

Abstract
Using a redox-inert methyl acceptor, we show that betaine-homocysteine S-methyltransferase (BHMT) requires a thiol reducing agent for activity. Short-term exposure of BHMT to reducing agent-free buffer inactivates the enzyme without causing any loss of its catalytic zinc. Activity can be completely restored by the re-addition of a thiol reducing agent. The catalytic zinc of BHMT is bound by three thiolates and one hydroxyl group. Thiol modification experiments indicate that a disulfide bond is formed between two of the three zinc-binding ligands when BHMT is inactive in a reducing agent-free buffer, and that this disulfide can be readily reduced with the concomitant restoration of activity by re-establishing reducing conditions. Long-term exposure of BHMT to reducing agent-free buffer results in the slow, irreversible loss of its catalytic Zn and a corresponding loss of activity. Experiments using the glutamate-cysteine ligase modifier subunit knockout mice Gclm (−/−), which are severely impaired in glutathione synthesis, show that BHMT activity is reduced about 75% in Gclm (−/−) compared to Gclm (+/+) mice.
Keywords: BHMT, cysteine, glutathione, methionine, glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse, oxidative stress
INTRODUCTION
Changes in the thiol redox status of the cell have been shown to elicit potent changes in cell behavior, including direct effects on protein function and gene expression. Studies have shown that redox reactive species can influence the activity of several enzymes involved in homocysteine (Hcy) and methionine (Met) metabolism. For example, cystathionine-β-synthase becomes activated in the presence of hydrogen peroxide [1]; whereas Met synthase is inactivated when its Co(I) becomes oxidized to Co(II) [2; 3] and the hepatic isoform of Met adenosyltransferase is inhibited by S-nitrosylation [4; 5]. Consistent with these results, pro-oxidants have been shown to increase the flux of sulfur from Met to Cys, and the latter into glutathione in human hepatoma cells [1] [6]. These data suggest that several enzymes of sulfur amino acid metabolism work in concert to enhance Cys biosynthesis as a mechanism to bolster glutathione synthesis when cells are under oxidative stress [7]. We thought that an elaboration of this regulation could include betaine-homocysteine S-methyltransferase (BHMT) since this enzyme is expressed at high levels in liver [8] and it is responsible for about half of the Met produced by the remethylation of Hcy [9]. Regulating the catalytic efficiency of BHMT by changing rates of production of reactive oxygen or reactive nitrogen species would be a direct and potentially potent way to partition Hcy to either Met or Cys biosynthesis.
BHMT catalyzes the transfer of a methyl group from betaine (Bet) to Hcy to produce Met and dimethylglycine, respectively. The reaction mechanism is bi-bi, with Hcy being the first substrate to bind and Met being the last product off [10], [11]. At the active site of BHMT there is a Zn atom that is coordinated in tetrahedral arrangement by three thiolate anions derived from Cys 217, 299 and 300 [12], and the hydroxyl group of Tyr77 [13]. When Hcy binds to BHMT it displaces Tyr77 as the fourth ligand to Zn, which in turn triggers conformational changes that creates the Bet binding site on the enzyme [13], [11]. The structure of human BHMT obtained from crystals grown in buffer devoid of reducing agents showed that two of the three active site thiolates (Cys217 and Cys 299) were connected by a disulfide bond [12] and the enzyme was devoid of Zn. Since Zn is required for BHMT activity and crystalizing the enzyme in reducing agent-free buffer caused the formation of a disulfide bond and loss of its catalytic Zn [12], it seemed reasonable to speculate that BHMT activity might be affected by factors that influence thiol redox status in vivo. The studies described herein show that the Cys residues involved in binding the catalytic Zn of BHMT might undergo a redox switch that could potentially modulate the flux through BHMT in response to changes in the thiol redox status of the cell.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Methylmethane thiosulfonate (MMTS), 4-(2-pryidylazo)resorcinol (PAR), 5’,5’-dithio-bis-(2-nitro)benzoic acid (DTNB), reduced glutathione hydrochloride, γ-glutamylcysteine trifluoroacetate, reduced cysteinylglycine, β-mercaptoethanol (βME), and L-amino acids were obtained from Sigma (St Louis, MO, USA). All other reagents were of the highest analytical or molecular grade available from commercial vendors. All spectroscopic measurements were performed using a Spectramax Plus spectrophotometer (Molecular Devices Corporation, Sunnyvale, CA, USA). [14C-methyl]-Bet and [14C-methylene]-dimethylsulfonioacetate were obtained from Moravek Biochemicals (Brea, CA) and Dupont NEN (Boston, MA), respectively.
Site-directed mutagenesis of human BHMT
Cys residues 104, 131, 186, 201, and 256 of human BHMT were successively changed to Ala by site directed mutagenesis (QuikChange™ kit; Stratagene, La Jolla, CA, USA) using the primers listed in table 1 and the pTBY4-hBHMT plasmid [14] as template. The mutant that had all of these Cys residues changed to Ala was designated hBHMT:5C→A. The sequences of all mutant cDNAs were verified by DNA sequencing.
Table 1.
Sequences of PCR primers used for the generation of Cys-to-Ala BHMT mutants1
| Primer | Sequence |
|---|---|
| FP-C104A | GCAGGAAGTCAATGAAGCTGCTgcaGACATCGCCC |
| RP-C104A | CGGGCGATGTCGgctGCAGCTTCATTGACTTCCTGCCC |
| FP-C131A | CCTTCATACCTTAGCgcCAAGAGTGAAACTGAAGTCAAAAAAGTATTTCTGC |
| RP-C131A | CAGTTTCACTCTTGgcGCTAAGGTATGAAGGTGTCTGACTCACTCC |
| FP-C186A | CCGGTAAACCTGTGGCAGCAACCATGgcCATTGGCCCAGAAGG |
| RP-C186A | GCAAATCTCCTTCTGGGCCAATGgcCATGGTTGCTGCC |
| FP-C201A | GGCGTGCCCCCCGGCGAGgcTGCAGTGCGCCTGGTGAAAGC |
| RP-C201A | GCTCCTGCTTTCACCAGGCGCACTGCAgcCTCGCCGGGGGG |
| FP-C256A | GGCTTACCACACTCCTGACgcCAACAAGCAGGGATTCATCG |
| RP-C256A | GGGAGATCGATGAATCCCTGCTTGTTGgcGTCAGGAGTGTGG |
All primers were synthesized by Integrated DNA Technologies and are listed 5’ to 3’. Lower-case letters denote a mutation, and bold letters denote the location of a new or destroyed restriction site. FP identifies primers complementary to the coding strand, and those complementary to the noncoding strand are identified as RP.
