

Abstract
Although the breast cancer susceptibility gene 1 (BRCA1) protein is predominantly nuclear, its localization can vary during the cell cycle in response to cellular insults. For example, in S-phase cells, BRCA1 forms subnuclear foci and localizes to the perinuclear region in response to DNA damage. The present study provides evidence that BRCA1 is transiently excluded from the nucleus during the early part of S phase in the absence of DNA damage. The percentage of MCF-7 human breast cancer cells predominantly expressing nonnuclear BRCA1 significantly correlates with the percentage of cells within early S phase. This redistribution of BRCA1 is partially sensitive to leptomycin B, indicating that CRM-1-mediated nuclear export is involved. Similar results were observed with MCF12A nonmalignant human mammary cells. The abilities of BAPTA-AM, an intracellular calcium chelator, to inhibit the change in BRCA1 localization, and of A23187, a calcium ionophore, and of thapsigargin to mimic nuclear exclusion of BRCA1, provide evidence for the involvement of calcium in this process. The calcium-mediated change in BRCA1 localization occurs in several cell lines, indicating that this effect is not cell line specific. BRCA2 localization is not affected by A23187. Furthermore, inhibition of calcium-calmodulin interaction and calcium-calmodulin dependent protein kinase II attenuates the calcium-mediated change in BRCA1 localization. These data suggest that BRCA1 nuclear export can be cell cycle-regulated by a calcium-dependent mechanism.
Keywords: BRCA1, nuclear export, leptomycin B, calcium, cell cycle, A23187, BAPTA-AM
1. Introduction
The breast cancer susceptibility gene 1 (BRCA1) encodes a tumor suppressor protein that functions in various cellular processes. These functions include regulation of centrosome duplication [1], acting as an E3 ubiquitin ligase [2], regulating cell cycle progression [3], repair of DNA damage [4] and serving as a transcriptional co-activator [5]. BRCA1 induces the expression of p21Waf1/Cip1 [3] and GADD45 [6] genes, which encode proteins involved in cell cycle control. This extant evidence is consistent with the interpretation that the ability of BRCA1 to regulate the cell cycle is based on its role as a transcription factor.
Both the expression and localization of BRCA1 can be cell cycle regulated. BRCA1 expression is detectable at the mid G1 phase of the cell cycle, increases to its maximal level during S phase, and remains elevated through M phase [7, 8]. During M phase, BRCA1 is found associated with γ-tubulin in centrosomes [9]. During S phase, BRCA1 localizes to subnuclear foci in a phosphorylation-dependent, DNA damage-responsive manner [10, 11]. In addition to forming nuclear dots, serine-988-phosphorylated BRCA1 relocalizes to a perinuclear region in S phase cells in response to ionizing or ultraviolet radiation [12]. Thus, the relative distribution of BRCA1 in the nucleus and cytoplasm appears to vary with the cell cycle.
The nuclear-cytoplasmic distribution of BRCA1 in a cell is determined by its nuclear import and export dynamics. BRCA1 has two nuclear localization sequences located between amino acid residues 503-508 and 606-615 [13]. These import sequences bind importin α, which subsequently binds to importin β. The complex is then actively imported into the nucleus and BRCA1 is released from the complex. BRCA1 also has two leucine-rich nuclear export sequences at residues 81-99 [14] and 22-30 [15]. The export sequences bind to the nuclear export receptor CRM-1 to actively export BRCA1 from the nucleus. The nuclear export of BRCA1 is sensitive to the antifungal agent, leptomycin B, which is an inhibitor of CRM-1. The presence of both nuclear import and nuclear export sequences within BRCA1 suggests that BRCA1 is capable of undergoing regulated nuclear-cytoplasmic shuttling. However, factors involved in modulating either the nuclear import or export of BRCA1 have not been reported and the only study to date that provides data consistent with regulated changes in BRCA1 localization was performed in cells which had been exposed to DNA damaging agents.
