

Abstract
The Dystrophin protein is encoded by a gene that, when mutated in humans, can cause Duchenne muscular dystrophy, a disease characterized by progressive muscle wasting. A number of Duchenne patients also exhibit poorly understood mental retardation, likely associated with loss of a brain-specific isoform. Furthermore, although Dystrophin isoforms and the related Utrophin protein have long been known to localize at synapses, their functions remain essentially unknown. In Drosophila, we find that the CNS-specific Dp186 isoform localizes to the embryonic and larval neuropiles, regions rich in synaptic contacts. In the absence of Dp186, evoked but not spontaneous presynaptic release is significantly enhanced. Increased presynaptic release can be fully rescued to wild-type levels by expression of a Dp186 transgene in the postsynaptic motoneuron, indicating that Dp186 likely regulates a retrograde signaling pathway. Potentiation of synaptic currents in the mutant also occurs when cholinergic transmission is inhibited or in the absence of Glass Bottom Boat (Gbb) or Wishful Thinking (Wit), a TGF-β ligand and receptor, respectively, both previously implicated in synaptic retrograde signaling. These results are consistent with the possibility that Dp186 modulates other non-Gbb/Wit-dependent retrograde signaling pathways required to maintain normal synaptic physiology.
Keywords: dystrophin, Drosophila, retrograde signaling, CNS, gbb, interneuronal synapse
Introduction
Mutations in the human dystrophin gene cause Duchenne muscular dystrophy (DMD), a common human genetic disease characterized by progressive muscle wasting (Hoffman et al., 1987). Dystrophin is part of a large multiprotein complex, the Dystrophin glycoprotein complex (DGC) (for review, see Lapidos et al., 2004), which stabilizes the muscle during contraction by linking the actin cytoskeleton to the extracellular matrix. Dystrophin and associated proteins bind a variety of signaling molecules (for review, see Rando, 2001), indicating that the DGC likely also acts as a scaffold for components of intercellular and intracellular signaling pathways.
One-third of DMD patients also present cognitive defects (for review, see Anderson et al., 2002), raising the possibility that, in addition to their roles in the musculature, dystrophin and its partially functionally redundant ortholog utrophin also have critical functions in the nervous system. Consistent with this, several Dystrophin isoforms and Utrophin are localized to synaptic regions in the mammalian brain, in the retina, and at the neuromuscular junction (NMJ) (for review, see Blake et al., 2002). Further indications that dystrophin-like proteins play roles at a variety of synapses include the observations that (1) Utrophin-deficient mice display NMJ structural abnormalities (Deconinck et al., 1997a; Grady et al., 1997a), (2) NMJs lacking Dystrophin and Utrophin display disrupted acetylcholine receptor (AChR) clustering (Deconinck et al., 1997b; Grady et al., 1997b), (3) Dystrophin-deficient Caenorhabditis elegans animals are hyperactive because of the delocalization of an acetylcholine transporter and the subsequent failure to clear acetylcholine (Kim et al., 2004), (4) absence of the postsynaptically localized Drosophila DLP2 Dystrophin isoform results in increased presynaptic neurotransmitter release at the larval NMJ (van der Plas et al., 2006), and (5) mice lacking the large dystrophin isoform required for muscle integrity exhibit deficits in both long-term spatial and recognition memory (Vaillend et al., 2004). These results suggest that alterations in synaptic function, resulting from dystrophin deficiency, may underlie DMD-associated cognitive defects. The precise roles of Dystrophin at any synapse, however, remain to be elucidated.
We have used Drosophila melanogaster, with its amenability to genetic analysis and highly orthologous single dystrophin gene, to examine the roles of Dystrophin at a central synapse. Here, we describe a role of the CNS-specific Dp186 isoform at an identified motoneuron–interneuron synapse. We found that decreased postsynaptic expression of Dp186 results in increased evoked, but not spontaneous, presynaptic neurotransmitter release, similarly to that observed in a previous study in which the absence of the Dystrophin DLP2 isoform from the muscle resulted in increased neurotransmitter release by the motoneuron. However, retrograde modulation of presynaptic release by Dp186 at this central synapse did not require the TGF-β signaling pathway previously implicated in DLP2-mediated modulation at the NMJ. Thus, although the Dp186 and DLP2 Dystrophin isoforms are required to maintain wild-type neurotransmitter release levels at the interneuronal synapse and NMJ, respectively, they apparently do so via different signaling pathways.
Materials and Methods
Fly stocks.
Flies were fed on apple juice agar supplemented with yeast. Wild type was Canton-S. RN2-Gal4 was used to selectively express UAS-driven transgenes in aCC and RP2 (Baines, 2003, 2004; Fujioka et al., 2003). Expression of RN2-Gal4 begins in early-stage 16 embryos, preceding the onset of synaptogenesis (Baines and Bate, 1998). B19-Gal4 bears the Gal4 open reading frame downstream of the choline acetyltransferase promoter sequence and is selectively expressed in cholinergic neurons (Salvaterra and Kitamoto, 2001). The gbb1 allele, described previously (Wharton et al., 1999), was rebalanced over Cyo::GFP to allow unequivocal identification of mutant larvae. A transheterozygous combination of witA12 and witB11 was used as described previously (McCabe et al., 2003). Ok6-Gal4 (Aberle et al., 2002) drives expression in most, if not all, larval motoneurons. UAS-mCD8-GFP (Lee and Luo, 1999), when driven by Ok6-Gal4, localizes to the membranes of cell bodies, axons, and dendrites of the motoneurons (Sanchez-Soriano et al., 2005). Elav-GAl4 drives expression throughout the embryonic and larval CNS (Luo et al., 1994). The chats2 allele was used to reduce cholinergic transmission. Recordings from chats2 were performed using early second instar larvae raised at 18°C, a temperature at which cholinergic signaling is significantly reduced (Salvaterra and McCaman, 1985). Df{3R}Exel6184, which uncovers the entire dys locus and several additional proximal genes, was obtained from the Bloomington Stock Center (Bloomington, IN).
Generation of Dp186 mutant, UAS-RNAi-Dp186, and UAS-Dp186 transgenic fly lines.
A P-element excision screen (Tower et al., 1993) was used to generate Dp186 mutant fly lines. The GE20705 P-element line (obtained from GenExel, Daejon, South Korea) was used as a starting point for the excision screen; the transposon is inserted 250 bp 5′ of the Dp186 initiator ATG (see Fig. 1A). Imprecise excisions, removing genomic sequences flanking the original P-element, were generated by crossing in a source of transposase. Two lines were used for additional analyses, dysDp186 166.3 and dysDp186 30.3 (see Fig. 1A). Sequence analyses indicated that dysDp186 166.3 bears a 1.2 kb deletion removing the Dp186 ATG codon and most of the unique first exon of Dp186 and dysDp186 30.3 bears a 0.9 kb deletion.
Figure 1.

The structure of the Drosophila dystrophin gene and the location of the deletions in the dysDLP2 E6, dysDp186 166.3, and dysDp18630.3 mutants. A, There are six known isoforms of dystrophin in Drosophila: DLP1, DLP2, DLP3, Dp186, Dp205, and Dp117; each are expressed from its own promoter. The location of the isoform-specific deletions in the dysDLP2E6, dysDp186 166.3, and dysDp186 30.3 mutants and the sequences used for the RNAi-Dp186 construct and the P-element used to generate the dysDp186 166.3 are shown. Exons are indicated as bars, and introns are indicated as horizontal lines. B, The unique and conserved protein domains of the DLP2 and Dp186 isoforms and the region used for the generation of the Dp186-specific rabbit polyclonal antibody (yellow star) are indicated. cys-rich, Cysteine rich.