BHMT purification, BHMT assay procedures and protein quantification
Wild type and mutant enzymes were over expressed in E. coli and purified using the Impact™ T7 system (New England Biolabs, Beverly, MA, USA) as previously described [14]. The standard Bet-Hcy dependent BHMT assay contains 5 mM D,L-Hcy, 2 mM [14C-methyl]-Bet (0.1 μCi) and 10 mM βME. Other exact details of this assay have been described elsewhere [15]. The assay for the BHMT-dependent methylation of L-aspartate (Asp) was the same except Hcy was replaced with Asp (5 mM) and Bet was replaced with 1 or 2 mM [14C-methylene]-dimethylsulfonioacetate (DMSA; 0.5 or 1.0 μCi). As specified, some reaction cocktails varied in the concentrations of substrate(s) and/or reducing agent. Enzyme solutions were made reducing agent-free by either exhaustive dialysis against multiple changes of the specified buffer over a 12 to 24h period, or by rapid gel filtration using HiTrap™ desalting columns (Amersham Pharmacia Biotech Piscataway, NJ, USA), as described for each experiment. The incubation periods for the standard Bet-Hcy assay ranged from 30 m to 1 h, whereas extended periods of incubation (3 to 6 h) were used when the DMSA-Asp assay was employed. The activity of the enzyme was linear over the incubation time periods used in these assays. Reaction blanks were devoid of enzyme and were less than 100 and 3000 dpm for the 14C-Bet and 14C-DMSA assays, respectively. Activity values were obtained by subtracting blank values from those obtained with enzyme. Assays were done in duplicate, and duplicate values did not vary greater than 5%. In the DMSA-Asp assays, an additional control assay was performed that contained enzyme, DMSA, and βME (or other reducing agent), but which was devoid of Asp. The amount of BHMT protein used in all experiments was quantified by a Bradford assay using BSA as a standard. Bradford analysis of pure human BHMT has been previously correlated to BHMT concentration determined by total amino acid analysis [16].
Preparation of Zn-depleted apoenzyme and repletion with exogenous Zn
Wild type BHMT was stripped of its Zn by treatment with MMTS followed by exhaustive dialysis against buffer containing 10 mM ethylenediaminetetraacetic acid (EDTA). The Cys-specific modification was then removed by dialysis against buffer containing βME. The exact procedure used was identical to that previously described by Millian and Garrow [16]. In tubes containing the standard reaction cocktail (10 mM βME) and apoBHMT, varying amounts of Zn chloride were added such that the final concentration of metal in the assay tube was 0.05, 0.1, 0.25, 0.5, 1, or 2 mM. Following the addition of protein, activity was then immediately quantified by the standard Bet-Hcy assay (1 h). Kact for Zn2+ binding was calculated by a one-site binding hyperbola nonlinear regression analysis using kaleidagraph software (Sinergy Software, Reading, PA, USA).
Spectroscopic determination of protein-bound Zn
The total Zn content of BHMT was determined following the MMTS- (1 mM) or hydrogen peroxide- (1 mM) stimulated release of the metal using the procedure described by Zhou et al. [17]. In brief, the amount of Zn released following thiol modification was quantified by the increase in absorbance at 500 nm when Zn complexes with PAR. The concentration of the Zn-PAR complex was calculated using a molecular extinction coefficient of 66,000 Lmol−1cm−1. The amount of protein used in the various experiments ranged from 0.16 to 0.28 mg (3.5–6.2 nmol).
Free sulfhydryl groups on hBHMT:5C→A before and after treatment with βME
Following purification, enzyme was routinely dialyzed against 100 mM potassium phosphate buffer (pH 7.6) containing 2 mM Zn chloride and 10 mM βME to make certain the enzyme was replete with metal. To measure the number of free thiols found on the enzyme stored in the presence of reducing agent, excess Zn and βME were rapidly removed from stored enzyme by HiTrap™ gel filtration desalting chromatography using 100 mM phosphate as the exchange buffer. The number of free sulfhydryl groups on the enzyme was then determined using DTNB modification. The formation of thio-nitrobenzoate anions was continuously monitored by measuring the change in absorbance at 412 nm (ε412 =13 600 L mol−1 cm−1) over time (25°C). Then, to measure the number of free thiols on enzyme stored for short periods of time in the absence of a reducing agent, enzyme was left on ice over the course of 1 hour and the number of free sulhydryl groups was determined by DTNB modification as described above. In a third experiment, reducing agent-free enzyme was re-reduced by the incubation with βME for 15 min, which was rapidly removed by HiTrap™ desalting chromatography prior to the determination of the total number of free thiols. For all experiments protein concentrations ranged from 4 to 5 µM, and DTNB was in excess (1.5 mM).
Measurement of protein-free thiol content and BHMT activity in Gclm(+/+) and Gclm(−/−) mouse livers
The effect of glutathione status on liver BHMT was studied using the mouse Gclm (−/−) as a model. Frozen Gclm (+/+) and Gclm (−/−) liver samples were kindly donated by Dr. Timothy P. Dalton (University of Cincinnati Medical Center, Cincinnati, OH, USA). Livers were homogenized in 3 volumes of degassed 30 mM potassium phosphate buffer (pH 7.6) containing 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride and 0.1 mM benzamidine using a teflon-glass potter homogenizer. Crude liver extracts were obtained by centrifugation at 16,000×g (10 m) and these supernatants were the starting material for the immediate determination of oxidized glutathione and protein-free thiol content, and for the measurement of BHMT activity. Protein concentration of the crude extract and each protein-containing fraction thereafter was determined by the Bradford assay using bovine serum albumin as the standard.
The total protein-free thiol content of liver was measured by employing a DTNB modification protocol. In brief, proteins in crude extracts were precipitated by the addition of ethanol (70% v/v final) and removed by centrifugation (16000 g, 10 m). Sixty µL of supernatant was then added to 940 µL of potassium phosphate buffer (pH 7.5) containing DTNB. Final concentrations of buffer and DTNB were 100 mM and 1.5 mM, respectively. The formation of thio-nitrobenzoate was assayed after 5 minutes by measuring absorbance at 412 nm. The amount of free thiols in liver extracts was extrapolated from a standard curve (r2 = 0.99). The curve used D,L-Hcy as the standard (2.5 to 100 µM). Hcy was prepared fresh from the thiolactone as previously described [15], and like the samples, was brought to 70% (v/v) ethanol. BHMT activities in crude extracts were immediately measured using the Bet-Hcy and DMSA-Asp assays, as described above.