Progression of the mammalian cell cycle is dependent on calcium signaling. Calcium transients occur during early G1, at the G1/S boundary, and during M phase and are dependent on the presence of extracellular and intracellular stores of calcium [16]. Cells in early G1 and near the G1/S transition are most sensitive to extracellular calcium, whereas cells at any point between G1 and S will exhibit a G0 state when intracellular calcium stores are depleted. In addition to regulating the cell cycle, calcium signaling also can regulate the nuclear transport of proteins. For example, increases in intracellular calcium result in translocation of histone deacetylase 5 (HDAC5) from the nucleus to the cytoplasm in cultured hippocampal neurons [17]. In contrast, increases in intracellular calcium result in nuclear accumulation of human transducer of regulated CREB (TORC) proteins [18] and nuclear factor of activated T-cells (NF-AT) [19]. Thus, a role for calcium in regulating the nuclear import and export dynamics of proteins has been established.
Because BRCA1 undergoes nuclear import and leptomycin B-sensitive nuclear export and its localization can be regulated during cell cycle progression, we investigated the role of calcium in the cell cycle-dependent localization of BRCA1. Here, we report that BRCA1 transiently localizes to a nonnuclear region during early S phase and this translocation, which is leptomycin B-sensitive, is dependent on calcium. This is the first report of cell cycle-dependent changes in BRCA1 nuclear-cytoplasmic distribution in the absence of DNA damaging agents and of the requirement for calcium in modulating the subcellular localization of BRCA1.
2. Materials and Methods
2.1 Cell culture
MCF-7 and T47D human breast cancer cells, MCF-12A nonmalignant human mammary epithelial cells, HeLa human cervical cancer cells were purchased from American Type Culture Collection (Manassas, VA, USA). MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen; Carlsbad, CA, USA) supplemented with 10% Fetal Clone III (Hyclone; Logan, UT, USA), 2% L-glutamine, 1% antibiotic-antimycotic (Invitrogen), non-essential amino acids, and insulin-transferrin-selenium-A (Invitrogen). MCF-12A cells were cultured in a 1:1 mixture of Dulbecco’s modified Eagle’s and Ham’s F12 media (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/ml human epidermal growth factor (Invitrogen), 100 ng/ml cholera toxin (Sigma; St. Louis, MO, USA), 0.01 mg/ml bovine insulin (Sigma) and 500 ng/ml hydrocortisone (Sigma). T47D (20), human mammary epithelial cells (kindly provided by Dr. Sakina Eltom; 21) and HeLa (15) cells were cultured as previously described. Cells were maintained at 37°C in 5% CO2/95% air.
2.2 Cell synchronization
To synchronize MCF-7 cells in G0/G1, cells were treated with 2.5 mM thymidine (Sigma) for 24 hours, released into fresh media for 12 hours and then treated with 400 μM mimosine (Sigma) for 12 hours. MCF-12A cells were treated with 0.5 mM mimosine for 24 hours. Cells were released from cell cycle arrest and harvested at times indicated in figures.
2.3 Fluorescence-activated cell sorting
Cells were plated at 2 × 106 cells per 100 mm dish, synchronized and harvested in cold phosphate-buffered saline (PBS) at indicated times. 1.5 × 106 cells/sample were fixed in 75% cold ethanol for 30 minutes. Cells were underlaid with serum and centrifuged at 2000 × g for 5 minutes at 4°C, washed in PBS and treated with 100 μg/ml RNase A (Sigma) for 15 minutes at 37°C. DNA was stained with 10 μg/ml propidium iodide (Sigma). The stained cells were analyzed by fluorescence-activated cell sorting (FACS) using a Becton-Dickinson FACScan Benchtop Analyzer. Collected data were analyzed with ModFit software (Verity Software; TopSham, ME, USA).
