

Abstract
Bacterial lipoproteins (LP) are a family of cell wall components found in a wide variety of bacteria. In this study, we characterized the response of HUCL, a telomerase-immortalized human corneal epithelial cell (HCEC) line, to LP isolated from Staphylococcus (S) aureus. S. aureus LP (saLP) prepared by Triton X-114 extraction stimulated the activation of NF-κB, JNK, and P38 signaling pathways in HUCL cells. The extracts failed to stimulate NF-κB activation in HUCL cells after lipoprotein lipase treatment and in cell lines expressing TLR4 or TLR9, but TLR2, indicating lipoprotein nature of the extracts. saLP induced the up-regulation of a variety of inflammatory cytokines and chemokines (IL-6, IL-8, ICAM-1) and antimicrobial molecules (hBD-2, LL-37, and iNOS), and homeostasis genes (Mn-SOD) at both the mRNA level and protein level. Similar inflammatory response to saLP was also observed in primarily cultured HCECs using the production of IL-6 as readout. Moreover, TLR2 neutralizing antibody blocked the saLP-induced secretion of IL-6, IL-8 and hBD2 in HUCL cells. Our findings suggest that saLP activates TLR2 and contributes to innate immune response in the cornea to S. aureus infection via production of proinflammatory cytokines and defense molecules.
1. Introduction
S. aureus is a leading cause of infectious keratitis associated with extended wear of contact lenses, accounting for approximately one-quarter of confirmed cases, with a gradual increase recently in the number of S. aureus keratitis in both the US and China [1–4]. Under normal condition, the cornea is highly resistant to infection despite its constant exposure to a wide array of microorganisms [5]. To cause infection, the opportunistic bacteria such as S. aureus are likely to confront first the epithelium with compromised barrier [6] such as that caused by contact lens wearing [7]. The epithelial cells that form the first line of defense against microbial pathogens, in turn must be able to recognize and respond appropriately to the invading pathogen [8]. It is now well-established fact that recognition of invading microorganisms by epithelial as well as other mammalian cells includes the action of the Toll-like receptor (TLR) family [8, 9].
TLRs play a central role in early innate immunity by sensing the presence of microbial pathogens and initiating a response to eliminate them [10, 11]. These receptors recognize the highly conserved structural motifs called pathogen-associated molecular patterns (PAMPs) [9]. At least 11 TLRs and their distinct ligands have been identified so far in mammals [12]. Among these receptors, TLR2 is activated by several bacterial products, including lipoproteins, peptidoglycan (PGN), and lipoteichoic acids (LTA), presumably in combination with TLR1 or TLR6 [9]. Unlike intestinal epithelial cells which express low levels of TLR2 [13], HCECs were found to express abundant TLR2 both at mRNA and protein levels. However, an early in vitro study found that HCECs did not respond to S. aureus peptidoglycan and suggested that the unresponsiveness of HCECs to this TLR2 ligand is a contributing factor to an immunosilent environment at the ocular surface [14]. Recently, Sun et al showed that the exposure of C57BL/6 mouse corneal epithelium to S. aureus was found to induce neutrophil recruitment to the corneal stroma and increased corneal thickness and haze in a TLR2 dependent manner [15]. Since interactions between bacteria and epithelium are one of the early events in the establishment of the infection [16], it is important to identify the bacterial virulence factors and host cell components that contribute to infection and inflammation.
We previously reported that HCECs recognize synthetic lipopeptide Pam3Cys and S. aureus exoproducts [17, 18], and, as such, suggested that TLR2 is a sentinel for the detection of Gram-positive bacteria such as S. aureus through recognition of lipoproteins. In the present study, we isolated the natural lipoproteins from S. aureus and investigated their stimulatory effects on signaling pathways and also the expression and production of proinflammatory cytokines and antimicrobial peptides (AMPs) and proteins, which are two arms of innate immune defense. Based on this study, we propose that lipoprotein plays a key role, through TLR2, in induction of S. aureus infection-associated inflammatory responses in the cornea.
2. Results
2.1. Isolation of saLP and stimulation of HCECs
We took advantage of temperature-dependent solubility of TX-114 in an aqueous solution and extracted lipids and lipoproteins from S. aureus homogenates, the extracts were then precipitated with methanol to obtain lipoproteins. We first tested lipoproteins isolated from S. aureus homogenates to determine whether they induce the production of proinflammatory cytokines in primary HCECs. Figure 1 shows that TX-114 extracts (0.5 μg of protein/ml) isolated from S. aureus, termed saLP, stimulated the production of IL-6; a 9 fold increase was observed in primary HCECs treated with the extracts when compared to the control.
Figure 1. Triton X-114 extract-induced IL-6 secretion in primary HCECs.
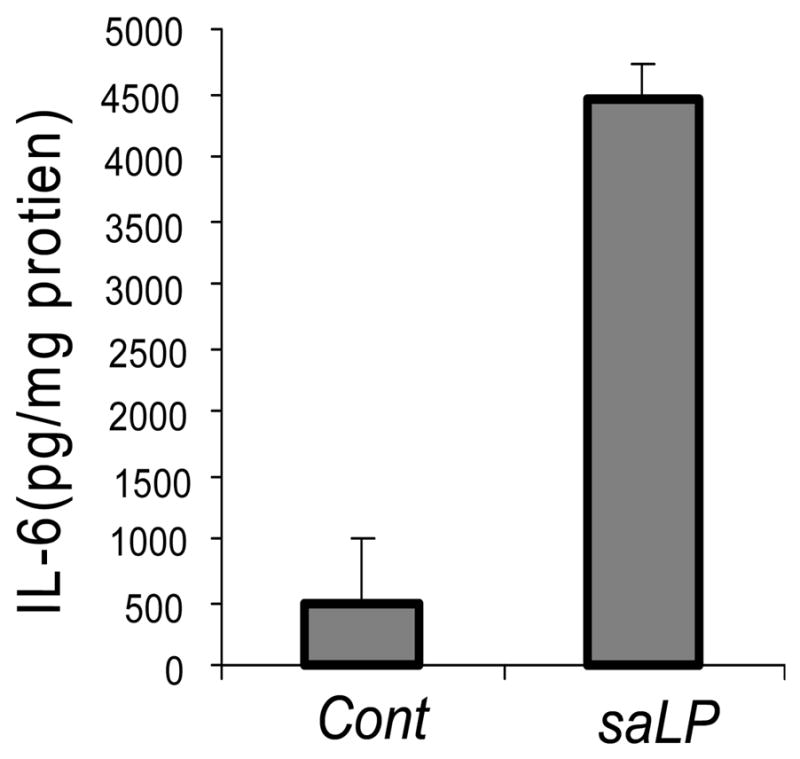
Primary HCECs were stimulated with (saLP) or without (Cont) Triton X-114 extracts of S. aureus (0.5 μg/ml protein) for 10 h. The supernatants were collected and IL-6 secretion was measured by ELISA. The amount of IL-6 in the culture media was normalized with the protein concentration of cell lysates. Results shown are representative of two independent experiments, *P < 0.001, N=3.