Statistical Analysis
The significance of the differences between Gclm(+/+) and Gclm(−/−) was determined using a two-tailed Student t-test. Statistical differences were considered significant when p<0.05.
Ability of different reducing agents to rescue the activity of oxidized BHMT
Wild type BHMT was made devoid reducing agent by dialysis against 100 mM potassium phosphate buffer (pH 7.6). Then, 300 µg of enzyme was added to DMSA-Asp reaction cocktails containing either 1 mM βME, dithiothreitol, tris(2-carboxyethyl)phosphine hydrochloride, reduced glutathione, γ-glutamylcysteine, cysteinylglycine or Cys. In case any reducing agent itself could be used as a methyl acceptor, control reactions were performed using the same concentration of enzyme, reducing agent and DMSA, but were devoid of Asp. These controls were in addition to the usual blanks that contained the complete reaction cocktail but no enzyme. In a second experiment, the effect of 0.05, 0.125, 0.25, 0.5, 1.0, 2.5 or 5.0 mM Cys on the DMSA-Asp dependent activity of BHMT was assessed.
RESULTS
BHMT methylates Asp using DMSA as the methyl donor
We had to find a non-thiol methyl acceptor for BHMT in order to determine whether this enzyme might be sensitive to the free thiol pool of the cell. We tested whether Asp, a structural homolog of Hcy, could function as methyl acceptor using either Bet or DMSA as the methyl donors. We had previously shown that DMSA, the sulfonium analog of Bet, conferred a 50-fold increase in the maximum catalytic rate of BHMT [15]. Therefore, we anticipated that this non-physiological methyl donor might improve the chances of BHMT performing an O-methylation of Asp, rather than the usual S-methylation of Hcy it catalyzes in vivo. To do this, wild type enzyme preparations were made reducing agent- free by desalting chromatography, and βME was either present or absent from the assay. The results obtained indicated that BHMT could methylate Asp when DMSA was used as the methyl donor, but only in the presence of the reducing agent βME (table 2). No activity toward Asp could be detected after prolonged incubation (6 h) with high levels of enzyme and 2 mM (0.1 μCi) Bet. Within experimental error, all of the enzyme activity lost upon removal of reducing agent could be rescued by the immediate addition of βME (table 2). In contrast, when enzyme was prepared reducing agent-free by dialysis over a 12 to 24 h period, only 70 to 85% of activity could be rescued upon the re-addition of βME (data not shown).
Table 2.
Effect of βME on Wild Type BHMT activity1
| Treatment | Ratio (Zn2+ : BHMT monomer) | βME (mM) | Specific Activity (nmol/h/mg of BHMT) | |
|---|---|---|---|---|
| Bet-Hcy | DMSA-Asp | |||
| Post-desalting | 1 : 1.03 | 5 | 2033 ± 76 | 25 ± 4.8 |
| Post-desalting | 1 : 0.98 | 0 | 1992 ± 43 | 0 |
| Re-addition of βME | 1 : 0.98 | 5 | 2094 ± 86 | 23.7 ± 2.9 |
Assays contained either 5 mM D,L-Hcy plus 2 mM Bet (0.1 µCi) or 5 mM L-Asp plus 1 mM DMSA (0.5 µCi). The Hcy-Bet assays (1 h) were performed with 0.67 nmol of BHMT, and the DMSA-Asp assays (6 h) were performed using 6.7 nmol of BHMT. Enzyme was made reducing agent-free by gel filtration chromatography using a HighTrap column. Zinc content was measured by detecting MMTS-stimulated zinc release.
The DMSA-Asp reaction was characterized further. The radioactive product (methylthioacetate) of the DMSA-Asp dependent BHMT-catalyzed reaction (which binds to the AG1 (OH−) resin) was not formed in the absence of enzyme. Additionally, the initial rate of product formation was directly dependent upon the duration of the assay and the amount of enzyme present and Asp (10 mM) inhibited BHMT activity (32%) when assayed using a reaction cocktail containing 250 µM Bet (0.1 μCi) and 100 µM Hcy, confirming that Asp has affinity for the Hcy binding site. Initial rate kinetic experiments were done as previously described [15] using subsaturating levels of DMSA (1 mM) to determine the characteristics of the DMSA-Asp methyltransferase activity. BHMT has an apparent Km toward Asp of approximately 10 mM, and an apparent Vmax of 493 nmol/hour/mg of protein. Combined, these results confirm that BHMT can perform the O-methylation of Asp, although the enzyme has very low affinity for this methyl donor and a greatly diminished catalytic efficiency.
BHMT retains its catalytic Zn during short-term exposure to reducing agent-free conditions
In order to determine if the loss of BHMT activity when enzyme was in buffer devoid of reducing agents was due to the loss of its catalytic Zn, we prepared Zn-deplete enzyme (apoBHMT) and quantified the amounts of Zn required to reactivate the enzyme. The data indicate that the reincorporation of Zn into apoBHMT was concentration-dependent, and it was necessary to add micromolar amounts exogenous Zn to fully reactivate nanomolar amounts of apoBHMT (figure 1A) (Kact = 356 ± 57 µM). In a separate experiment we preincubated (120 min.) an equal amount of apoenzyme with either 12.5 or 25 µM Zn and observed a small (~10%) increase in activity at both concentrations (not shown). These experiments indicate that the rapid and complete loss of DMSA-Asp dependent BHMT activity when enzyme is assayed in the absence of βME could not be due to the total loss of its catalytic Zn. That is, if nmolar amounts of protein were to release its nmolar amounts of Zn while in the absence of reducing agent, then, as extrapolated from the experimental results discussed above, when reducing agent is reintroduced, the very low amounts of Zn released into solution would not be able to efficiently reincorporate back into the enzyme fast enough over the time course of an assay to result in any measurable activity.
Figure 1.
