonance microimaging of individual amyloid plaques in Alzheimer’s transgenic mice. J Neurosci. 2005;25:10041–10048. doi: 10.1523/JNEUROSCI.2588-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 19.Kandimalla KK, Curran GL, Holasek SS, Gilles EJ, Wengenack TM, Ramirez-Alvarado M, Poduslo JF. Physiological and Biophysical Factors that Influence Alzheimer’s Disease Amyloid Plaque Targeting of Native and Putrescine Modified Human Amyloid β 40. J Pharmacol Exp Ther. 2006;318:17–25. doi: 10.1124/jpet.105.095711. [DOI] [PubMed] [Google Scholar]
- 20.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 21.Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SP, Falangola MF, Nixon RA, Duff K, Helpern JA. Visualization of β-amyloid plaques in a transgenic mouse model of Alzheimer’s disease using MR microscopy without contrast reagents. Magn Reson Med. 2004;52:538–544. doi: 10.1002/mrm.20196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic B. Transport across the blood-brain barrier and differential cerebrovascular sequestration of circulating Alzheimer’s amyloid-β peptide in aged Rhesus versus aged Squirrel monkeys. Vascul Pharmacol. 2002;38:303–313. doi: 10.1016/s1537-1891(02)00198-2. [DOI] [PubMed] [Google Scholar]
- 24.Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, Li Y, Boppana M, De Leon MJ. Reduced hippocampal metabolism in MCI and AD - Automated FDG-PET image analysis. Neurol. 2005;64:1860–1867. doi: 10.1212/01.WNL.0000163856.13524.08. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen PT, Holschneider DP, Maarek JMI, Yang J, Mandelkern M. Statistical parametric mapping applied to an autoradiographic study of cerebral activation during treadmill walking in rats. Neuroimage. 2004;23:252–259. doi: 10.1016/j.neuroimage.2004.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poduslo JF, Curran GL, Peterson JA, McCormick DJ, Fauq AH, Khan MA, Wengenack TM. Design and chemical synthesis of a magnetic resonance contrast agent with enhanced in vitro binding, high blood-brain barrier permeability adn in vivo targeting ot Alzheimer’s disease amyloid plaques. Biochem. 2004;43:6064–6075. doi: 10.1021/bi0359574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poduslo JF, Wengenack TM, Curran GL, Wisniewski T, Sigurdsson EM, Macura SI, Borowski BJ, Jack CR. Molecular contrast enhanced magnetic resonance imaging of Alzheimer’s amyloid plaques. Neurobiol Dis. 2002;11:315–329. doi: 10.1006/nbdi.2002.0550. [DOI] [PubMed] [Google Scholar]
- 28.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 29.Redwine JM, Kosofsky B, Jacobs RE, Games D, Reilly JF, Morrison JH, Young WG, Bloom FE. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci U S A. 2003;100:1381–1386. doi: 10.1073/pnas.242746599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roney C, Kulkarni P, Arora V, Antich P, Bonte F, Wu A, Mallikarjuana NN, Manohar S, Liang HF, Kulkarni AR, Sung HW, Sairam M, Aminabhavi TM. Targeted nanoparticles for drug delivery through the blood-brain barrier for Alzheimer’s disease. J Control Release. 2005;108:193–214. doi: 10.1016/j.jconrel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, Mathews P, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T. Blocking the apolipoprotein E/β-amyloid interaction reduces β-amyloid toxicity and decreases β-amyloid load in transgenic mice. Am J Pathol. 2004;165:937–948. doi: 10.1016/s0002-9440(10)63355-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sigurdsson EM. Histological staining of amyloid-beta in mouse brains. Methods Mol Med. 2005;299:299–308. doi: 10.1385/1-59259-874-9:299. [DOI] [PubMed] [Google Scholar]
- 33.Sigurdsson EM. Immunotherapy for conformational disorders. Curr Pharm Des. 2006;12:2569–2585. doi: 10.2174/138161206777698837. [DOI] [PubMed] [Google Scholar]