2.4 Immunocytochemistry and fluorescence microscopy
Cells were plated onto coverslips at 4 × 104/well in 6-well culture dishes, synchronized and MCF-7 cells were released from cell cycle arrest for 0, 4, 8, and 12 hours before harvesting. MCF-12A cells were released from cell cycle arrest for 0 and 3 hours. Cells were fixed in 3:1 ethanol: acetic acid or 2% paraformaldehyde and incubated with 1:100 dilution of monoclonal anti-BRCA1 IgG (Ab-1; Calbiochem; San Diego, CA, USA) and counterstained with Cy-3 conjugated donkey anti-mouse IgG (1:500; Jackson Immunoresearch Laboratories; West Grove, PA, USA), with 1:200 dilution of polyclonal anti-BRCA1 (AbC; BD Biosciences; San Jose, CA, USA) and counterstained with Cy-3 conjugated donkey anti-mouse IgG (1:500; Jackson Immunoresearch Laboratories) or with 1:50 anti-BRCA2 (Santa Cruz Biotechnology; Santa Cruz, CA, USA) and counterstained with FITC-conjugated donkey anti-goat IgG (1:250; Jackson Immunoresearch Laboratories), DNA was stained with 4’,6-diamidino-2-phenylindole (DAPI; Sigma). Coverslips were mounted in Aqua Polymount (PolySciences, Inc; Warrington, PA, USA) and cells visualized on a Leica DMR-B or Olympus BX-41 fluorescent microscope.
2.5 shRNA studies
Control shRNA or BRCA1-targeted shRNA plasmids (SuperArray Biosciences; Frederick, MD, USA) were transfected into MCF-7 cells using GeneJammer (Stratagene: Cedar Creek, TX, USA). Twenty-four hours post transfection, cells were synchronized by thymidine-mimosine block and released into the cell cycle for 4 hours. Cells were probed for BRCA1 with Ab-1 as described above.
2.6 DNA constructs and transfections
pEGFPC1-BRCA1 has been previously described [15]. pEGFPC1BRCA1 nes-, in which leucine 28, phenylalanine 93 and leucine 95 were changed to alanines, was generated as described [15] using pEGFPC1BRCA1 L28A as template.
2.7 Inhibitors and agonists
A23187 and W-7 were purchased from EMD Chemicals (Gibbstown, NJ, USA). Thapsigargin and BAPTA were obtained from Invitrogen. KN-62 and leptomycin B were from Sigma.
2.8 Statistical analysis
Statistical analyses were performed using GraphPad Prism version 4 for Windows (GraphPad Software; San Diego, CA, USA).
3. Results
3.1 Nonnuclear localization of BRCA1 correlates with early S phase of the cell cycle
Because the nuclear import and export of BRCA1 in MCF-7 cells has been previously described [13, 14], we chose these cells to evaluate the cell cycle effect on BRCA1 localization. To do so, we synchronized MCF-7 cells in G0/G1 using a thymidine-mimosine block. This treatment resulted in ~76% of the cells arrested in G0/G1, based on FACS analysis (Fig. 1A; 0 hr). Upon release of cells from this block by the removal of mimosine, cells progressed into S phase within 4 hours resulting in ~ 68% of cells in S phase. Eight hours post release, an equal percentage of cells remained in S phase. However, division of S phase into five equivalent compartments and analysis of these data with ModFit software (Verity Software) demonstrate that at 4 hours, the cells were in the early part of S (the first two channels of a 5-channel ModFIT analysis). At 8 hours, the cells had progressed to later stages of S and by 12 hours, cells had progressed into G2/M with 33% remaining in S phase.
Fig. 1. BRCA1 transiently localizes outside the nucleus during cell cycle progression.