To reduce Dp186 expression levels in a tissue-specific manner, we generated transgenic fly lines that express double-stranded (ds) RNA targeting Dp186 unique sequences under Gal4 control. The UAS-RNAi-Dp186 construct contains the unique Dp186 sequences from base pairs 74–714 (GenBank accession number NM_169863) cloned into a pUAST (Brand and Perrimon, 1993) derivative bearing the mub intron between the Dp186-specific sequences (van der Plas et al., 2006). Multiple independent transgenic lines were generated using standard P-element transformation techniques, and two lines, 3 and 8, were used in these studies.
To express Dp186 in a tissue-specific manner, we generated fly lines expressing the full-length Dp186 cDNA sequence under Gal4 control in the pUAST P-element vector. Multiple transgenic lines were obtained, and two lines, 6.1 and 12.1, were used in these studies.
Immunohistochemistry.
Anti-Dp186 rabbit antisera were raised against a histidine-tagged fusion protein containing unique Dp186 sequences (amino acids 15–170 of GenBank accession number AAK15257) (van der Plas et al., 2006). Immunohistochemistry was performed as described previously (Dekkers et al., 2004). Anti-Dp186 (1:2500), anti-Synapsin (1:1000) (Klagges et al., 1996), anti-Bruchpilot nc82 antibody (1:50; Developmental Studies Hybridoma Bank, Iowa City, IA) (Hofbauer, 1991), anti-green fluorescent protein (GFP; 1:1000; Roche Diagnostics, Almere, The Netherlands), Alexa Fluor 488 Phalloidin (1:200; Invitrogen, Breda, The Netherlands), and Alexa Fluor-conjugated secondary antibodies (1:300; Invitrogen) were used as indicated. The samples were visualized and photographed using confocal and/or standard epifluorescence microscopy.
Dissection of larvae.
First (within 4 h of hatching) and second instar larvae were dissected, and central neurons were accessed as described previously (Baines and Bate, 1998). Larvae were viewed using a water-immersion lens (total magnification, 800×) combined with Nomarski optics (BX51 WI microscope; Olympus Optical, Tokyo, Japan).
Electrophysiology.
Whole-cell voltage-clamp recordings were performed using thick-walled borosilicate glass electrodes (GC100TF-10; Harvard, Edenbridge, UK), fire-polished to resistances of between 15 and 20 MΩ. Recordings were made using an Axopatch-1D amplifier controlled by pClamp 8.1 via a Digidata 1322A analog-to-digital converter (Molecular Devices, Sunnyvale, CA). Cells were identified based on their invariant size and dorsal position in the ventral nerve cord. After breakthrough, currents were measured for a period of time ranging from at least 2 min to no longer than 4 min from a minimum of eight cells for each genotype tested. Although currents can be recorded for >5 min, cells often begin to show increased leak currents after this duration of whole-cell recording. Traces were filtered at 2 kHz and sampled at 20 kHz. Spontaneous miniature currents were recorded in the presence of 0.1 μm tetrodotoxin (TTX; Alomone Labs, Jerusalem, Israel). Miniature currents were identified based on a fast rise and slower decay and on having amplitudes of at least twice background (2–3 pA). Amplitudes of both evoked and miniature currents were measured using Minianalysis 6.0.3 (Synaptosoft, Decatur, GA). Composite averaged current amplitude was calculated by grouping all individual currents recorded in each genotype tested. All recordings were performed at room temperature (22–24°C). External saline consisted of (in mm) 135 NaCl, 5 KCl, 4 MgCl2 · 6H2O, 2 CaCl2 · 2H2O, 5 N-Tris[hydroxymethyl]methyl-2-amonoethanesulfonic acid, and 36 sucrose, pH 7.15. The internal patch solution consisted of (in mm) 140 KCH3SO3, 2 MgCl2 · 6H2O, 2 EGTA, 5 KCl, and 20 HEPES, pH 7.4.
Statistics.
Data were compared using a nonpaired t test. Results were deemed significant at *p ≤ 0.05 or **p ≤ 0.01. All values shown are mean ± SEM.
Results
The Dystrophin isoform Dp186 is expressed in the embryonic and larval synapse-rich neuropiles
The mammalian dystrophin gene is a large, highly conserved gene that encodes a number of isoforms expressed from distinct promoters (for review, see Blake et al., 2002). The D. melanogaster dystrophin (dys) gene encodes six known isoforms that, similarly to their mammalian orthologs, are expressed predominantly in the musculature and nervous system (Greener and Roberts, 2000; Neuman et al., 2001, 2005; Dekkers et al., 2004) (Fig. 1A). All Drosophila isoforms bear the highly conserved cysteine-rich Dystrophin C-terminal region (Fig. 1). The N termini of three large isoforms (DLP1, DLP2, and DLP3) include the actin-binding domain and spectrin repeats found in the large mammalian protein, whereas the N termini of the smaller isoforms Dp186, Dp205, and Dp117 do not resemble those of the smaller mammalian isoforms. The Drosophila DLP2 isoform is expressed predominantly in the musculature and accumulates at the sarcomeres and at the postsynaptic side of the NMJ (van der Plas et al., 2006). Previous RNA in situ analyses revealed that Dp186 is highly expressed in the embryonic CNS (Neuman et al., 2001; Dekkers et al., 2004). We raised rabbit polyclonal antisera against sequences unique to Dp186 (see Materials and Methods) and examined the localization of the Dp186 protein during the embryonic and larval stages. Dp186 protein was first detected in the embryonic CNS at stage 13 and is expressed throughout embryonic development, localizing to regions near the longitudinal connectives (Fig. 2B). These regions likely represent the synapse-rich dorsal neuropile, because the presynaptic Bruchpilot protein, recognized by monoclonal antibody nc82 (Kittel et al., 2006; Wagh et al., 2006), is also present there (Fig. 2A,C).
Figure 2.