TLR-induced inflammatory cytokine production is known to involve NF-κB activation. We next examined dose-dependent activation of NF-κB in HUCL cells stimulated by saLP. As shown in Figure 2A, while 0.1 μg/ml saLP was sufficient to trigger NF-κB activation in 1 h, indicated by the increase in IκB-α phosphorylation, 0.5 μg/ml saLP treatment resulted in maximum level of IκB-α phosphorylation and its apparent degradation, we used 0.5 μg/ml saLP to challenge HUCL cells and assessed the time course of NF-κB activation in HUCL (Fig. 2B). IκB-α phosphorylation and degradation was detected within 30 minutes, reached the highest level at 2 h, and then declined to a level similar to the control 4 h post stimulation (Fig. 2B). In addition to the NF-κB, saLP also elicited the activation of other TLR mediated proinflammatory signaling pathways JNK and p38 in a time-dependent manner (Fig. 2B).
Figure 2. saLP-induced activation of NF-κB as well P38 and JNK pathways.

(A) HUCL cells were stimulated with different concentrations of saLP for 1h and total protein was extracted and subjected to SDS-PAGE, followed by phospho-IκB-α (pIκB-α) and IκB-α immunoblotting. (B) HUCL cells were stimulated with 0.5 μg/ml saLP for indicated time (hours) and cells were lysed for Western blot analysis with antibodies against phospho-IκB-α (pIκB-α), IκB-α, phospho-p38 (pP38), phospho-JNK (pJNK). Total ERK was used as protein loading control. Results shown are representative of three independent experiments.
2.2 The stimulatory activity of saLP is lipoprotein lipase sensitive
To determine whether the stimulatory activity in TX-114 extracts was due to lipoproteins, HUCL cells were challenged with saLP digested with 8500 units/ml lipoprotein lipase at 37°C for 6 h. The prolonged incubation of saLP at 37°C for 6 h resulted in slightly reduced NF-κB activation, the presence of lipoprotein lipase however resulted in the loss of stimulatory activity of saLP (Fig. 3A), suggesting the structural integrity of lipoproteins is required for saLP to elicit HCEC activation.
Figure 3. Lipoprotein lipase sensitive and TLR-dependent activation of NF-κB induced by saLP.
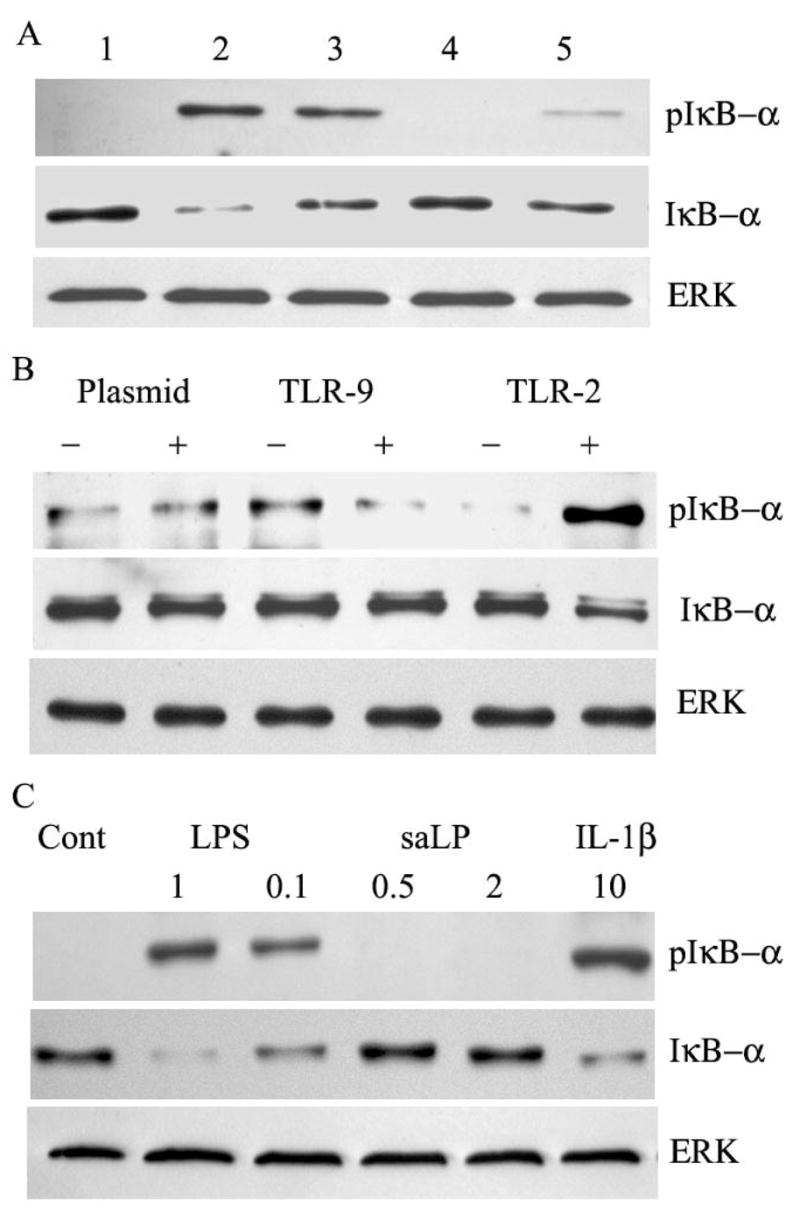
(A) HUCL cells were challenged with saLP (lane 2), saLP digested with (lane 5) or without (lane lane 3) lipoprotein lipase (8500unit/ml, 37°C, 6 h); or lipoprotein lipase (lane 4), along with HUCL cells without treatment as the control (Lane1). After 1 h incubation, total protein was extracted for Western blotting to detect phospho-IκB-α and IκB-α. (B) Stable HEK cell lines transfected with empty (plasmid), TLR2, or TLR9 plasmids were stimulated with (+) or without (−) 0.5 μg/ml saLP. After 1 h incubation, cells were lysed and subjected to phospho-IκB-α and IκB-α. (C) Stable Hela cell line expressing TLR4, CD14 and MD2 were stimulated with LPS (1 and 0.1 μg/ml), or saLP (0.5 or 2 μg/ml), with the controls including unstimulated (Cont) or 10 ng/ml IL-1β. After 1 h incubation, cells were lyased and subjected to Western blotting to detect phospho-IκB-α and IκB-α. Total ERK was used as protein loading control. The results are representative for two independent experiments.