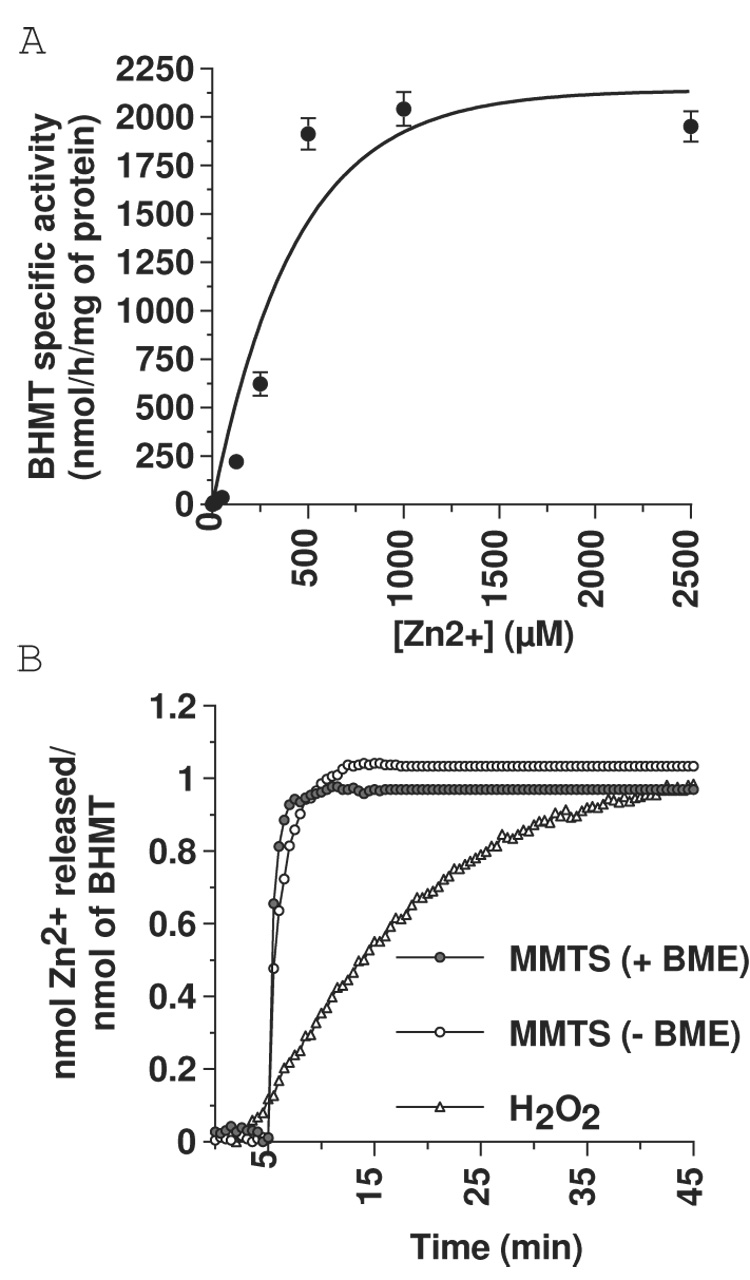
A) Repletion of apoBHMT with exogenous zinc (Zn). Filled circles represent specific activity of apoenzyme (0.97 nmol) after repletion with increasing concentrations of exogenous Zn chloride. Error bars represent mean ± SD. Triplicates were tested for each point. B) Stimulated release of Zn from WT BHMT by 1mM methylmethane thiosulfonate (white circles) or 1mM hydrogen peroxide (white triangles) in reducing agent-free buffer and stimulated release of Zn from WT BHMT in buffer containing reducing agents (grey circles). Stimulated release of zinc from the enzyme was measured using the Zn chelate, 4-(2-pryidylazo)resorcinol (PAR), as described in Experimental Procedures.
As a direct approach to determine whether enzyme in reducing agent-free buffer loses all activity because of a complete loss of its catalytic Zn, we measured total Zn bound to dialyzed BHMT. This was done by monitoring the change in absorbance at 500 nm as it was chelated by PAR following its release from the enzyme by treatment with two different sulfur modifying reagents, MMTS or H2O2. There was a rapid release of Zn from reducing agent free preparations of enzyme following treatment with MMTS or H2O2 (figure 1b). Zn was released from desalted protein at a 0.98 : 1 (zinc : BHMT monomer) ratio, and an almost identical 1.02 : 1 ratio was found when Zn was released from BHMT that had been maintained in the presence of reducing agents, indicating that even in the absence of a reducing agent, the enzyme was still nearly replete with Zn (figure 1b).
Long term exposure of BHMT to reducing agent-free conditions results in a progressive loss of Zn and a concomitant irreversible loss of activity
We then tested whether prolonged exposure of BHMT to reducing agent free buffer results in a slow loss of Zn and activity. We characterized the effects of long-term exposure to reducing agent free buffer on the ability of the enzyme to bind Zn. We tested also the level of activity that could be rescued upon the re-addition of βME. We rapidly removed βME from an enzyme preparation by gel filtration and we immediately measured the Zn content and DMSA-Asp and Bet-Hcy dependent activities of this reducing agent-free sample. These measurements were then continued for 7 days. The Bet-Hcy dependent activity and Zn content of the parent preparation, which was not subjected to gel filtration, were also measured. Between measurements enzyme was stored at 4 °C.
The enzyme that was stored in 10 mM βME retained its Zn and suffered no loss of activity over time (Figure 2). In contrast, enzyme stored in the absence of reducing agent lost Zn and activity. At no time point did the enzyme stored in reducing agent-free buffer have activity in the reducing agent-free DMSA-Asp assay (not shown), even when the enzyme was completely replete with Zn (t = 0).
Figure 2.

Time-dependent loss of BHMT activity when stored in the absence of a thiol reducing agent. Enzyme (0.67 nmol) was assayed using the Bet-Hcy substrate pair 100% activity corresponds to a specific activity of 2050 nmol Met/hour/mg of BHMT. The MMTS-stimulated release of Zn from enzyme (3.5 nmol) was measured using PAR, and the Zn:BHMT monomer ratio is given in parenthesis for each selected time point. Exact details are given in Experimental Procedures.