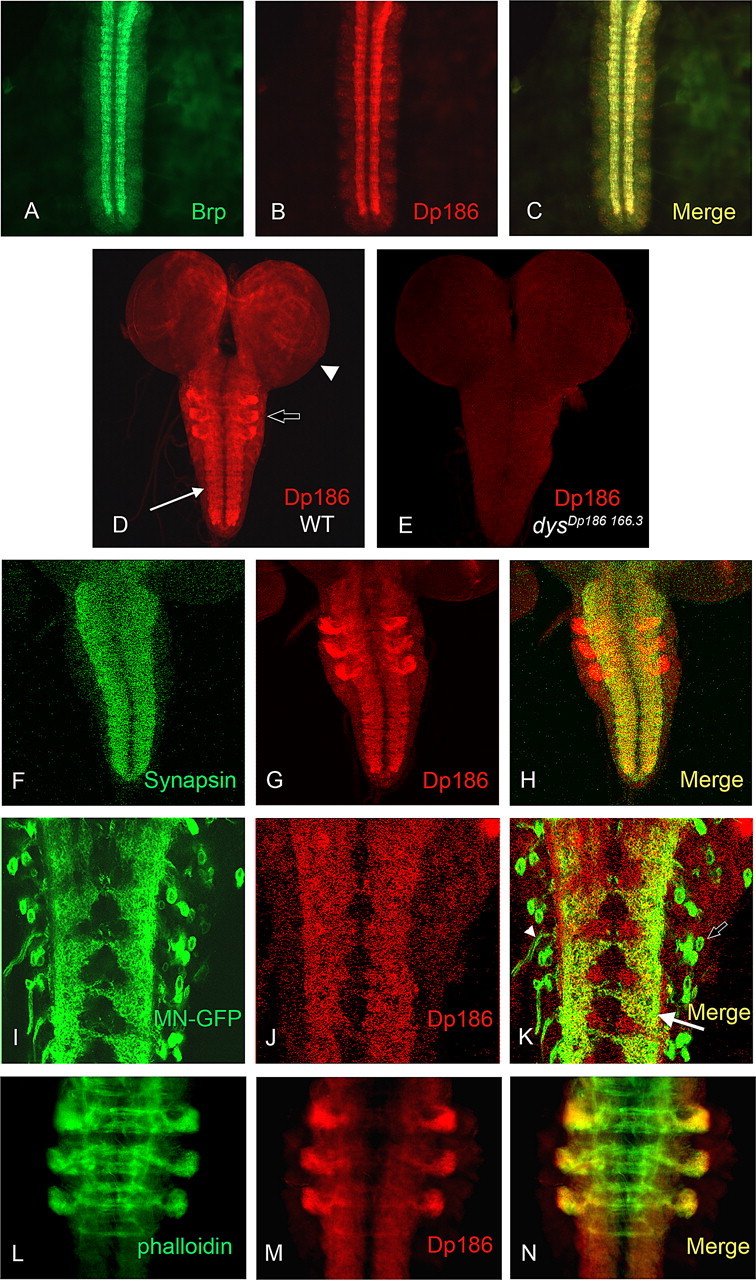
Dp186 protein is expressed in synapse-rich regions of the embryonic and larval neuropiles. A–N, Dissected stage 16 embryo ventral nerve cords (A–C), entire third instar larval CNSs (D, E), or only the neuropile (F–H) or a portion thereof (I–N) are shown. A, The synapse-rich embryonic neuropile as detected by monoclonal antibody nc82, which recognizes the presynaptic Bruchpilot protein (Brp). B, Anti-Dp186 staining of the ventral nerve cord. C, The merge of the channels displayed in A and B. D, Dp186 is expressed in the neuropile (arrow), in the brain lobes (arrowhead), and in three bilaterally symmetric clusters within the thoracic segments (open arrow; see also L–N) in third instar larvae. WT, Wild type. E, Dp186 expression cannot be detected in the mutant dysDp186 166.3. F, G, The presynaptic Synapsin protein (F) and Dp186 (G) are expressed throughout the larval neuropile. H, The merge of the channels shown in F and G. I–K, mCD8-GFP protein driven by OK6-Gal4 in motoneurons (MN-GFP; I) colocalizes with Dp186 protein (J) in motoneuron dendrites (arrow), but not in cell bodies (open arrow) or axons (arrowhead) (K). L–N, Alexa Fluor 488-conjugated phalloidin (L), which binds to F-actin, overlaps with Dp186 (M) in the three bilaterally symmetric thoracic clusters (N). Anterior is up in all preparations.
In the third instar larval CNS, Dp186 protein is found in the neuropile and accumulates in distinct areas in the two brain lobes (Fig. 2D). In addition, the protein can be observed in three bilaterally symmetric clusters, likely within the thoracic neuromeres, which are located at the lateral sides of the neuropile. Staining is not evident in the dysDP186 166.3 mutant (Fig. 2E). We performed double labeling of anti-Dp186 with an antibody recognizing the presynaptically localized Synapsin protein (Klagges et al., 1996). We found that Dp186 and Synapsin are present in close proximity in the synapse-rich neuropile (Fig. 2 F–H), but Synapsin does not appear to as be highly expressed in the sets of lateral clusters within the presumptive thoracic neuromeres as Dp186 (Fig. 2 F–H). We also examined a postsynaptic marker localized to motoneuron dendrites in double labelings with anti-Dp186. UAS-mCD8-GFP driven by a motoneuron-specific Gal4 driver (OK6-Gal4) results in the localization of GFP to the membranes of motoneuron cell bodies, dendrites, and axons (Landgraf et al., 2003; Sanchez-Soriano et al., 2005). Double labeling with anti-Dp186 shows colocalization of GFP and Dp186 proteins (Fig. 2 I–K), indicating that Dp186 is present at interneuron–motoneuron synapses. To further characterize Dp186 localization in the presumptive thoracic neuromeres, we used fluorescently tagged phalloidin to stain F-actin in the larval neuropile and found that Dp186 colocalized with F-actin most strongly in the lateral cluster extremities (Fig. 2 L–N).
Lack of Dp186 results in an increase in synaptic currents in motoneurons
To study the role of the Dp186 dystrophin isoform during CNS development, we generated mutant fly lines that lack Dp186 protein. We used a P-element mobilization strategy starting with a P-element, GE20705, inserted 250 bp upstream of the Dp186 ATG initiator codon to generate Dp186-specific deletions (see Materials and Methods) (Fig. 1). Two lines were generated, dysDp186 166.3 and dysDp186 30.3, which both lack detectable levels of Dp186 protein (Fig. 2E) and mRNA (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) but have wild-type levels of the Dystrophin DLP2, Dp117, and Dp205 isoform mRNAs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
The lack of the large muscle-specific DLP2 Dystrophin isoform results in a significant increase in evoked neurotransmitter release at the Drosophila NMJ (van der Plas et al., 2006). To determine whether synaptic transmission in the CNS is similarly regulated by the Dp186 CNS-specific Dystrophin isoform, voltage-clamp recordings (Vh −60 mV) were made from either the aCC or RP2 motoneurons (no differences were observed between these neurons). In wild-type backgrounds, such recordings show inward synaptic currents that are relatively long lived (500–1000 ms) and have an average amplitude of 76 ± 3.3 pA (Fig. 3 A–C). These excitatory synaptic currents, which are cholinergic in nature and action potential dependent, result from the synchronous activity of interneurons that are presynaptic to the motoneurons (Baines and Bate, 1998; Baines, 2003). These currents arise from the combined synaptic output of more than one interneuron and are not the result of single action potentials (Baines, 2003). Identical recordings in dysDp186 166.3, an allele that bears a deletion encompassing most of the unique first Dp186 exon, revealed that synaptic currents were significantly increased in amplitude (127 ± 4.4 pA; p ≤ 0.01) (Fig. 3B,C). Cumulative probability plots of individual synaptic currents, which better show the range of current amplitudes recorded, revealed that there is a significant increase in amplitudes of the majority of the individual currents measured, compared with the wild-type control (Fig. 3D). Synaptic current amplitude was also significantly increased in dysDp186 30.3, a second independent imprecise excision allele lacking Dp186 (117 ± 3.5 pA; p ≤ 0.01) (Fig. 3C). Similarly high currents were observed both in dysDp186 166.3/+ heterozygotes (152 ± 3.4 pA, p ≤ 0.01) and in Df(3R)Exel6184/+ heterozygotes (139 ± 2.4 pA; p ≤ 0.01); this deficiency uncovers the entire Dystrophin locus. Together, these data indicate that Dp186 is haploinsufficient in maintaining the wild-type electrophysiology of this interneuronal synapse. In contrast, synaptic currents in dysDLP2E6, which lacks the DLP2 muscle-specific isoform but expresses Dp186 at wild-type levels (van der Plas et al., 2006), were no different in amplitude compared with wild type (75 ± 2.5 pA; p > 0.05) (Fig. 3C). Evoked synaptic currents at the NMJ were, however, significantly elevated in dysDLP2E6 (van der Plas et al., 2006). The frequency of action potential-dependent synaptic currents recorded in aCC/RP2 was not significantly altered in either dysDp186 166.3 or dysDp186 30.3 compared with wild type (21.1 ± 4.2, 19.3 ± 3.2, and 21 ± 3.8 currents per minute, respectively; p > 0.05), suggesting that the degree of synaptic connectivity of these two motoneurons with presynaptic interneurons remains unaltered.