To rule out the possibility that the stimulatory effects of the isolated saLP is due to the contamination of LPS and other TLR lignads, we used cell lines that were constructed to assess the nature of TLR ligands. For LPS, a HeLa cell line expressing functional TLR4 was used and challenged with saLP along with LPS or IL-1β as positive controls (Fig. 3C). While both LPS and IL-1β induced IκB-α phosphorylation and degradation, saLP, up to 2 μg/ml, failed to elicit any responses in Hela cells expressing functional TLR4. Similarly, 0.5 μg/ml saLP was capable to stimulate IκB phosphorylation and degradation in HEK cells expressing functional TLR2, but not TLR9 (Fig. 3B). Taken together, these data indicated that the TX-114 extracts are lipoproteins that induce NF-kB and MAPK activation in HCECs through TLR2.
2.3 saLP stimulates the expression of proinflammatory cytokines and innate defense molecules in HCECs
Activation of TLRs is known to induce the expression of an array of proinflammatory cytokines/chemokines and a variety of other genes with antimicrobial and protective functions in myeloid and epithelial cells. Using semi-quantitative RT-PCR, we assessed the expression of some of the genes in HCECs in response to saLP challenge (Fig. 4). Exposure of HUCL cells to 0.5 μg/ml saLP resulted in up-regulation of IL-6, IL-8 and TNF-α in a time-dependent manner, IL-6 expression reached the maximum level at 1 h, remained elevated at 2 h post stimulation, IL-8 and TNF-α expression reached the maximum level at 2 h post stimulation and declined afterwards. saLP also induced the expression of antimicrobial genes hBD2, LL-37, iNOS, as well as a homeostasis gene Mn-SOD. The levels of GAPDH remained largely unchanged in control and challenged cells during the course of the study.
Figure 4. saLP-elicited expression of proinflammatory cytokine/chemokine, antimicrobial molecules, and Mn-SOD in HUCL cells.
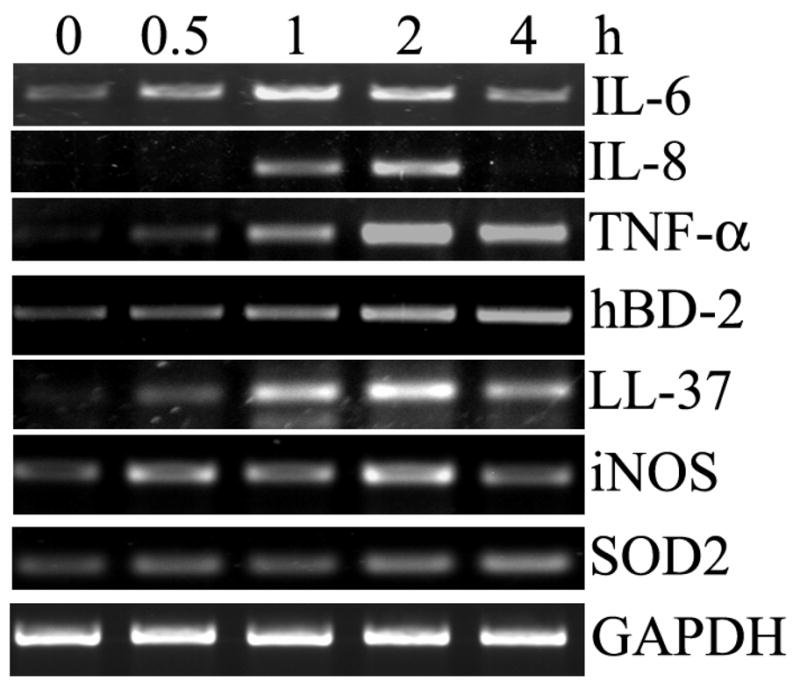
HUCL were incubated with 0.5 μg/ml saLP for the indicated times (hours) and the total cellular RNA was extracted at the end of incubation and subjected to RT-PCR analysis with primers amplifying human IL-6, IL-8, TNF-α, hBD-2, LL-37 iNOS, Mn-SOD with GAPDH as the internal control. The results are representative of three independent experiments.
Having demonstrated that saLP stimulated transcriptional expression of proinflammatory cytokines in HCECs, we next measured the production of IL-6 and IL-8 in HUCL cells in response to saLP challenge. As shown in figure 5, the increased amounts of IL-6 and IL-8 secreted into the culture media were induced by increased concentration of saLP used; saLP as low as 0.05 μg/ml was able to induce the production of both IL-6 and IL-8. Significant increase for both IL-6 and IL-8 was observed when 0.2 and 0.5 μg/ml saLP were used; the treatment with 0.5 μg/ml saLP resulted in 8.6-fold and 7.7-fold increases in IL-6 and IL-8, respectively when compared to the control, unstimulated cells.
Figure 5. saLP concentration dependent induction of IL-6 and IL-8 production in HUCL cells.
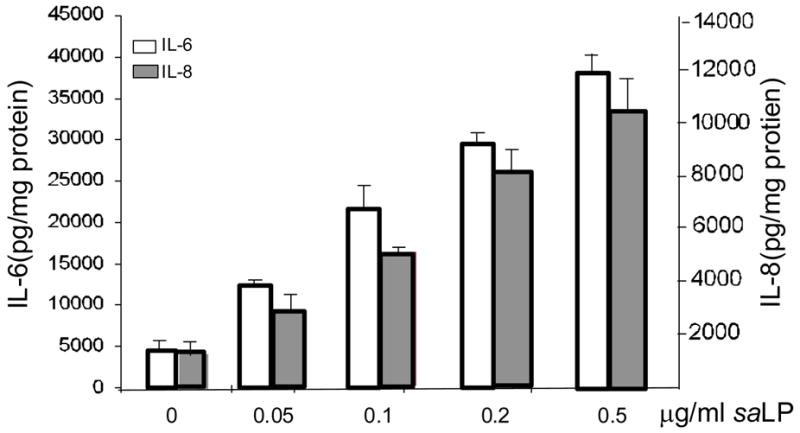
HUCL cells were challenged with increasing concentrations of saLP for 8 h along with HUCL cells without stimulation as the control. Accumulated levels of IL-6 and IL-8 in cell culture supernatant were measured by ELISA. Statistical analysis was performed using one way ANOVA (N=3; P=0.0015 for IL-6 and 0.002 for IL-8). Data are representative of two independent experiments.