(A) MCF-7 cells were synchronized in G0/G1 with a thymidine-mimosine block and harvested for FACS analysis at 0, 1, 2, 4, 8, and 12 hours after release from cell cycle arrest. Histograms shown are representative of 3 experiments. Numbers are the mean ± s.e. of percentage of cells in each phase of the cell cycle (n=3). (B) Immunocytochemical analysis of BRCA1 in MCF-7 cells synchronized in G0/G1 and released for 0, 4, and 8 hours. Cells were fixed in ethanol: acetic acid. BRCA1 was detected using antibody Ab-1, which was generated against amino acid residues 1-304. Nuclei were detected by staining DNA with DAPI. Magnification, 400x. (C) Graphical representation of the percentage of MCF-7 cells exhibiting predominantly nonnuclear immunostaining of BRCA1 from immunocytochemical studies described in B. (mean ± s.e.; n=3 experiments in which at least 200 cells/time point were evaluated; *, p<0.05). (D) Immunocytochemical analysis of MCF-7 cells transfected with negative control shRNA or BRCA1-targeted shRNA. Transfected cells were synchronized by thymidine-mimosine block and released into the cell cycle for 4 hours. BRCA1 was detected using Ab-1 as described in (B). Magnification, 400x. Transfected cells were evaluated for fluorescence intensity of BRCA1. Graph depicts the average fluorescence intensity. (mean ± s.e. of cells in 6 fields, *** p< 0.001) (E) Immunocytochemical analysis of BRCA1 as described in (B) except that cells were fixed in 2% paraformaldehyde. Magnification, 400x.
To assess the intracellular distribution of BRCA1 at times when the majority of cells were either in G0/G1, early S, or late S, immunocytochemistry was performed on cells at 0, 4, and 8 hours post mimosine removal. Immediately after removal of mimosine (0 hr), the majority of cells (72%) displayed both a nuclear and cytoplasmic distribution of BRCA1 (Fig. 1B and C); the remaining 28% of cells displayed a predominantly nonnuclear localization pattern for BRCA1. In contrast, 4 hours after removal of the G0/G1 block, the percentage of cells exhibiting a nonnuclear accumulation of BRCA1 increased to 63%. The percentage of cells with a nonnuclear BRCA1 staining pattern decreased by 8 hours and the majority of the cells (70%) displayed a nuclear and cytoplasmic distribution of BRCA1 with only 30% of the cells displaying a predominantly nonnuclear accumulation. At 12 hours, a staining pattern similar to that at 8 hours was observed with 23% of cells exhibiting nonnuclear accumulation of BRCA1 (data not shown).
To engender confidence that the detected protein undergoing the localization changes was BRCA1, several additional experiments were performed. First, BRCA1-targeted shRNA or control shRNA was transiently transfected into MCF-7 cells prior to synchronization, fixation and immunofluorescence (Fig. 1D). The introduction of BRCA1 shRNA resulted in a significant reduction in fluorescence intensity of BRCA1 when the cells were probed with BRCA1 antibody Ab-1 relative to that in cells transfected with control shRNA. This indicated that the signal was specific for BRCA1.
Second, to ensure that the results were not an artifact due to fixation conditions, immunocytochemistry of BRCA1, using Ab-1, was repeated in MCF-7 cells fixed in 2% paraformaldehyde (Fig. 1E). Similar to the results obtained in Figure 1B, cells at the 4-hour time point exhibited a predominantly nonnuclear distribution of BRCA1. By 8 hours, BRCA1 was again accumulating in the nucleus. These results indicate that the fixation conditions were not altering BRCA1 localization.
Thirdly, immunocytochemistry of MCF-7 cells was repeated using a second BRCA1 antibody, AbC. This antibody was generated against amino acid residues 768-793 in exon 11 of BRCA1 and has been reported to provide a strong signal with little background in immunocytochemistry [22]. Similar to the results obtained with Ab-1, which was generated against amino acids 1-304 of BRCA1, the detected protein localized predominantly to the nucleus when cells were arrested at G0/G1 (Fig 2; 0 hour). Four hours post-release from cell cycle arrest, a time when cells were in early S phase, the antibody detected a protein that was mostly outside the nucleus. By eight hours, the protein again was detected in the nucleus. Thus, both antibodies generated against BRCA1 result in similar localization changes. Collectively, these data support a cell cycle-dependent change in localization of BRCA1 during early S phase.
Fig 2. Nonnuclear BRCA1 is detected with an antibody generated against amino acid residues 768-793.