Figure 3.

Synaptic currents in motoneurons are increased in the absence of Dp186. A, Whole-cell voltage-clamp recordings (Vh −60 mV) from aCC/RP2 reveal large inward currents that are the result of action potential-dependent (evoked) release of presynaptic ACh (Baines et al., 2001). B, These currents are significantly increased in amplitude in Dp186 mutants (dysDp186 166.3 is shown). C, The average amplitude of synaptic currents is significantly increased in both dysDp18630.3 and dysDp186 166.3 compared with both Canton-S wild type and the homozygous DLP2 loss-of-function allele (dysDLP2E6). Values shown are 76 ± 3.3, 117 ± 3.5, 127 ± 4.4, and 75 ± 2.5 pA, respectively. **p ≤ 0.01 compared with both wild type and dysDLP2E6. Currents were recorded from at least eight cells of each genotype. D, Cumulative probability plots of individual excitatory currents reveals a clear increase in amplitude for the large majority of currents recorded in dysDp186 166.3 compared with wild type. WT, Wild type.
There are a number of conceivable mechanisms that might underlie an increase in evoked synaptic transmission between presynaptic interneurons and aCC/RP2. Two possibilities are that there is an increase in evoked release of ACh from presynaptic terminals or an increased postsynaptic sensitivity to this neurotransmitter. To distinguish between these two possibilities, recordings were repeated in the presence of TTX (0.1 μm). All evoked transmitter release was blocked under these conditions (Fig. 4A). Analysis of the TTX-insensitive miniature synaptic currents (mEPSC) in aCC/RP2 showed no difference in amplitude between dysDp186 166.3, dysDp18630.3, and wild-type controls (Fig. 4B,C). This result is consistent with a lack of change in postsynaptic sensitivity to neurotransmitter in the mutant. The frequency of mEPSCs was, however, significantly increased in both dysDp186 166.3 and dysDp18630.3 compared with the control (Fig. 4D). An increase in mEPSC frequency is consistent with, and indeed predictive of, a heightened probability of presynaptic vesicle release.
Figure 4.

Loss of Dp186 results in increased mEPSC frequency but no change in amplitude. A, In the presence of TTX (0.1 μm), evoked excitatory currents are abolished, leaving only those currents that are elicited by the spontaneous release of vesicles from the presynaptic interneurons (mEPSCs). Currents from Canton-S wild type and dysDp186 166.3 are shown. B, Analysis of averaged mEPSC amplitudes shows no change because of the absence of Dp186 (dysDp18630.3 and dysDp186 166.3) compared with Canton-S wild type. Values shown are 9.5 ± 0.4, 8.9 ± 0.3, and 9.3 ± 0.3 pA, respectively. p > 0.05. C, Cumulative probability plots of individual mEPSC currents for both wild type and dysDp186 166.3 show no differences in amplitude distribution. D, The frequency of mEPSCs is significantly greater in both dysDp18630.3 and dysDp186 166.3 compared with wild type. Values shown are 9.2 ± 1.7, 16.3 ± 3.1, and 14.6 ± 3.0 events per minute. *p ≤ 0.05. Currents were recorded from at least eight cells of each genotype. WT, Wild type.
RNA interference and rescue experiments reveal a predominantly postsynaptic role for Dp186
Our data indicate that Dp186 is required for normal synaptic signaling between interneurons and motoneurons in the CNS. To better localize the site of action of Dp186, we adopted two approaches. First, we used targeted expression of an RNA interference (RNAi) transgene to reduce the endogenous levels of Dp186 protein specifically either presynaptically or postsynaptically. Second, we attempted to rescue the Dp186 mutant electrophysiological phenotype by tissue-specific expression of a wild-type Dp186 transgene. Targeted expression of both RNAi and rescue constructs was achieved through use of two well characterized Gal4 drivers. B19-Gal4 drives expression in all cholinergic neurons, including those interneurons that are presynaptic to aCC/RP2 (Baines, 2004). RN2-Gal4 is an even-skipped promoter-Gal4 transgene that drives expression in the aCC and RP2 motoneurons (Fujioka et al., 2003; Baines, 2004). The efficiency of the RNAi was determined by quantitative reverse transcription-PCR, and Dp186 expression levels were found to be reduced to one-third to one-half of wild-type levels when the UAS-RNAi-Dp186 constructs were expressed throughout the larval neuropile by use of the pan-neuronal Elav-Gal4 driver (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Expression of the Dp186-RNAi transgene was sufficient to phenocopy the Dp186 mutant phenotype: synaptic currents were significantly increased, when expressed in either presynaptic cholinergic neurons or in the postsynaptic motoneurons aCC/RP2 (Fig. 5A). The increase in synaptic currents observed was, however, significantly greater when the RNAi transgene was expressed in the postsynaptic aCC/RP2 than in the presynaptic interneuron (presynaptic vs postsynaptic, p ≤ 0.01). It should be noted that synaptic currents are potentiated by the presence of the Dp186-RNAi transgene alone (i.e., in the absence of Gal4), suggesting that its “leaky” pan-neuronal expression might, in part, contribute to the effect observed when using either the presynaptic B19-Gal4 or postsynaptic RN2-Gal4 driver. However, Gal4-dependent expression results in clearly significantly increased synaptic current amplitude relative to the UAS control level.
Figure 5.