The expression of iNOS, ICAM-1, and Mn-SOD at the protein levels were also assessed after saLP stimulation in HUCL cells (Fig 6A). While low levels of ICAM-1 was detected in unstimulated HUCL cells, a much elevated expression of the protein at a steady state was apparent at 4 h and lasted up to 24 h post saLP stimulation. Concomitant with mRNA expression, the expression of iNOS at the protein level was significantly up-regulated in saLP challenged HUCL cells, the up-regulated iNOS expression was observed as early as 2 h and reach at the highest level at 12 h. The level of Mn-SOD which may counteract the effect of NO generated by iNOS to protect cells from oxidative damage was also up-regulated.
Figure 6. saLP-induced protein expression of iNOS, ICAM-1, Mn-SOD and accumulation of LL-37 in HUCL cells.
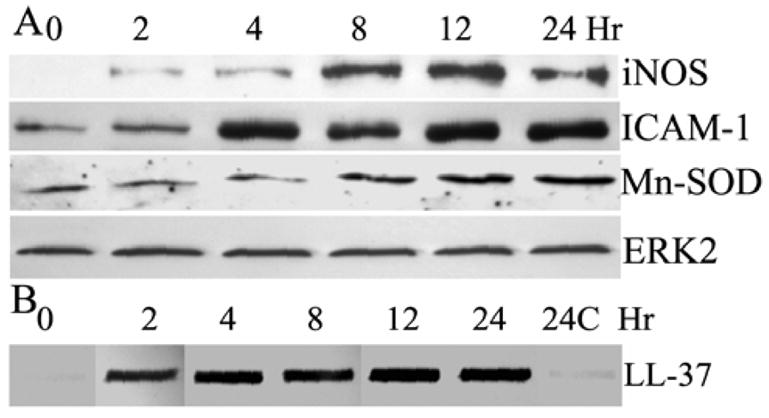
HUCL cells were stimulated with 0.5 μg/ml saLP for the indicated time (hours). At the end of incubation, the culture media were collected and cells were lysed. The cell lysates were subjected to Western blotting analysis with antibodies against ICAM-1, iNOS and Mn-SOD (A). The collected culture media were assayed for LL-37 production by slot-blot (B) with both media collected from untreated cells at beginning (0 h) and after 24 h of culture (24C) as the controls. The results are representative for two independent experiments.
Accumulation of antimicrobial peptide LL-37 in the culture media of saLP challenged cells was also assessed using slot blot (Fig. 6B). Using synthetic LL-37 as standard, we estimated the amount of LL-37 in the culture media of HUCL cells. Consistent with its expression at the mRNA levels (Fig. 4), the rapid accumulation of ~530 ng/ml LL-37 in the culture media of HUCL cells was observed 2 h post saLP stimulation, and significant amount, ~700 ng/ml LL-37 remained during the course of saLP stimulation (up to 24 h). Very low level of LL-37 was detected in the medium collected from the control, unstimulated cells cultured for 24 h.
2.4. The innate response of HCECs to saLP challenge is TLR2-dependent
To determine whether saLP-induced HCEC response was TLR2-dependent, HCECs were challenged with 0.5 μg/ml saLP in the presence of TLR2 neutralizing antibody or nonspecific isotype matched control antibody and the production of IL-6, IL-8 and hBD-2 in the culture media were assessed. While presence of control antibody resulted in slightly higher levels of IL-6 and IL-8, TLR2 neutralizing antibody significantly inhibited saLP induced production of IL-6 and IL-8 (Fig. 7), resulting in levels of the cytokines that were not significantly higher than that of the control. Similarly, saLP treatment resulted in much elevated production of hBD2 in the culture media, and TLR neutralizing antibody but not the control antibody, attenuated saLP-induced hBD2 accumulation in HUCL cells.
Figure 7. saLP induced IL-6, IL-8, and hBD-2 secretion in HUCL cells were TLR2 dependent.
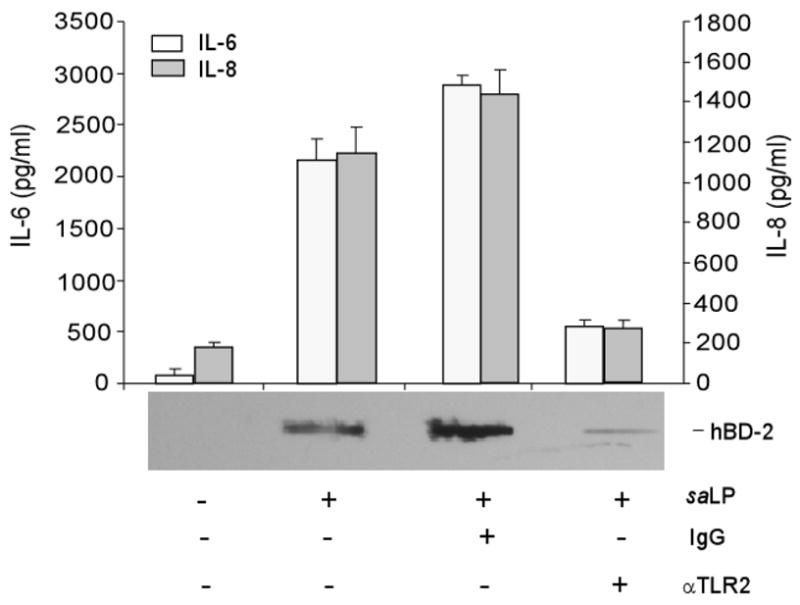
HUCL cells were pre-incubated with 20 μg/ml anti-TLR2 or mouse IgGα as the control antibody for 1 h. Antibody-pretreated cells were incubated with 0.5 μg/ml saLP for 8h. Culture supernatant was analyzed for IL-6 and IL-8 production by ELISA or for hBD2 accumulation by slot-blot. Statistical analysis of IL-6 and IL-8 secretion was performed using one way ANOVA (N=3, P=0.0048 for IL-6 and 0.0056 for IL-8). Data shown are representative of two independent experiments.