Immunocytochemical analysis of BRCA1 in MCF-7 cells as described in Fig. 1B, except an antibody generated against amino acids 768-793 (AbC) of BRCA1 was used. Magnification, 400x.
To further evaluate whether a relationship exists between cell cycle progression into S phase and BRCA1 localization, we compared the percentage of cells in early S phase with the percentage of cells with predominantly nonnuclear BRCA1 staining as a function of time following release from G0/G1 block (Fig. 3). Changes in the two parameters paralleled each other at 4, 8, and 12 hours. Calculation of a Pierson correlation coefficient from the data points revealed a value for r2 of 0.9683 (p = 0.016), indicative of a significant correlation between percentage of cells in early S phase and percentage of cells with nonnuclear BRCA1.
Fig. 3. Nonnuclear accumulation of BRCA1 correlates with early S phase.
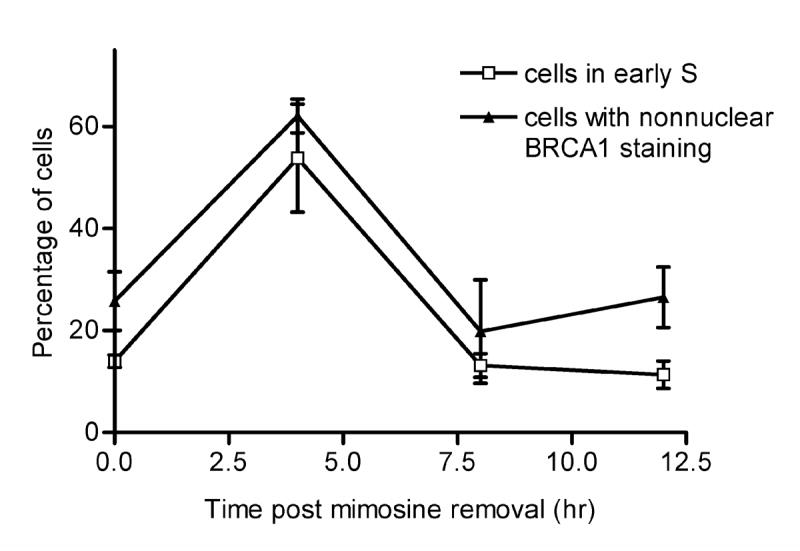
Time course (0, 4, 8, and 12 hours) of the percentage of MCF-7 cells with nonnuclear BRCA1 and of the percentage of MCF-7 cells in early S phase (compartments 1 and 2 of a 5-channel ModFIT analysis of FACS data) after removal of mimosine. Each point is the mean ± s.e. of three experiments. Data for the nonnuclear accumulation of BRCA1 and for the percentage of cells in early S phase were obtained from independent experiments.
3.2 Nonnuclear localization of BRCA1 is blocked by leptomycin B
To determine whether cell cycle-dependent changes in BRCA1 distribution are dependent on CRM-1-mediated nuclear export, we tested the effect of leptomycin B, an inhibitor of CRM-1, on the early S phase accumulation of BRCA1 outside the nucleus. Synchronized MCF-7 cells were released from cell cycle arrest in the absence or presence of 10 ng/ml leptomycin B. Immunofluorescence analysis of BRCA1 expression showed that the decrease in the nuclear BRCA1 observed in cells four hours after release from cell cycle arrest was less evident in cells treated with leptomycin B (Fig. 4). These results suggest that the increase in nonnuclear BRCA1 is dependent, at least in part, on CRM-1-mediated nuclear export.
Fig. 4. Nonnuclear localization of BRCA1 is partially blocked by leptomycin B.