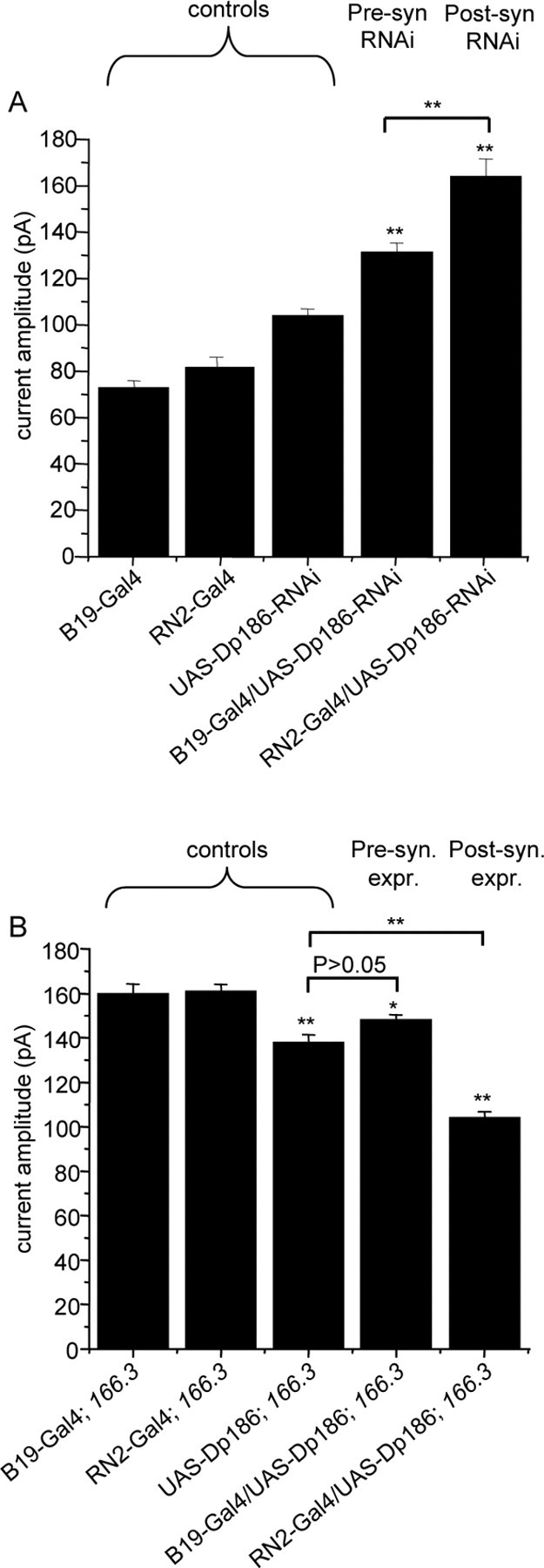
Dp186 is required in postsynaptic motoneurons. A, Expression of dsRNA specifically targeting Dp186 is sufficient to increase action potential-dependent synaptic currents when expressed in postsynaptic motoneurons aCC/RP2 (RN2-Gal4) and, to a lesser extent, in presynaptic cholinergic neurons (B19-Gal4). Averaged current amplitudes are plotted for parental control lines (B19- and RN2-Gal4s and UAS-RNAi-Dp186) and expression lines (B19-Gal4/UAS-RNAi-Dp186 and RN2-Gal4/UAS-RNAi-Dp186). Values shown are 72 ± 3.1, 82 ± 4.2, 104 ± 3.4, 131 ± 4.0, and 164 ± 7.6 pA, respectively (**p ≤ 0.01 compared with UAS RNAi-Dp186). B, Rescue of the Dp186 (dysDp186 166.3) mutant phenotype by postsynaptic expression of a UAS-Dp186 transgene (RN2-Gal4/UAS-Dp186). Values shown are 160 ± 4.2, 16 1 ± 3.1, 138 ± 3.4, 148 ± 2.4, and 104 ± 2.8 pA, respectively (*p ≤ 0.05; **p ≤ 0.01 compared with Gal4 controls). Currents were recorded from at least eight cells in each genotype. The 166.3 is used for dysDP186 166.3. Pre-syn, Presynaptic; Post-syn, postsynaptic; expr., expression.
Significant rescue of the Dp186 mutant phenotype by expression of a UAS-Dp186 transgene was, in contrast, only observed after expression in the postsynaptic aCC/RP2 motoneurons and not when expression was limited to the presynaptic cholinergic interneurons compared with the UAS-transgene control (Fig. 5B) (p ≤ 0.01). Thus, the reduction in current amplitude observed after presynaptic expression of Dp186 was not significantly different from that seen in the UAS control, which is presumably again the result of basal Gal4-independent expression of UAS-Dp186. Together, our data are consistent with a predominantly postsynaptic role for Dp186 in the regulation of the strength of central motoneuron synaptic excitation; however, given the effect of presynaptic Dp186-directed dsRNA expression observed, we cannot rule out Dp186 roles in the interneurons or their upstream partners (see Discussion).
Gbb/Wit signaling is not required for increased synaptic transmission in the Dp186 mutant
The observation that Dp186 is required in postsynaptic motoneurons to regulate the efficacy of presynaptic neurotransmitter release is consistent with the existence of a retrograde signal. Studies have shown that such a signaling mechanism, based on the secretion of the bone morphogenetic protein (BMP) ligand Gbb, is active at both the NMJ and at this motoneuron–interneuron synapse in the CNS (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003; Baines, 2004). A more recent study has, however, refined the action of gbb signaling at the NMJ by showing that it is required to establish the competence of the motoneuron to respond to another non-gbb retrograde signal that itself is presumably responsive to synaptic change (Goold and Davis, 2007). To determine whether the increase in synaptic currents observed in aCC/RP2 in the Dp186 mutant is reliant on gbb retrograde signaling, we performed whole-cell recordings in larvae lacking both Dp186 and gbb.
As reported previously (Baines, 2004), synaptic currents in aCC/RP2 were significantly reduced in homozygous gbb1 mutants compared with heterozygous controls (54 ± 1.5 pA vs 79.5 ± 2.2) (Fig. 6A). In the presence of only a single copy of the wild-type gbb gene, the absence of Dp186 (gbb1/Cyo; dysDp186 166.3) was sufficient to increase synaptic currents to a level (128 ± 2.6 pA) (Fig. 6A) comparable to that observed in the Dp186 mutant with wild-type levels of gbb expression (Fig. 3C). Recordings from aCC/RP2 in the doubly homozygous gbb1; dysDp186 166.3 mutant similarly display increased synaptic currents relative to the gbb1 control, albeit to lower absolute values (82 ± 3.2 pA; p ≤ 0.01) (Fig. 6A). These results are consistent with the effect of Gbb and removal of Dp186 being additive and as such acting through separate noninteracting pathways. This is because the presence of Gbb increases synaptic current amplitude by ∼28 pA (wild-type − gbb1 values), whereas removal of Dp186 results in an increase of ∼79 pA (dysDp186 166.3 − wild type). The combined total of 107 pA is comparable to the value we observed after removal of Dp186 in the presence of gbb (128 pA). We therefore conclude that, although the absolute levels of synaptic current increases because of the absence of Dp186 are limited in the absence of gbb1, potentiation of synaptic currents in the absence of Dp186 is unlikely to require gbb-dependent signaling. Gbb retrograde signaling at this interneuronal synapse may instead represent a parallel pathway required for other aspects of synaptic maturation or function.
Figure 6.

aCC/RP2 synaptic current increase in Dp186 mutants is independent of Gbb/Wit signaling and ongoing cholinergic neurotransmission. A, Loss of dysDp186 166.3 in the absence of gbb potentiates synaptic current amplitude to a level that is similar in relative magnitude to that observed in the presence of a single wild-type copy of gbb [i.e., compare (gbb1; dysDp186 166.3 with gbb1) with (gbb1/CyO; dysDp186 166.3 with gbb1/CyO)]. Values shown are 127 ± 4.4, 79.5 ± 2.2, 128 ± 2.6, 54 ± 1.5, and 82 ± 3.2 pA. B, Postsynaptic (Post-syn) expression of UAS-Dp186 (lines 6.1 and 12.1) results in significant gbb-independent increases in synaptic current amplitudes. Values shown are 91.7 ± 2.1, 137 ± 2.0, 175 ± 4.2, 144 ± 2.9, 159 ± 3.6, and 137 ± 2.5 pA. C, wit is not required for normal synaptic current amplitudes in an otherwise wild-type (WT) background, nor is it required for the increased amplitudes observed after postsynaptic expression of Dp186-directed dsRNA. Values shown are 97 ± 3.2, 93 ± 2.1, 115 ± 4.4, and 220 ± 4.2 pA. D, Postsynaptic overexpression of either Gbb or Dp186-RNAi results in increased synaptic currents in aCC/RP2 in second instar larvae raised at 18°C. In comparison, only postsynaptic expression of Dp186-RNAi is able to potentiate synaptic currents in a chats2 background in which cholinergic neurotransmission is significantly reduced. This reduction is evident by comparing the current amplitude of RN2-Gal4 with either that of UAS-Gbb;chats2 or UAS-Dp186-RNAi;chats2. In comparison, postsynaptic overexpression of Gbb is unable to potentiate synaptic currents in the chats2 background. Values shown are 172 ± 2.6, 241 ± 4.2, 240 ± 4.9, 112 ± 12.0, 94 ± 8.8, 113 ± 5.7, and 149 ± 4.4 pA. **p ≤ 0.01.