3. Discussion
In the present study, we reported the isolation of TX-114 soluble components from S. aureus and confirmed the identity of the stimulatory element in TX-114 extract as lipoproteins using lipopeptide lipase and cells expressing functional TLR2, TLR9, and TLR4. We demonstrated that HCECs in culture possess the ability to recognize lipoproteins from S. aureus, resulting in the activation of NF-κB and MAPK signaling pathways. Exposure of HCECs to saLP also resulted in the gene expression as well as protein production of proinflammatory cytokines and chemokines (IL-6, IL-8 and ICAM-1), antimicrobial peptides/genes (hBD2, LL-37 and iNOS), and gene with protective role (Mn-SOD) in HCECs. Moreover, we showed that TLR2 neutralizing antibody attenuated the production of IL-6 and IL-8 and the accumulation of antimicrobial peptide hBD2. Thus, lipoproteins are a major PAMP of S. aureus that are recognized by TLR2 in HCECs and are responsible, at least in part, for the stimulation of corneal inflammation and innate defense to S. aureus infection.
Upon contact with the ocular surface with compromised epithelial barrier, S. aureus activates the host cells such as the epithelia and initiates inflammatory response that is essential for containing infection in the cornea [8, 19]. S. aureus cell wall-associated and secreted proteins (e.g., lipoproteins, protein A and hemolysins)[20–22] and cell wall components (e.g., peptidoglycan and lipoteichoic acid) [23, 24] have been shown to cause inflammatory responses and, as such, may contribute to S. aureus keratitis. TLR2 has been shown to play a crucial role in the host response to S. aureus [15]. Several S. aureus components including lipoteichoic acid (LTA) [25], peptidoglycan [26], and lipoproteins[20, 27] were reported to be the ligands of TLR2. However, it is controversial whether LTA and peptidoglycan serve as PAMPs recognized by a cell surface TLR2 [27–29]. We showed previously that HCECs express abundant TLR2 but do not recognize LTA [17]. In addition to LTA, TLR2 is known to be a predominant receptor utilized by lipoproteins derived from various microorganisms, including both gram-negative bacteria, such as Borrelia burgdorferi, Escherichia coli, Neisseria gonorrhoea, and Porphyromonas gingivalis and Gram positive bacteria S. aureus [20]. Consistent with a recent reports showing that not LTA but lipoproteins are dominant, immunobiologically active TLR2 ligand in S. aureus [20, 27], we showed in the present study that the lipoproteins isolated from S. aureus stimulate epithelial cells, leading to the activation of TLR-mediated MAPK and NF-κB signaling pathways, and expression of an array of molecules associated with innate immune response. Thus, we conclude that S. aureus lipoproteins are a major activator of TLR2 in HCECs. Furthermore, we showed that the stimulatory activity of the isolated S. aureus lipoproteins is sensitive to mammalian lipoprotein lipase; thus, the enzymes might be used as adjunctive therapeutic modalities to selectively target TLR2-mediated inflammatory response in the cornea upon ocular infection.
Recent studies have shown that rather than being a passive barrier, the epithelium is an active participant in the host response to infection through not only the expression and production of proinflammatory cytokines and chemokines that recruit inflammatory cells to the site of infection, but also the expression of antimicrobial peptides and proteins (AMPs) such as β-defensins and LL-37 [30–32]. Consistent with previous study using synthetic TLR2 ligand Pam3Cys [18], we showed that saLP stimulated production of IL-6 and IL-8 in a concentration dependent manner. The use of TLR2 neutralizing antibody revealed that the production of these cytokines induced by saLP is TLR2-dependent. Similarly, the secretion of hBD-2 in saLP-treated cells is also sensitive to TLR2 inhibition, consistent with our previous study showing that synthetic ligand Pam3Cys-stimulated hBD2 productio0n in HCECs is TLR2-dependent [18]. hBD2 is a member of the β-defensin family and a major infection- and/or inflammation- inducible AMP genes in epithelial cells. Interestingly, hBD2 is known for its low bactericidal activity against Gram-positive bacteria (e.g. S. aureus) [33, 34], other AMPs and antimicrobial genes such as LL-37 are therefore likely also up-regulated in HCECs challenged with S. aureus or TLR2 ligands. Indeed, using RT-PCR and slot blot, we showed rapid induction of LL-37 expression and its abundant production in HUCL cells challenged with saLP. We estimate that 0.53 μg/ml (130 nM) LL-37 was accumulated within 2 h of saLP challenge and the highest levels reached 700 ng/ml (170 nM), representing 1.7 × 10−10 moles of LL-37 released from 1 ×106 cells cultured in 1 ml media, the concentration of LL-37 in vivo at the bacterium-HCEC contact site may be much higher than in the culture media of cells. Thus, the elevated levels of LL-37 at the proximity to epithelial cells near infection site may be sufficient to kill the invading pathogens, especially through its synergetic action with other antimicrobial gene products, including eye components lysozyme, up-regulated hBD2 and oxidative oxygen/nitrogen species found on the ocular surface. To that end, we also observed up-regulation of iNOS at the mRNA and more apparently the protein levels in saLP challenged HUCL cells. An early study of mouse model of P. aeruginosa infection revealed that iNOS in infected BALB/c cornea is greatly up-regulated at day 1 post infection and iNOS-derived NO is required for bacterial killing/stasis in this P. aeruginosa resistant strain. Our study suggests that corneal epithelial cells might be a source of NO in infected site. Although the general conjecture is that the hematopoietic cell population represents the major source of system or local NO, a recent study revealed that the dramatically elevated levels of NO during septic shock are not produced by hematopoietic cells such as monocytes/macrophages but rather by parenchymal cells in liver, kidney and gut [35]. Thus, we suggest that epithelial produced NO, LL37 and/or hBD2, are part of innate defense mechanisms in the cornea against bacterial infection.
While generation of the reactive oxygen/nitrogen species, which include hydrogen peroxide (H2O2) and NO, in epithelial cells may aid the innate killing of invaded pathogens, the ROS are also harmful to the host. The genes possess protective role might also be induced upon TLR activation in epithelial cells. Thus, the up-regulation of Mn-SOD in response to saLP challenge may play a role in defending cells against toxic superoxide O2− and other oxidative stress [36]. Furthermore, it was suggested that Mn-SOD was associated with mucosal defense against H. pylori infection [37]. TLR-mediated expression of Mn-SOD has been reported in macrophages challenged with different TLR ligands [38, 39] and our study is the first to indicate that it is up-regulated in epithelial cells upon TLR2 activation. Our observation that saLP induces the expression of Mn-SOD suggests that TLR activation leads to a protective mechanism to reduce cell damage that might be caused by host inflammatory response.