Loss and gain of DMSA-Asp activity is coincident with the reversible formation of a disulfide bond between two of the three thiolates that coordinate Zn
Site-directed mutagenesis was used to investigate whether the loss of the DMSA-Asp activity of BHMT when in the absence of a reducing agent was due to the oxidation of an essential thiol within the protein. When we individually mutated each of the 5 Cys residues not involved in Zn binding to Ala (Cys104, 131, 186, 201 and 256), we found that all of the resulting mutants were essentially as active as the wild type enzyme when in the presence of βME with the DMSA-Asp assay. Like the wild type enzyme, these mutants had no DMSA-Asp activity in the absence of βME. Moreover, another mutant protein was made that had all 5 of these Cys residues mutated to Ala (hBHMT:5C→A), and it too behaved like wild type enzyme in the presence and absence of βME. The specific activities of these mutants, as determined using the standard Bet-Hcy dependent BHMT assay, ranged from 84 to 95% of the wild type enzyme. These results indicated that the 5 Cys residues that are not involved in Zn binding are not essential residues for catalysis and they do not participate in the redox phenomenon reported here.
Using hBHMT:5C→A, we determined whether there was a change in the oxidation state of any of the three Cys residues involved in Zn binding (Cys217, 299 and 300) when the enzyme was subjected to a reducing agent-free environment. Using a DTNB modification protocol, it was determined that reduced enzyme had 2.7 modifiable thiols per monomer. Two thiols were rapidly modified and a third became modified more slowly (figure 3). When enzyme was first dialyzed against reducing agent free buffer, 0.98 nmol of thiol were modified per nmol of BHMT subunit indicating that only one Cys per BHMT molecule was modifiable. Upon addition of βME, the enzyme regained 3 nmol of modifiable thiol(ate) per nmol of BHMT (figure 3). These data indicate that two of the three thiolates that bind Zn can undergo reversible disulfide bond formation.
Figure 3.
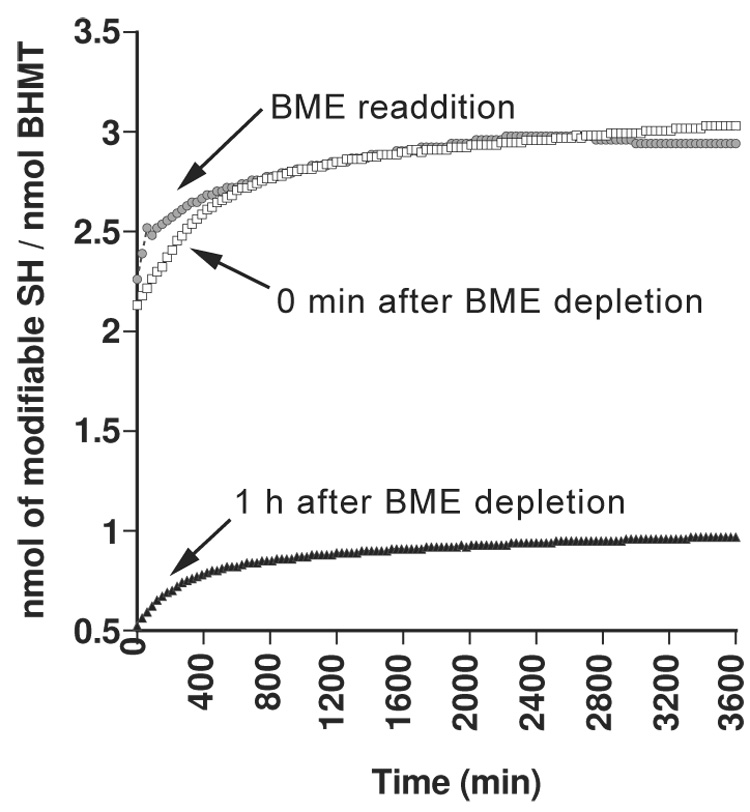
Modifiable sulfhydryl groups on reduced and oxidized BHMT:5C→A enzyme. The number of free sulfhydryl groups per nmol of purified protein was measured using 5’,5’-dithio-bis-(2-nitro)benzoic acid (DTNB). The BHMT solution was made devoid of reducing agent by desalting chromatography and the number of modifiable –SH groups were measured right after βME depletion (white squares), one hour after βME depletion (gray dots) and after re-addition of βME followed by desalting chromatography (black triangles).
BHMT activity is sensitive to the free thiol content of the liver
In order to study whether the redox sensitivity of BHMT observed in vitro was a physiological phenomenon, we assayed hepatic BHMT activity in Gclm (−/−) and Gclm (+/+) mice. Gclm (−/−) are genetically modified mice whose GCLM gene has been disrupted [18]. This gene encodes for a 38 kDa enzyme subunit that modifies the catalytic properties of the glutamate-Cys ligase [19]; [20], an enzyme critical for glutathione synthesis. The lack of this enzyme was recently reported to cause an 87% decrease in total liver glutathione levels, and also confers higher sensitivity of the fetal fibroblast derived from this mice to oxidative stress [18].
BHMT activity was measured in these livers using the DMSA-Asp assay. It is important to note that the reducing equivalents required to sustain the DMSA-Asp assay had to be supplied by the liver extracts, which were prepared in degassed reducing agent-free buffer. Compared to the Gclm (+/+) control mice, the Gclm (−/−) had a significant (p < 0.005), 87% reduction in total protein-free thiol content (table 3), consistent with earlier results [18]. Notably, the DMSA-Asp dependent BHMT activity was 75% lower in Gclm (−/−) than Gclm (+/+) mice (p < 0.005). Nevertheless, the Bet-Hcy dependent BHMT activity was essentially identical between groups (p<0.1), indicating that disruption of the GCLM protein, and thus glutathione synthesis, did not influence BHMT expression. Combined, these results suggest that the loss of DMSA-Asp dependent activity in Gclm (−/−) was due to the lower level of free thiols in those livers.
Table 3.
Measurement of total non-protein sulfhydryl groups and BHMT activity in the liver of GCLM knockout mice.
| Gclm(+/+) | Gclm(−/−) | |
|---|---|---|
| Total non-protein SH1 (nmol/mg of liver potein) | 118±25 | 8.1± 2.02 |
| Bet-Hcy activity1 (nmol /hour/mg BHMT) | 128± 5.8 | 113 ± 133 |
| DMSA-Asp activity1 (pmol /hour/mg BHMT) | 477±46 | 119 ± 242 |
Values are means ± SEM; n=3 for each (−/−) value and n=2 for each (+/+) value.
Significantly different from the (+/+) group (p < 0.005) using a two-tailed Student’s t-test.