To further investigate requirements for gbb in the dysDp186 166.3 phenotype, we examined the consequences of overexpressing Dp186 in the presence or absence of gbb. Surprisingly, postsynaptic expression of UAS-Dp186 in a wild-type background resulted in significant potentiation of synaptic currents (Fig. 6B). Thus, either loss or gain of Dp186 function results in increased synaptic currents indicating that specific absolute levels of Dp186 are required for normal synaptic function. That the RNAi knockdown of Dp186 (which is only 50–70% effective) also results in larger synaptic currents than the complete removal of Dp186 supports this hypothesis. Although this result was unexpected, it nevertheless allowed us to further test for gbb requirements. Consistent with the effects of dysDp186 166.3 in the absence of gbb (Fig. 6A), potentiation of synaptic currents attributable to overexpression of Dp186 was also independent of gbb (Fig. 6B). Moreover, overexpression of Dp186 in the gbb null background potentiated synaptic currents to levels similar to those seen with loss of Dp186 in an otherwise wild-type background (Fig. 6A,B), indicating that Dp186 levels must be tightly regulated for its wild-type function. Together, these data strongly suggest that Dp186 regulation of synaptic function does not act through the previously described Gbb-dependent retrograde signaling mechanism at this synapse (Baines, 2004).
Retrograde signaling at the NMJ requires the type II TGF-β receptor Wit (McCabe et al., 2003; Goold and Davis, 2007). To determine whether Wit is required in retrograde signaling at this interneuron–motoneuron synapse, we recorded synaptic currents in the absence of Wit (Fig. 6C). In contrast to the requirement for Wit at the NMJ, central synaptic transmission was unaffected by the absence of this receptor. Moreover, postsynaptic expression of dsRNA directed against Dp186, which increased synaptic currents in the wild-type background (Fig. 5A), also did so in the absence of wit (Fig. 6C).
Finally, we tested the possibility that Dp186-dependent regulation of presynaptic neurotransmitter release is activity dependent, as was previously shown for Gbb at this synapse (Baines, 2004). Cholinergic neurotransmission was significantly reduced by the utilization of a temperature-sensitive allele of choline acetyltransferase (chats2). Compared with controls containing the wild-type cha gene, synaptic currents recorded in chats2 second instar larvae raised at 18°C are significantly reduced (Fig. 6D), consistent with the demonstrated reduction in enzyme activity at this temperature (Salvaterra and McCaman, 1985). Overexpression of gbb in the chats2 background fails to potentiate synaptic currents; however, postsynaptic expression of Dp186-directed dsRNA significantly increased synaptic currents in this same background (Fig. 6D). This strongly suggests that the increase in neurotransmitter release caused by the absence of Dp186 was not dependent on cholinergic neurotransmission and, as such, differs from that observed when gbb is overexpressed postsynaptically. These data also suggest that gbb and Dp186 act through independent, but possibly parallel, pathways.
Discussion
The cognitive impairments displayed by many DMD patients (for review, see Anderson et al., 2002; Culligan and Ohlendieck, 2002) and the localization of Dystrophin to synapse-rich regions (for review, see Blake and Kroger, 2000) are consistent with Dystrophin roles in the CNS. Mutations primarily in the sequences encoding the highly conserved common C-terminal domain of Dystrophin have been correlated with human mental deficits (Moizard et al., 1998), suggesting that mutation of one of the shorter CNS-specific Dystrophin isoforms may underlie mental impairment. Mental defects have, however, also been observed in patients (Hinton et al., 2000) and in the mdx mouse model (Vaillend et al., 2004), lacking only the large Dystrophin isoform. Therefore, the relative roles of the various Dystrophin isoforms in normal CNS function remain unclear.
We show in this study that the Drosophila Dp186 isoform is expressed in the CNS and is required to maintain normal synaptic physiology. It is found in the synapse-rich neuropile and in its absence, evoked presynaptic neurotransmitter release is significantly increased at an identified interneuron–motoneuron central synapse, without apparent change in postsynaptic receptor field sensitivity. Transgenic RNAi and rescue experiments indicate that Dp186 is required primarily in the postsynaptic motoneuron to maintain wild-type presynaptic release levels. The effects of altering postsynaptic levels of Dp186 on presynaptic neurotransmitter release are consistent with a role for Dp186 in retrograde signaling, but, as we show, this signaling is not dependent on gbb, previously implicated in interneuronal retrograde signaling, or wit, shown to act at the NMJ.
Dystrophin Dp186 accumulates at synaptic regions of the CNS
During embryogenesis, the Dp186 protein is found close to the longitudinal axon bundles of the ventral nerve cord. The presence of the presynaptic protein Bruchpilot (Kittel et al., 2006; Wagh et al., 2006) in this domain indicates that it is rich in synaptic contacts that include the motoneuron dendrites and their interneuronal inputs. In the third instar larval neuropile, Dp186 is also in close proximity to the presynaptic Synapsin protein (Klagges et al., 1996). Double stainings performed with anti-Dp186 and membrane-associated GFP (mCD8-GFP) (Lee and Luo, 1999) expressed in motoneurons reveal colocalization, further suggesting that Dp186 is synaptically localized. The density of synaptic contacts in these regions precluded us from evaluating the precise degree of colocalization between Dp186 and these presynaptic markers at the level of light microscopy; however, these data support our hypothesis that Dp186 is synaptically localized.
Dystrophin Dp186 is required for wild-type synaptic physiology
In a previous study, we observed that the absence of the large postsynaptically expressed Dystrophin DLP2 isoform resulted in increased presynaptic neurotransmitter release at the Drosophila NMJ (van der Plas et al., 2006). To address whether Dp186 might play a similar role at interneuronal synapses, we generated mutant lines lacking Dp186 protein and evaluated the electrophysiology of a well characterized synapse between the aCC/RP2 motoneurons and their presynaptic cholinergic interneurons (Baines et al., 2001; Baines, 2003). Unlike the NMJ motoneurons, the presynaptic cholinergic neurons cannot be directly stimulated to allow evaluation of evoked responses. However, in the preparation used, endogenous evoked responses, which form part of the motor pattern generator, occur at defined frequencies from late embryogenesis onward (Baines et al., 2002), allowing the recording of endogenous evoked responses.
Recordings of such endogenous evoked currents in Dp186 mutants reveal that they are significantly increased in amplitude, but not frequency, relative to wild-type controls and a mutant lacking DLP2. Recordings performed in the presence of TTX, which allows measurement of spontaneous mEPSC events in the absence of evoked responses, indicate that the postsynaptic AChR field is apparently unaffected by the absence of Dp186. Together with the increased frequency of mEPSC observed in the Dp186 mutant, these findings support the hypothesis that evoked presynaptic neurotransmitter release is significantly elevated in the absence of Dp186.