In summary, our data presented in this study support a role played by the corneal epithelium in the innate defense against pathogen challenge. Understanding the molecular events of lipoprotein-epithelial interactions at different stage of bacterial infection and the inflammatory consequences may permit the development of novel, specific therapies that can promote innate defense and prevent some of the destructive consequences of ocular infections.
4. Methods
4.1. Reagents and antibodies
Triton X-114 and n-octyl-β-glucopyranoside were purchased from Biochemika. Lipoprotein lipase was purchased from Sigma. Anti-phospho-IκB-α antibody, anti-IκB-α antibody, anti-phospho-p38 antibody, anti-p38 antibody, anti-phospho-JNK, anti-JNK antibody were purchased from Cell Signaling Technology (Beverly, MA); anti- hBD-2 antibodies were obtained from SantaCruz Biotechnology; anti-LL-37 antibody was from PANATecs; anti-iNOS and anti-ICAM-1 antibody were from BD transduction laboratory; mouse TLR-2-neutralizing mAb (2392) was a gift from Genentech (San Francisco, CA).
4.2. Isolation of bacterial lipoproteins
saLP was isolated according to the method described by Hashimoto et al [20]. S. aureus (strain 8325-4) was grown in a chemically defined medium which is known to enhance the production of cell-wall–associated products at 37°C with constant shaking (150 rpm) until the late-logarithmic phase (OD600 ≤ 1). Bacteria were separated from culture medium by centrifugation at 12,000g for 30 minutes. To prepare saLP from bacteria, the pellets were suspended in TS buffer (NaCl in Tris-HCL solution with protease inhibitor) and sonicated. The bacteria suspension (0.9 ml) was mixed with 0.1 ml of 20% (vol/vol) TX-114, the mixture was incubated at 4°C for 2 h and then centrifuged at 10,000 × g for 10 min at 4°C to remove insoluble materials. The supernatant was transferred to a new tube and incubated at 37°C for 10 min, followed by 10,000 × g, 5 min centrifugation at 37°C for phase separation. The upper, aqueous phase was discarded, An equal volume of TS buffer was added to the TX-114 phase, and the mixture was extracted two more times. After the third round of TX-114 extraction, saLP were precipitated from the TX-114 phase by mixing with 9 volumes of methanol, incubated at −20°C overnight, and then centrifuge at 4°C. The saLP precipitant was dissolved in PBS containing 10 mM n-octyl-β-glucopyranoside (OG/PBS). To confirm the stimulatory activity in TX-114 extract is due to saLP, aliquots of TX-114 extracts were also treated with or without 8500 unit/ml lipoprotein lipase in 37°C for 6h and used to challenge HUCL cells.
4.3. HCEC culture and lipoprotein challenge
Both a human telomerase-immortalized corneal epithelial (HUCL) cells and primary HCECs were used for the study. Primary HCECs were isolated from human donor corneas obtained from Michigan Eye Bank. Epithelial sheet was separated from underlying stroma after overnight dispase treatment. The dissected epithelial sheet was trypsinized and the epithelial cells were collected by centrifugation (500g, 5minutes). HCECs were cultured in KGM (supplemented with growth factors) in T25 flasks coated with fibronectin-collagen (FNC, Athena Environmental Service, Inc.) and used at passage 4. Before treatment, HUCL or primary cells were cultured in keratinocyte basic medium (KBM; biowhittaker, Walkersville, MD) overnight (growth factor-starvation). At the time of treatment, culture medium was replaced with fresh KBM containing various concentrations of isolated saLP, the cells were further cultured for indicated time and then lysed with Trizon solution (Invitrogen) for RT-PCR, or RIPA buffer (150 mM NaCl, 100 mM Tris-HCl [pH 7.5], 1% deoxycholate, 0.1% SDS, 1% Triton X-100, 50 mM NaF, 100 mM sodium pyrophosphate, 3.5 mM sodium orthovanadate, proteinase inhibitor cocktails, and 0.1 mM phenylmethylsulfonyl fluoride [PMSF]) for Western blotting analysis, and the supernatant were collected and, afer removal of cell debries used for ELISA or slot blotting. Protein concentration of cell lysates was determined by MicroBCA.
4.4. saLP-challenge of cells expressing TLR2, TLR9, and TLR4
Human embryonic kidney epithelial cells expressing functional TLR2 or TLR9 along with plasmid cell lines [40] were seeded in six-well dishes. After overnight starvation in DMEM, they were treated with 0.5 μg/ml saLP. Hela cells expressing TLR4 and MD2[41] were treated with 0.5 μg/ml saLP along with LPS or IL-1β as positive control for 1 h and then lysed for Western blotting using anti-IκB-α and anti-phospho- IκB-α antibodies.
4.5. Western Blot Analysis
Various amount of protein lysates were electrophoresed on 5–15% SDS-PAGE, transferred onto nitrocellulose membrane, blocked with 5% nonfat milk in Tris buffer (pH 7.6) with 0.05% Tween 20, and then incubated with a primary antibody (e.g., anti-iNOS, ICAM-1, anti-IκB-α, JNK, p38, ERK and their phosphorylated forms) overnight at 4°C. After washing, the membranes were incubated with the secondary antibody conjugated with a HRP and detected by a chemiluminescence system (Supersignal, Pierce).
4.6. RNA Extraction and RT-PCR Analysis
RNA was isolated from HUCL cells with Trizol (Invitrogen) and 2 μg of total RNA were reverse-transcribed with a first-strand synthesis system for RT-PCR (SuperScript; Invitrogen). cDNA was amplified by PCR with specific primers with amplifying conditions previously determined to be optimal for each gene (Table 1). The PCR products were subjected to electrophoresis on 1% agarose gels containing ethidium bromide. Stained gels were captured by a digital camera.
Table 1.