- 34.Sigurdsson EM, Knudsen EL, Asuni A, Sage D, Goni F, Quartermain D, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-β derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sigurdsson EM, Scholtzova H, Mehta P, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer’s disease associated pathology in transgenic mice. Am J Pathol. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sigurdsson EM, Wadghiri YZ, Blind JA, Knudsen EL, Asuni A, Sadowski M, Turnbull DH, Wisniewski T. In vivo magnetic resonance imaging of amyloid plaques in mice with a non-toxic Aβ derivative. Neurobiol Aging. 2004;25(S2):57. [Google Scholar]
- 37.Sigurdsson EM, Wadghiri YZ, Sadowski M, Elliot JI, Li Y, Scholtzova H, Tang CY, Aguilanldo JG, Pappolla M, Duff K, Wisniewski T. In vivo magnetic resonance of amyloid plaques in Alzheimer’s disease model mice. In: Hyman B, editor. The Living Brain and Alzheimer’s disease. Springer-Verlag; Berlin: 2004. [Google Scholar]
- 38.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generating free radicals. Proc Natl Acad Sci (USA) 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith MA, Hirai K, Hsiao K, Pappolla M, Harris PL, Siedlak SL, Tabaton M, Perry G. Amyloid beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 40.Thal DR, Rub U, Schultz C, Sassin I, Ghebremedhin E, Del TK, Braak E, Braak H. Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59:733–748. doi: 10.1093/jnen/59.8.733. [DOI] [PubMed] [Google Scholar]
- 41.Vanhoutte G, Dewachter I, Borghgraef P, Van LF, Van der LA. Noninvasive in vivo MRI detection of neuritic plaques associated with iron in APP[V717I] transgenic mice, a model for Alzheimer’s disease. Magn Reson Med. 2005;53:607–613. doi: 10.1002/mrm.20385. [DOI] [PubMed] [Google Scholar]
- 42.Wadghiri YZ, Sigurdsson EM, Sadowski M, Elliot JI, Li Q, Scholtzova H, Tang CY, Aguinaldo JG, Pappolla M, Duff K, Turnbull D, Wisniewski T. Detection of Alzheimer’s amyloid in transgenic mice using magnetic resonance micro-imaging. Magn Reson Med. 2003;50:293–302. doi: 10.1002/mrm.10529. [DOI] [PubMed] [Google Scholar]
- 43.Wadghiri YZ, Sigurdsson EM, Wisniewski T, Turnbull D. MR Imaging of amyloid plaques in transgenic mice. In: Sigurdsson EM, editor. Amyloid Proteins: Methods and Protocols. Humana Press Inc; Totowa, NJ: 2005. pp. 365–379. [DOI] [PubMed] [Google Scholar]
- 44.Wengenack TM, Curran GL, Poduslo JF. Targeting Alzheimer amyloid plaques in vivo. Nat Biotech. 2000;18:868–872. doi: 10.1038/78482. [DOI] [PubMed] [Google Scholar]
- 45.Wisniewski T, Frangione B. Immunological and anti-chaperone therapeutic approaches for Alzheimer’s disease. Brain Pathol. 2005;15:72–77. doi: 10.1111/j.1750-3639.2005.tb00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wisniewski T, Mosconi L, Wadghiri YZ, Blind JA, Tsui W, Knudsen E, Asuni A, Sadowski M, Turnbull DH, de Leon M, Sigurdsson E. In vivo MRI detection of amyloid plaques in AD transgenic using gadolinium labeled non-toxic Abeta homologous ligands followed by voxel-based analysis. Society for Neuroscience Abstracts. 2005 [Google Scholar]
- 47.Worsley KJ, Marrett S, Neelin P, Vandal AC, Friston KJ, Evans AC. A unified statistical approach for determining significant signals in images of cerebral activation. Hum Brain Mapping. 1996;4:58–73. doi: 10.1002/(SICI)1097-0193(1996)4:1<58::AID-HBM4>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 48.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J, Yarowsky P, Gordon MN, Di Carolo G, Munireddy S, van Zijl PCM, Mori S. Detection of amyloid plaques in mouse models of Alzheimer’s disease by magnetic resonance imaging. Magn Reson Med. 2004;51:452–457. doi: 10.1002/mrm.10730. [DOI] [PubMed] [Google Scholar]