Not significantly different from the (+/+) group (p<0.1) using a two tailed Student’s t-test
Cys, but not other physiological thiols can reduce oxidized BHMT
The mouse study discussed above support our in vitro experiments indicating that flux through BHMT might be sensitive to the thiol redox status the enzyme is exposed to in vivo. However, all of the in vitro experiments presented used βME as the reducing agent. To test the ability of other reductants to rescue the DMSA-Asp dependent activity of oxidized BHMT, including physiological thiols, we treated oxidized enzyme with βME, dithiothreitol, tris(2-carboxyethyl)phosphine hydrochloride, reduced glutathione, reduced cysteinylglycine, or reduced γ-glutamylcysteine. Each reducing agent was tested at 1 mM and only βME, dithiothreitol and Cys could rescue enzyme activity to any appreciable degree (figure 4). In experiments where Cys was varied from 25 µM to 5 mM, the activation was linear to 1 mM (figure 4, inset). Concentrations of Cys at 2.5 or 5 mM resulted in the precipitation of enzyme and complete loss of activity.
Figure 4.
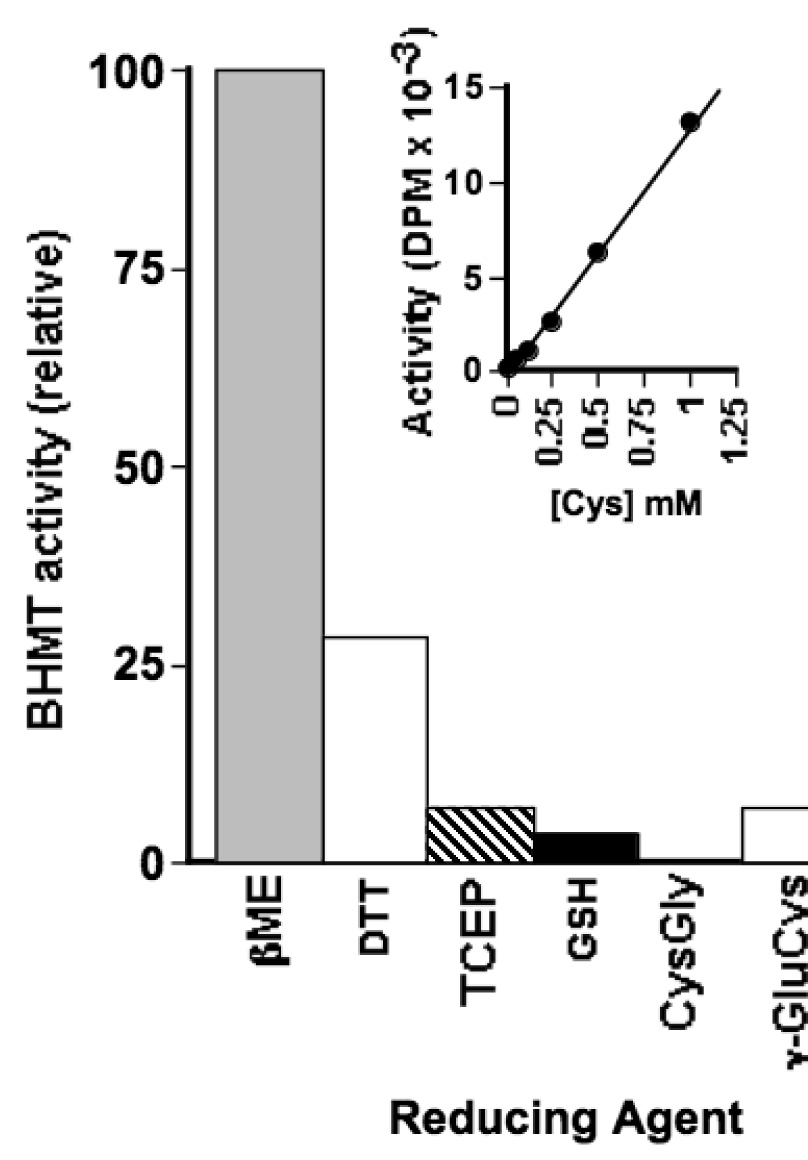
Relative ability of different thiol and non-thiol reducing agents to activate oxidized BHMT. Wild type enzyme (6.6 nmol) was assayed (6 h) using the DMSA-Asp substrate pair as described in the Experimental Procedures. Inset: Effect of Cys concentration on BHMT activity. Wild type enzyme (15 nmol) was assayed (6 h) using the DMSA-Asp substrate pair as described in the Experimental Procedures.
DISCUSSION
Using non-physiological and redox-inert substrates (DMSA and Asp) for BHMT, it was determined that this enzyme has an absolute requirement of a thiol as reducing agent for activity in vitro. When enzyme preparations were rapidly made reducing agent-free, two of the three thiolates that bind the catalytic Zn form a disulfide bond causing the complete loss of activity. The loss of activity following short-term exposure to reducing agent-free conditions does not result in a substantial loss of metal. When reducing conditions are rapidly restored, full activity is regained with the reduction of the disulfide bond at the Zn binding site, indicating that thiol redox fluctuations can switch BHMT activity on and off. Long term exposure of pure enzyme to reducing agent-free conditions results in the slow loss of its catalytic Zn. This treatment also causes irreversible loss of activity unless exogenous Zn as well as a thiol reducing agent are added to rescue function. When we measured BHMT activity in crude extracts of mouse livers genetically deficient in glutathionine using the DMSA-Asp substrate pair in reducing agent-free buffer, the level was about 75% lower than control mice with normal glutathione concentrations. These data show that fluctuations in the the major thiol redox buffer of the liver cell can affect BHMT function, but it does not indicate which reducing agent(s) in vivo can support BHMT function. Rather than a small molecule reductant, another explanation would be that BHMT may require an ancillary protein (e.g. thioredoxin) to keep its Zn-binding Cys residues reduced, and that the activity of this ancillary protein is inhibited in the Gclm−/− mice model. If such an ancillary protein were required, of course it would not have been present in our in vitro experiments, which used highly purified BHMT. It is then tempting to speculate, when reviewing our in vitro data, that BHMT is sensitive to the concentration of reduced Cys in the cell, the rate-limiting substrate for glutathione synthesis. We have shown that Cys is the best physiological thiol (other than Hcy, see Table 2) capable of keeping the active site of BHMT reduced, and therefore the enzyme competent to perform catalysis. It is important to note that normal Cys levels in rodent liver is between 100 and 200 micromolar, which is at least ten- to twentyfold higher than Hcy. Furthermore, BHMT does not methylate Cys (not shown), nor did 1 mM Cys inhibit the Bet-Hcy activity of the enzyme using our standard assay conditions (5 mM D,L-Hcy). In support of this work, earlier studies by Finkestein et al. showed that BHMT activity in crude rat liver extract (using only 20 µM Hcy) was stimulated (20%) by the addition of 50 µM Cys [10]. What is interesting is that their assay system used 1 mM DTT, which we show is not as good as βME at keeping the Zn-binding site reduced, but undoubtedly keeps the Hcy reduced in the assay buffer. Interestingly, 50 µM cystine in their assay system resulted in a 59% reduction of activity. Combined, the in vitro data suggest that the Cys-to-cystine ratio might directly influence the redox state of the active site of BHMT.