Sequences and Product Sizes of PCR Primers
| Gene | Primer | Sequence | Cycles/T° C* | Produt Size(bp) |
|---|---|---|---|---|
| IL-6 | Forward
Reverse |
CTCCTTCTCCACAAGCGCCTTC
GCGCAGAATGAGATGAGTTGTC |
24/55 | 583 |
| IL-8 | Forward
Reverse |
GCAGTTTTGCCAAGGAGTGCT
GCATCTGGCAACCCTACAACA |
30/55 | 347 |
| TNF-α | Forward
Reverse |
GAAAGCATGATCCGGGACGTG
GATGGCAGAGAGGAGGTTGAC |
26/60 | 510 |
| iNOS | Forward
Reverse |
ACCTTCAAGGCAGCCTGTGAGACGT
AGTAGAACCTGGGCTTCAGAATGGG |
35/55 | 700 |
| Mn-SOD | Forward
Reverse |
GAGTTGCTGGAAGCCATCAAACGT
GTATCTTTCAGTTACATTCTCCCA |
26/55 | 306 |
| hBD-2 | Forward
Reverse |
CCAGCCATCAGCCATGAGGGT
GGAGCCCTTTCTGAATCCGCA |
35/62 | 255 |
| LL-37 | Reverse
Forward |
GTCCCCATACACCGCTTCAC
ATCATTGCCCAGGTCCTCAG |
40/62 | 251 |
| GAPDH | Forward
Reverse |
CACCACCAACTGCTTAGCAC
CCCTGTTGCTGTAGCCAAAT |
30/55 | 515 |
4.7. Determination of IL-6 and IL-8 Secretion
HUCL cells were plated in a six-well dish, after growth factor starvation, cells were treated with saLP for 8h and culture media were harvested. For measurement of IL-6 and -8, ELISA detection kits were used according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). The amount of IL-6 and -8 in culture media was normalized with the total amount of cellular protein. Results were expressed as mean picograms of cytokine per milligram cell lysates or per ml of culture medium ± S.D, and analyzed using analysis of variance (ANOVA, one way) for multiple comparisons. A value of p < 0.05 was considered statistically significant.
4.8. Application of neutralizing TLR-2 antibody
To determine the involvement of TLR2, HUCL cells were plated 4 × 105 cells/well in 12-well plates. After overnight growth factor starvation, cells were pre-treated with 20 μg/ml anti-TLR2 antibody or mouse IgGa (isotype control) for 1 hour and then challenged with 0.5 μg/ml saLP for 8h. The culture supernatants were harvested and centrifuged for ELISA or slot blot analyses to determine cytokine and hBD2 expression.
4.9. Slot blot determination of hBD2 and LL-37
Accumulation of hBD-2 and LL-37 in the culture media was detected by slot blot [18]. Briefly, 100 μl supernatant was applied to a nitrocellulose membrane (0.2 μm; Bio-Rad) by vacuum using a slot-blot apparatus (Bio-Rad). The membrane was fixed by incubating with 10% formalin for 2 hours at room temperature, followed by blocking in Tris-buffered saline (TBS) containing 5% nonfat powdered milk for 1 h at room temperature. The membrane was then incubated overnight at 4°C with rabbit anti-human hBD-2 or LL-37 antibody diluted 1:1000 in TBS containing 5% nonfat powdered milk. After washing, the membrane was incubated for 1 h at room temperature with goat anti-rabbit IgG conjugated to horseradish peroxidase diluted 1:2000 with 5% nonfat powdered milk. Immunoreactivity was visualized with Supersignal reagents (Pierce).
Acknowledgments
This work was supported by the NIH grants RO1-EY14080 and EY10869 (FSY), Fight for Sight and Midwest Eye banks (A.K) and by an unrestricted grant from the Research to Prevent Blindness to the Department of Ophthalmology, Wayne State University School of Medicine. The authors are grateful to Dr. Douglas T. Golenbock(University of Massachusetts Medical School, Worcester, MA) for providing TLR2 and TLR9 expressing HEK cells and Dr. Fabio Re (University of Tennessee, Memphis, TN ) for providing Hela cells expressing functional TLR4 and MD2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sun X, Deng S, Li R, Wang Z, Luo S, Jin X, Zhang W. Distribution and shifting trends of bacterial keratitis in north China (1989–98) Br J Ophthalmol. 2004;88:165–166. doi: 10.1136/bjo.2002.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrakis G, Alfonso EC, Miller D. Shifting trends in bacterial keratitis in south Florida and emerging resistance to fluoroquinolones. Ophthalmology. 2000;107:1497–1502. doi: 10.1016/s0161-6420(00)00179-2. [DOI] [PubMed] [Google Scholar]
- 3.Jett BD, Gilmore MS. Host-parasite interactions in Staphylococcus aureus keratitis. DNA Cell Biol. 2002;21:397–404. doi: 10.1089/10445490260099683. [DOI] [PubMed] [Google Scholar]
- 4.Cohen E, Laibson P, Arentsen J, Clemons C. Corneal ulcers associated with cosmetic extended wear soft contact lenses. Ophthalmology. 1987;94:109–114. doi: 10.1016/s0161-6420(87)33491-8. [DOI] [PubMed] [Google Scholar]
- 5.Kurpakus-Wheater M, Kernacki KA, Hazlett LD. Maintaining corneal integrity how the “window” stays clear. Prog Histochem Cytochem. 2001;36:185–259. [PubMed] [Google Scholar]
- 6.Limberg MB. A review of bacterial keratitis and bacterial conjunctivitis. Am J Ophthalmol. 1991;112:2S–9S. [PubMed] [Google Scholar]
- 7.Fleiszig SM. The Glenn A. Fry award lecture 2005. The pathogenesis of contact lens-related keratitis. Optom Vis Sci. 2006;83:866–873. doi: 10.1097/01.opx.0000250045.85499.55. [DOI] [PubMed] [Google Scholar]
- 8.Yu FS, Hazlett LD. Toll-like Receptors and the Eye. Invest Ophthalmol Vis Sci. 2006;47:1255–1263. doi: 10.1167/iovs.05-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 10.Yeh WC, Chen NJ. Immunology: Another toll road. Nature. 2003;424:736–737. doi: 10.1038/424736a. [DOI] [PubMed] [Google Scholar]
- 11.Barton GM, Medzhitov R. Toll-Like Receptor Signaling Pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 12.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004;6:1382–1387. doi: 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Melmed G, Thomas LS, Lee N, Tesfay SY, Lukasek K, Michelsen KS, Zhou Y, Hu B, Arditi M, Abreu MT. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut. J Immunol. 2003;170:1406–1415. doi: 10.4049/jimmunol.170.3.1406. [DOI] [PubMed] [Google Scholar]