Tissue-specific rescue experiments revealed that postsynaptic, but not presynaptic, expression of Dp186 in the dysDP186 mutant background rescued presynaptic release to wild-type levels. Therefore, Dp186 is apparently predominantly required postsynaptically. Our results from the transgenic RNAi approach are less unambiguous but do show that the largest increase in presynaptic release occurred when postsynaptic Dp186 expression levels were decreased. Presynaptic expression of Dp186-RNAi, however, also resulted in increased release, albeit to lower levels. A possible explanation is that decreased Dp186 expression in first-order interneurons that drive motoneurons might, in turn, elevate their own excitation from second-order interneurons (to which they are postsynaptic). The resultant increased activity in these first-order interneurons might manifest itself by increasing the synaptic excitation of downstream motoneurons. Whereas many details of the circuitry that form the motor pattern generator are lacking, first-order interneurons that synapse directly with motoneurons are indeed reliant on second-order interneurons for synaptic excitation (Carhan et al., 2004). At present, markers suitable for evaluating whether Dp186 is present at these upstream synapses are not available.
Dystrophin and the retrograde control of presynaptic release
The requirement for Dp186 in motoneurons for normal function of presynaptic cholinergic interneurons is consistent with the regulation of neurotransmitter release by a retrograde signal derived from the targets. Recent studies have shown that presynaptic release at these synapses is regulated, at least in part, by BMP signaling (Baines, 2004). Moreover, increasing expression of the BMP ortholog Gbb in postsynaptic motoneurons is sufficient to significantly increase synaptic current amplitudes, achieving levels similar to those observed in the Dp186 null mutants (Baines, 2004). We therefore examined the effects of eliminating Gbb signaling on the Dp186 electrophysiological phenotype. In the absence of gbb, the relative increase in synaptic currents caused by the lack of expression of Dp186 was similar to that seen in the dysDp186 mutant alone, suggesting that Dp186 regulates presynaptic neurotransmitter release independently of gbb. The failure of the loss of gbb to suppress the increases in central synaptic currents observed after overexpression of Dp186 also indicates the apparent independence of these two pathways.
Previous studies have implicated wit in the proper morphological development of the NMJ synapse and retrograde signaling there (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003) and in establishing the competence of motoneurons to respond to muscle-derived retrograde signals (Goold and Davis, 2007). Potentiation of synaptic transmission at the NMJ as a result of postsynaptic loss of the DLP2 Dystrophin isoform was previously shown to require wit (van der Plas et al., 2006). In the CNS, we find that there is seemingly no corresponding requirement for wit for increased neurotransmitter release at the Dp186-deficient synapse. Furthermore, contrary to what was observed at the NMJ, wit is apparently not required for wild-type electrophysiology. Clearly then, although the absence of a specific Dystrophin isoform at either the NMJ or a central synapse results in increased neurotransmitter release, the underlying mechanisms likely differ, perhaps reflecting the use of different retrograde signaling pathways.
We observe that dysDp186-dependent potentiation of central synaptic currents persists when cholinergic neurotransmission is significantly reduced. This, again, is in contrast to the effect of postsynaptic overexpression of gbb under the same conditions. There are two implications of these data. First, they further support the hypothesis that Dp186 and gbb independently regulate central synaptic transmission. Second, these data predict the existence of two types of retrograde signaling pathways regulating presynaptic function, one activity-dependent and the other, constitutive. Indeed, our data are consistent with the possibility that Dp186 controls the ongoing release of a negative regulator of synaptic function. Conceivably, the strength of synaptic transmission will be regulated by the interplay of these and other mechanisms. The identity of the signal regulated by Dp186 is not known, but it may be one or more of the six related BMP signaling molecules present in Drosophila: Dpp, Screw, Activin, Activin-like protein, Myoglianin, and Maverick (Lo and Frasch, 1999; Keshishian and Kim, 2004); or it is possibly unrelated to BMP/TGF-β signaling.
It is not currently clear by what mechanisms reduced postsynaptic expression levels of Dp186 potentiate presynaptic neurotransmitter release. The increased mEPSC frequency, but not amplitude, observed in the mutant is consistent with an increase in quantal content caused by heightened probability of release. Additional possibilities include increased synchronicity of release from multiple interneurons that drive these motoneurons. Alternatively, individual interneurons might have increased numbers of presynaptic motoneuron contacts. We consider the latter possibility less probable because we observe that evoked synaptic current amplitude, but not frequency, is affected in the Dp186 mutant. This indicates that the synaptic connectivity between interneurons and motoneurons is apparently normal in the absence of Dp186.
In summary, the Drosophila Dystrophin Dp186 isoform is required predominantly postsynaptically for wild-type neurotransmitter release levels at an identified cholinergic central synapse. Our previous study of the DLP2 isoform revealed that its absence from the muscle increased the probability of presynaptic release at the glutamatergic NMJ (van der Plas et al., 2006). Therefore, these two postsynaptically localized Dystrophin isoforms are required for the appropriate regulation of presynaptic release at two different types of synapses, each using a different neurotransmitter. Furthering our understanding of the role of the Dystrophin isoforms in synaptic transmission in Drosophila should yield insights into evolutionarily conserved roles of dystrophin in the nervous system and perhaps shed light on the poorly understood mental retardation presented by a significant subset of DMD patients.