There is indirect evidence that supports the idea that changes in intracellular Cys concentrations can affect flux through the BHMT reaction. Finkelstein et al. showed an increase in BHMT activity in rat liver extracts isolated from animals that consumed diets supplemented with Cys compared to an unsupplemented control group [21], [22]. Like the study cited above, the assay for BHMT employed in these studies used 1 mM DTT, which we show here is not as good as βME at keeping the active site of BHMT reduced, and so the stimulation they observed could have been the result of higher levels of reduced Cys in the liver extracts of Cys-supplemented animals. Thus, intracellular concentrations of reduced Cys may act as a metabolic sensor that control the dithiolate-disulfide switch of BHMT and thereby regulate flux through the BHMT reaction.
A redox switch regulatory mechanism similar to the one found in BHMT has been described in the human mitochondrial branched chain aminotransferase. This enzyme has two Cys residues located at its active site. Under oxidative conditions a disulfide bond between this two Cys residues is formed while the enzyme becomes concomitantly inactivated. Similarly to what we found with BHMT, this inactivation can be reversed by re-addition of the reducing agent DTT [23]. A disulfide-dithiol switch also modulates the activity of inducible nitric oxide synthase (iNOS). Within the Zn binding motif of the iNOS a reversible intramolecular disulfide can be formed that inactivates the protein by avoiding its ligand-induced dimerization [24].
If the formation of a disulfide bond between two Zn binding thiolates is responsible for the loss of BHMT activity when the enzyme is in reducing agent free buffer, and yet Zn remains bound to the enzyme, what are the potential mechanisms that could explain this behavior? One hypothesis is that disulfide formation causes small structural changes in the catalytic site affecting the efficiency or oblating Hcy binding, or subsequent conformational changes required to generate the Bet binding site. Another hypothesis, which is not mutually exclusive to a microstructural change, is that the formation of the disulfide bond, regardless of how Zn remains precisely coordinated to the enzyme, results in a change in the electronics at the Zn catalytic center. As it has been proposed by Matthews [25], the net –2 charge of the Zn-tetrathiolate complex improves the reactivity of the Hcy thiolate by facilitating its dissociation from the Zn, a required step for the subsequent nucleophilic attack of the methyl donor. Thus, a reduction in the net negative charge of this complex would greatly diminish the capacity of the Hcy thiolate to dissociate, which in turn would either significantly reduce or abolish methyltransferase activity. Indeed, the substitution of Cys217 for a ligand of neutral charge in BHMT (Cys217His) resulted in an enzyme that could bind Zn although it had no detectable activity [14].
As indicated above, during very short term exposure (< 1 h) of BHMT to reducing agent-free conditions the enzyme loses negligible Zn. However, continued incubation of the enzyme in these conditions results in the slow loss of Zn from the protein (figure 3). When reducing conditions are restored by the re-addition of βME, the amount of enzyme activity that can be rescued roughly correlates to the amount of enzyme that has retained Zn. The longer oxidized, Zn-replete enzyme goes without reduction, the loss of its metal becomes more likely. One possible explanation of how Zn is lost from the active site after long periods of time in the absence of reducing agents would be that a specific disulfide arrangement triggers Zn expulsion. In support of this interpretation, the crystal structure of BHMT in the oxidized and reduced states shows that the structure of BHMT obtained from crystals grown in the absence of reducing agent was devoid of Zn and a disulfide bond was present between residues 217 and 299 [12]. This observation is at odds with our short term studies with BHMT in the absence of reducing agent where a disulfide is formed, but the enzyme retains Zn for a considerable period of time. One possibility that could explain these divergent observations is that the crystal structure only revealed the most thermodynamically stable disulfide between at least two (217–299 or 217–300), or perhaps three (299–300) possibilities. It could be that the 217–299 disulfide arrangement may not be the kinetically preferred one formed when enzyme is first exposed to reducing agent free conditions, but only after longer periods of time without reduction is there a shift to the more thermodynamically stable disulfide (217–299), which is accompanied by the concomitant expulsion of Zn.
In summary, we propose that a thiol redox switch may regulate BHMT activity in vivo. The enzyme becomes inactive when two of its three Zn-binding thiolates participate in the formation of a disulfide bond. At least one conformation of the oxidized enzyme can retain Zn for a considerable period of time, but there is a loss of activity due to changes in microstructure around the Zn site and/or a decrease in the negative charge at the Zn center. The loss of activity by autoxidation can be rescued upon the successful reduction of the disulfide bond, a process that regenerates the native coordination of the metal to the enzyme. We present in vitro evidence that suggests that the dithiolate-disulfide switch may be controlled, at least in part, by the level of reduced Cys in the cell. Previous studies have demonstrated that under oxidative stress conditions liver sulfur amino acid metabolism is shunted toward Cys biosynthesis as a mechanism to increase the levels of glutathione and protect cells from oxidative damage [1], [7]. Inactivation of BHMT leads homocysteine metabolism in the same direction, increasing the redox buffering capacity of liver cells, which in the process re-establishes normal reduced Cys levels leading to the autocorrection of normal flux through BHMT. The work reported herein on BHMT, in combination with previous reports [1], [2], [4; 5] on other enzymes strongly implicate redox regulation at the Hcy locus of metabolism.
ACKNOWLEDGEMENTS
We thank Dr. Timothy P. Dalton for providing with the Gclm−/− and Gclm+/+ mice livers. We thank Drs. Sandra Szegedi and Andew P. Breksa for many helpful discussions about this work. This material is based upon work supported by NIDDK under Award No. DK52501 and the Illinois Agricultureal Experiment Station (Project # ILLU-698-352).