- 14.Ueta M, Nochi T, Jang MH, Park EJ, Igarashi O, Hino A, Kawasaki S, Shikina T, Hiroi T, Kinoshita S, Kiyono H. Intracellularly expressed TLR2s and TLR4s contribution to an immunosilent environment at the ocular mucosal epithelium. J Immunol. 2004;173:3337–3347. doi: 10.4049/jimmunol.173.5.3337. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Hise AG, Kalsow CM, Pearlman E. Staphylococcus aureus-induced corneal inflammation is dependent on Toll-like receptor 2 and myeloid differentiation factor 88. Infect Immun. 2006;74:5325–5332. doi: 10.1128/IAI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee EJ, Evans DJ, Fleiszig SM. Role of Pseudomonas aeruginosa ExsA in penetration through corneal epithelium in a novel in vivo model. Invest Ophthalmol Vis Sci. 2003;44:5220–5227. doi: 10.1167/iovs.03-0229. [DOI] [PubMed] [Google Scholar]
- 17.Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Ophthalmol Vis Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar A, Zhang J, Yu FS. Toll-like receptor 2-mediated expression of beta-defensin-2 in human corneal epithelial cells. Microbes Infect. 2006;8:380–389. doi: 10.1016/j.micinf.2005.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson A, Pearlman E. Toll-like Receptors in the Cornea. The ocular surface. 2005;3:S187–189. doi: 10.1016/s1542-0124(12)70252-5. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto M, Tawaratsumida K, Kariya H, Aoyama K, Tamura T, Suda Y. Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. Int Immunol. 2006;18:355–362. doi: 10.1093/intimm/dxh374. [DOI] [PubMed] [Google Scholar]
- 21.Callegan MC, Engel LS, Hill JM, O’Callaghan RJ. Corneal virulence of Staphylococcus aureus: roles of alpha-toxin and protein A in pathogenesis. Infect Immun. 1994;62:2478–2482. doi: 10.1128/iai.62.6.2478-2482.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar A, Tassopoulos AM, Li Q, Yu FS. Staphylococcus aureus protein A induced inflammatory response in human corneal epithelial cells. Biochem Biophys Res Commun. 2007;354:955–961. doi: 10.1016/j.bbrc.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol. 2003;170:1625–1629. doi: 10.4049/jimmunol.170.4.1625. [DOI] [PubMed] [Google Scholar]
- 24.You L, Kruse FE, Bacher S, Schmitz ML. Lipoteichoic acid selectively induces the ERK signaling pathway in the cornea. Invest Ophthalmol Vis Sci. 2002;43:2272–2277. [PubMed] [Google Scholar]
- 25.Menzies BE, Kenoyer A. Signal transduction and nuclear responses in Staphylococcus aureus-induced expression of human beta-defensin 3 in skin keratinocytes. Infect Immun. 2006;74:6847–6854. doi: 10.1128/IAI.00389-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- 27.Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, Krikae F, Kirikae T, Gotz F. Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol. 2006;177:3162–3169. doi: 10.4049/jimmunol.177.5.3162. [DOI] [PubMed] [Google Scholar]
- 28.Travassos LH, Girardin SE, Philpott DJ, Blanot D, Nahori MA, Werts C, Boneca IG. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5:1000–1006. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dziarski R, Gupta D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun. 2005;73:5212–5216. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chromek M, Slamova Z, Bergman P, Kovacs L, Podracka L, Ehren I, Hokfelt T, Gudmundsson GH, Gallo RL, Agerberth B, Brauner A. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12:636–641. doi: 10.1038/nm1407. [DOI] [PubMed] [Google Scholar]
- 31.Bowdish DM, Davidson DJ, Hancock RE. Immunomodulatory properties of defensins and cathelicidins. Curr Top Microbiol Immunol. 2006;306:27–66. doi: 10.1007/3-540-29916-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beisswenger C, Bals R. Antimicrobial peptides in lung inflammation. Chem Immunol Allergy. 2005;86:55–71. doi: 10.1159/000086651. [DOI] [PubMed] [Google Scholar]
- 33.Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. 1997;387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 34.Schroder JM, Harder J. Human beta-defensin-2. Int J Biochem Cell Biol. 1999;31:645–651. doi: 10.1016/s1357-2725(99)00013-8. [DOI] [PubMed] [Google Scholar]
- 35.Bultinck J, Sips P, Vakaet L, Brouckaert P, Cauwels A. Systemic NO production during (septic) shock depends on parenchymal and not on hematopoietic cells: in vivo iNOS expression pattern in (septic) shock. Faseb J. 2006;20:2363–2365. doi: 10.1096/fj.06-5798fje. [DOI] [PubMed] [Google Scholar]
- 36.Smoot DT, Elliott TB, Verspaget HW, Jones D, Allen CR, Vernon KG, Bremner T, Kidd LC, Kim KS, Groupman JD, Ashktorab H. Influence of Helicobacter pylori on reactive oxygen-induced gastric epithelial cell injury. Carcinogenesis. 2000;21:2091–2095. doi: 10.1093/carcin/21.11.2091. [DOI] [PubMed] [Google Scholar]
- 37.Gotz JM, van Kan CI, Verspaget HW, Biemond I, Lamers CB, Veenendaal RA. Gastric mucosal superoxide dismutases in Helicobacter pylori infection. Gut. 1996;38:502–506. doi: 10.1136/gut.38.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rakkola R, Matikainen S, Nyman TA. Proteome analysis of human macrophages reveals the upregulation of manganese-containing superoxide dismutase after toll-like receptor activation. Proteomics. 2007 doi: 10.1002/pmic.200600582. [DOI] [PubMed] [Google Scholar]
- 39.Tsan MF, Clark RN, Goyert SM, White JE. Induction of TNF-alpha and MnSOD by endotoxin: role of membrane CD14 and Toll-like receptor-4. Am J Physiol Cell Physiol. 2001;280:C1422–1430. doi: 10.1152/ajpcell.2001.280.6.C1422. [DOI] [PubMed] [Google Scholar]
- 40.Latz E, Franko J, Golenbock DT, Schreiber JR. Haemophilus influenzae type b-outer membrane protein complex glycoconjugate vaccine induces cytokine production by engaging human toll-like receptor 2 (TLR2) and requires the presence of TLR2 for optimal immunogenicity. J Immunol. 2004;172:2431–2438. doi: 10.4049/jimmunol.172.4.2431. [DOI] [PubMed] [Google Scholar]
- 41.Re F, Strominger JL. Monomeric recombinant MD-2 binds toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem. 2002;277:23427–23432. doi: 10.1074/jbc.M202554200. [DOI] [PubMed] [Google Scholar]