Footnotes
This work was supported by The Wellcome Trust (R.A.B.) and “Pionier” Grant 900-02-003 from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (J.N.N.). We thank E. Buchner, M. Fujioka, J. B. Jaynes, K. A. Wharton, and the Bloomington Stock Center for fly stocks and antibodies and B. van Veen, M. Bansraj, A. de Jong, and L. Dekkers for help with the experiments. We also acknowledge J. Plomp, S. Potikanond, G. Pilgram, and the reviewers for providing insightful suggestions on this manuscript as well as B. Gerber and R. Stocker for their kind advice.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Anderson JL, Head SI, Rae C, Morley JW. Brain function in Duchenne muscular dystrophy. Brain. 2002;125:4–13. doi: 10.1093/brain/awf012. [DOI] [PubMed] [Google Scholar]
- Baines RA. Postsynaptic protein kinase A reduces neuronal excitability in response to increased synaptic excitation in the Drosophila CNS. J Neurosci. 2003;23:8664–8672. doi: 10.1523/JNEUROSCI.23-25-08664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA. Synaptic strengthening mediated by bone morphogenetic protein-dependent retrograde signaling in the Drosophila CNS. J Neurosci. 2004;24:6904–6911. doi: 10.1523/JNEUROSCI.1978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Bate M. Electrophysiological development of central neurons in the Drosophila embryo. J Neurosci. 1998;18:4673–4683. doi: 10.1523/JNEUROSCI.18-12-04673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Uhler JP, Thompson A, Sweeney ST, Bate M. Altered electrical properties in Drosophila neurons developing without synaptic transmission. J Neurosci. 2001;21:1523–1531. doi: 10.1523/JNEUROSCI.21-05-01523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Seugnet L, Thompson A, Salvaterra PM, Bate M. Regulation of synaptic connectivity: levels of Fasciclin II influence synaptic growth in the Drosophila CNS. J Neurosci. 2002;22:6587–6595. doi: 10.1523/JNEUROSCI.22-15-06587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake DJ, Kroger S. The neurobiology of Duchenne muscular dystrophy: learning lessons from muscle? Trends Neurosci. 2000;23:92–99. doi: 10.1016/s0166-2236(99)01510-6. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Carhan A, Reeve S, Dee CT, Baines RA, Moffat KG. Mutation in slowmo causes defects in Drosophila larval locomotor behaviour. Invert Neurosci. 2004;5:65–75. doi: 10.1007/s10158-003-0028-y. [DOI] [PubMed] [Google Scholar]
- Culligan K, Ohlendieck K. Diversity of the brain dystrophin-glycoprotein complex. J Biomed Biotechnol. 2002;2:31–36. doi: 10.1155/S1110724302000347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck AE, Potter AC, Tinsley JM, Wood SJ, Vater R, Young C, Metzinger L, Vincent A, Slater CR, Davies KE. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J Cell Biol. 1997a;136:883–894. doi: 10.1083/jcb.136.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997b;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- Dekkers LC, van der Plas MC, van Loenen PB, den Dunnen JT, van Ommen GJ, Fradkin LG, Noordermeer JN. Embryonic expression patterns of the Drosophila dystrophin-associated glycoprotein complex orthologs. Gene Expr Patterns. 2004;4:153–159. doi: 10.1016/j.modgep.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Fujioka M, Lear BC, Landgraf M, Yusibova GL, Zhou J, Riley KM, Patel NH, Jaynes JB. Even-skipped, acting as a repressor, regulates axonal projections in Drosophila. Development. 2003;130:5385–5400. doi: 10.1242/dev.00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997a;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997b;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Greener MJ, Roberts RG. Conservation of components of the dystrophin complex in Drosophila. FEBS Lett. 2000;482:13–18. doi: 10.1016/s0014-5793(00)02018-4. [DOI] [PubMed] [Google Scholar]
- Hinton VJ, De Vivo DC, Nereo NE, Goldstein E, Stern Y. Poor verbal working memory across intellectual level in boys with Duchenne dystrophy. Neurology. 2000;54:2127–2132. doi: 10.1212/wnl.54.11.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer A. Würzburg, Germany: University of Würzburg; 1991. Eine Bibliothek monoklonaler Antikörper gegen das Gehirn von Drosophila melanogaster. [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Keshishian H, Kim YS. Orchestrating development and function: retrograde BMP signaling in the Drosophila nervous system. Trends Neurosci. 2004;27:143–147. doi: 10.1016/j.tins.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Kim H, Rogers MJ, Richmond JE, McIntire SL. SNF-6 is an acetylcholine transporter interacting with the dystrophin complex in Caenorhabditis elegans. Nature. 2004;430:891–896. doi: 10.1038/nature02798. [DOI] [PubMed] [Google Scholar]
- Kittel RJ, Wichmann C, Rasse TM, Fouquet W, Schmidt M, Schmid A, Wagh DA, Pawlu C, Kellner RR, Willig KI, Hell SW, Buchner E, Heckmann M, Sigrist SJ. Bruchpilot promotes active zone assembly, Ca2+ channel clustering, and vesicle release. Science. 2006;312:1051–1054. doi: 10.1126/science.1126308. [DOI] [PubMed] [Google Scholar]
- Klagges BR, Heimbeck G, Godenschwege TA, Hofbauer A, Pflugfelder GO, Reifegerste R, Reisch D, Schaupp M, Buchner S, Buchner E. Invertebrate synapsins: a single gene codes for several isoforms in Drosophila. J Neurosci. 1996;16:3154–3165. doi: 10.1523/JNEUROSCI.16-10-03154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf M, Jeffrey V, Fujioka M, Jaynes JB, Bate M. Embryonic origins of a motor system: motor dendrites form a myotopic map in Drosophila. PLoS Biol. 2003;1:E41. doi: 10.1371/journal.pbio.0000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- Lo PC, Frasch M. Sequence and expression of myoglianin, a novel Drosophila gene of the TGF-beta superfamily. Mech Dev. 1999;86:171–175. doi: 10.1016/s0925-4773(99)00108-2. [DOI] [PubMed] [Google Scholar]
- Luo L, Liao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 1994;8:1787–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- Marques G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O'Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Moizard MP, Billard C, Toutain A, Berret F, Marmin N, Moraine C. Are Dp71 and Dp140 brain dystrophin isoforms related to cognitive impairment in Duchenne muscular dystrophy? Am J Med Genet. 1998;80:32–41. doi: 10.1002/(sici)1096-8628(19981102)80:1<32::aid-ajmg6>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Neuman S, Kaban A, Volk T, Yaffe D, Nudel U. The dystrophin/ utrophin homologues in Drosophila and in sea urchin. Gene. 2001;263:17–29. doi: 10.1016/s0378-1119(00)00584-9. [DOI] [PubMed] [Google Scholar]
- Neuman S, Kovalio M, Yaffe D, Nudel U. The Drosophila homologue of the dystrophin gene - introns containing promoters are the major contributors to the large size of the gene. FEBS Lett. 2005;579:5365–5371. doi: 10.1016/j.febslet.2005.08.073. [DOI] [PubMed] [Google Scholar]
- Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- Salvaterra PM, Kitamoto T. Drosophila cholinergic neurons and processes visualized with Gal4/UAS-GFP. Brain Res Gene Expr Patterns. 2001;1:73–82. doi: 10.1016/s1567-133x(01)00011-4. [DOI] [PubMed] [Google Scholar]
- Salvaterra PM, McCaman RE. Choline acetyltransferase and acetylcholine levels in Drosophila melanogaster: a study using two temperature-sensitive mutants. J Neurosci. 1985;5:903–910. doi: 10.1523/JNEUROSCI.05-04-00903.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Soriano N, Bottenberg W, Fiala A, Haessler U, Kerassoviti A, Knust E, Lohr R, Prokop A. Are dendrites in Drosophila homologous to vertebrate dendrites? Dev Biol. 2005;288:126–138. doi: 10.1016/j.ydbio.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Tower J, Karpen GH, Craig N, Spradling AC. Preferential transposition of Drosophila P elements to nearby chromosomal sites. Genetics. 1993;133:347–359. doi: 10.1093/genetics/133.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillend C, Billard JM, Laroche S. Impaired long-term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin-deficient Dmd(mdx) mouse. Neurobiol Dis. 2004;17:10–20. doi: 10.1016/j.nbd.2004.05.004. [DOI] [PubMed] [Google Scholar]
- van der Plas MC, Pilgram GS, Plomp JJ, de Jong A, Fradkin LG, Noordermeer JN. Dystrophin is required for appropriate retrograde control of neurotransmitter release at the Drosophila neuromuscular junction. J Neurosci. 2006;26:333–344. doi: 10.1523/JNEUROSCI.4069-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagh DA, Rasse TM, Asan E, Hofbauer A, Schwenkert I, Durrbeck H, Buchner S, Dabauvalle MC, Schmidt M, Qin G, Wichmann C, Kittel R, Sigrist SJ, Buchner E. Bruchpilot, a protein with homology to ELKS/CAST, is required for structural integrity and function of synaptic active zones in Drosophila. Neuron. 2006;49:833–844. doi: 10.1016/j.neuron.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Wharton KA, Cook JM, Torres-Schumann S, de Castro K, Borod E, Phillips DA. Genetic analysis of the bone morphogenetic protein-related gene, gbb, identifies multiple requirements during Drosophila development. Genetics. 1999;152:629–640. doi: 10.1093/genetics/152.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]