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R. The Yin-Yang of cobalamin biochemistry. Chem Biol. 1997;4:175–186. doi: 10.1016/s1074-5521(97)90286-6. [DOI] [PubMed] [Google Scholar]
- 3.Ludwig ML, Matthews RG. Structure-based perspectives on B12-dependent enzymes. Annu Rev Biochem. 1997;66:269–313. doi: 10.1146/annurev.biochem.66.1.269. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Gongora E, Ruiz F, Mingorance J, An W, Corrales FJ, Mato JM. Interaction of liver methionine adenosyltransferase with hydroxyl radical. Faseb J. 1997;11:1013–1019. doi: 10.1096/fasebj.11.12.9337154. [DOI] [PubMed] [Google Scholar]
- 5.Avila MA, Mingorance J, Martinez-Chantar ML, Casado M, Martin-Sanz P, Bosca L, Mato JM. Regulation of rat liver S-adenosylmethionine synthetase during septic shock: role of nitric oxide. Hepatology. 1997;25:391–396. doi: 10.1002/hep.510250222. [DOI] [PubMed] [Google Scholar]
- 6.Vitvitsky V, Mosharov E, Tritt M, Ataullakhanov F, Banerjee R. Redox regulation of homocysteine-dependent glutathione synthesis. Redox Rep. 2003;8:57–63. doi: 10.1179/135100003125001260. [DOI] [PubMed] [Google Scholar]
- 7.Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R. Evidence for heme-mediated redox regulation of human cystathionine beta- synthase activity. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 8.Sunden SL, Renduchintala MS, Park EI, Miklasz SD, Garrow TA. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch Biochem Biophys. 1997;345:171–174. doi: 10.1006/abbi.1997.0246. [DOI] [PubMed] [Google Scholar]
- 9.Finkelstein JD, Martin JJ. Methionine metabolism in mammals. Distribution of homocysteine between competing pathways. J Biol Chem. 1984;259:9508–9513. [PubMed] [Google Scholar]
- 10.Finkelstein JD, Harris BJ, Kyle WE. Methionine metabolism in mammals: kinetic study of betaine-homocysteine methyltransferase. Arch Biochem Biophys. 1972;153:320–324. doi: 10.1016/0003-9861(72)90451-1. [DOI] [PubMed] [Google Scholar]
- 11.Castro C, Gratson AA, Evans JC, Jiracek J, Collinsova M, Ludwig ML, Garrow TA. Dissecting the catalytic mechanism of betaine-homocysteine S-methyltransferase by use of intrinsic tryptophan fluorescence and site-directed mutagenesis. Biochemistry. 2004;43:5341–5351. doi: 10.1021/bi049821x. [DOI] [PubMed] [Google Scholar]
- 12.Evans J, Huddler D, Jiracek J, Castro C, Millian N, Garrow T, Ludwig M. Betaine-homocysteine methyltransferase. Zinc in a distorted barrel. Structure (Camb) 2002;10:1159. doi: 10.1016/s0969-2126(02)00796-7. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez B, Pajares MA, Martinez-Ripoll M, Blundell TL, Sanz-Aparicio J. Crystal structure of rat liver betaine homocysteine s-methyltransferase reveals new oligomerization features and conformational changes upon substrate binding. J Mol Biol. 2004;338:771–782. doi: 10.1016/j.jmb.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Breksa AP, 3rd, Garrow TA. Recombinant human liver betaine-homocysteine S-methyltransferase: identification of three cysteine residues critical for zinc binding. Biochemistry. 1999;38:13991–13998. doi: 10.1021/bi991003v. [DOI] [PubMed] [Google Scholar]
- 15.Garrow TA. Purification, kinetic properties, and cDNA cloning of mammalian betaine- homocysteine methyltransferase. J Biol Chem. 1996;271:22831–22838. doi: 10.1074/jbc.271.37.22831. [DOI] [PubMed] [Google Scholar]
- 16.Millian NS, Garrow TA. Human betaine-homocysteine methyltransferase is a zinc metalloenzyme. Arch Biochem Biophys. 1998;356:93–98. doi: 10.1006/abbi.1998.0757. [DOI] [PubMed] [Google Scholar]
- 17.Zhou ZS, Peariso K, Penner-Hahn JE, Matthews RG. Identification of the zinc ligands in cobalamin-independent methionine synthase (MetE) from Escherichia coli. Biochemistry. 1999;38:15915–15926. doi: 10.1021/bi992062b. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Dieter MZ, Chen Y, Shertzer HG, Nebert DW, Dalton TP. Initial characterization of the glutamate-cysteine ligase modifier subunit Gclm(−/−) knockout mouse: novel model system for a severely compromised oxidative stress response. J Biol Chem. 2002;15:15. doi: 10.1074/jbc.M209372200. [DOI] [PubMed] [Google Scholar]
- 19.Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 20.Misra I, Griffith OW. Expression and purification of human gamma-glutamylcysteine synthetase. Protein Expr Purif. 1998;13:268–276. doi: 10.1006/prep.1998.0897. [DOI] [PubMed] [Google Scholar]
- 21.Finkelstein JD, Martin JJ, Harris BJ. Effect of dietary cystine on methionine metabolism in rat liver. J Nutr. 1986;116:985–990. doi: 10.1093/jn/116.6.985. [DOI] [PubMed] [Google Scholar]
- 22.Finkelstein JD, Martin JJ, Harris BJ. Methionine metabolism in mammals. The methionine-sparing effect of cystine. J Biol Chem. 1988;263:11750–11754. [PubMed] [Google Scholar]
- 23.Conway ME, Yennawar N, Wallin R, Poole LB, Hutson SM. Identification of a peroxide-sensitive redox switch at the CXXC motif in the human mitochondrial branched chain aminotransferase. Biochemistry. 2002;41:9070–9078. doi: 10.1021/bi020200i. [DOI] [PubMed] [Google Scholar]
- 24.Li D, Hayden EY, Panda K, Stuehr DJ, Deng H, Rousseau DL, Yeh SR. Regulation of the monomer-dimer equilibrium in inducible nitric-oxide synthase by nitric oxide. J Biol Chem. 2006;281:8197–8204. doi: 10.1074/jbc.M507328200. [DOI] [PubMed] [Google Scholar]
- 25.Matthews RG, Goulding CW. Enzyme-catalyzed methyl transfers to thiols: the role of zinc. Curr Opin Chem Biol. 1997;1:332–329. doi: 10.1016/s1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]