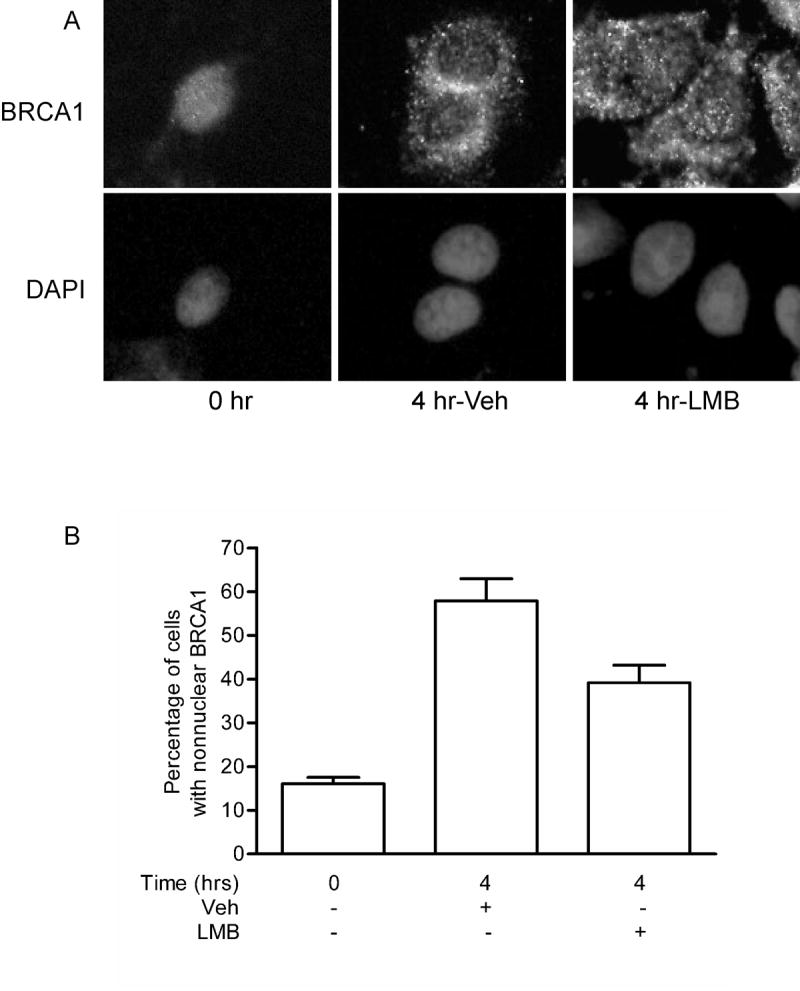
(A) MCF-7 cells synchronized by treatment with thymidine and mimosine were released from cell cycle arrest in the presence or absence of 10 ng/ml leptomycin B. Cells were fixed in ethanol:acetic acid and BRCA1 content was probed with Ab-1. Nuclei were detected by staining DNA content with DAPI. Magnification, 400x. (B) Graphical representation of the percentage of cells with predominantly nonnuclear BRCA1. (mean ± s.d.; at least 10 fields per treatment were analyzed).
3.3 Cell cycle-dependent changes in BRCA1 localization occur in noncancerous mammary epithelial cells
We next determined whether the early S phase change in BRCA1 localization also occurred in noncancerous cells. To evaluate the change in BRCA1 localization, MCF-12A nonmalignant human mammary epithelial cells were treated with 0.5 mM mimosine for 24 hours, which resulted in 68% of cells arrested in G0/G1 phase of the cell cycle (Fig. 5A). Three hours after release from the mimosine block, 52% of cells had progressed into the S phase and the majority of these cells were in earlier stages of S phase (Fig. 5A). Similar to MCF-7 cells, immunocytochemical analysis of BRCA1 staining in MCF-12A cells showed a change in the distribution of BRCA1 from diffusely nuclear and nonnuclear immediately after removal of mimosine (0 hr) to predominantly nonnuclear 3 hours after release from mimosine block (Fig. 5B and C). This shift was limited in cells treated with leptomycin B, which affirms the importance of nuclear export in the S phase nuclear exclusion of BRCA1. Furthermore, these data demonstrate that the early S phase redistribution of BRCA1 between the nucleus and cytosol is not specific to malignant cells.
Fig. 5. Noncancerous mammary epithelial cells exhibit cell cycle-dependent change in BRCA1 localization.
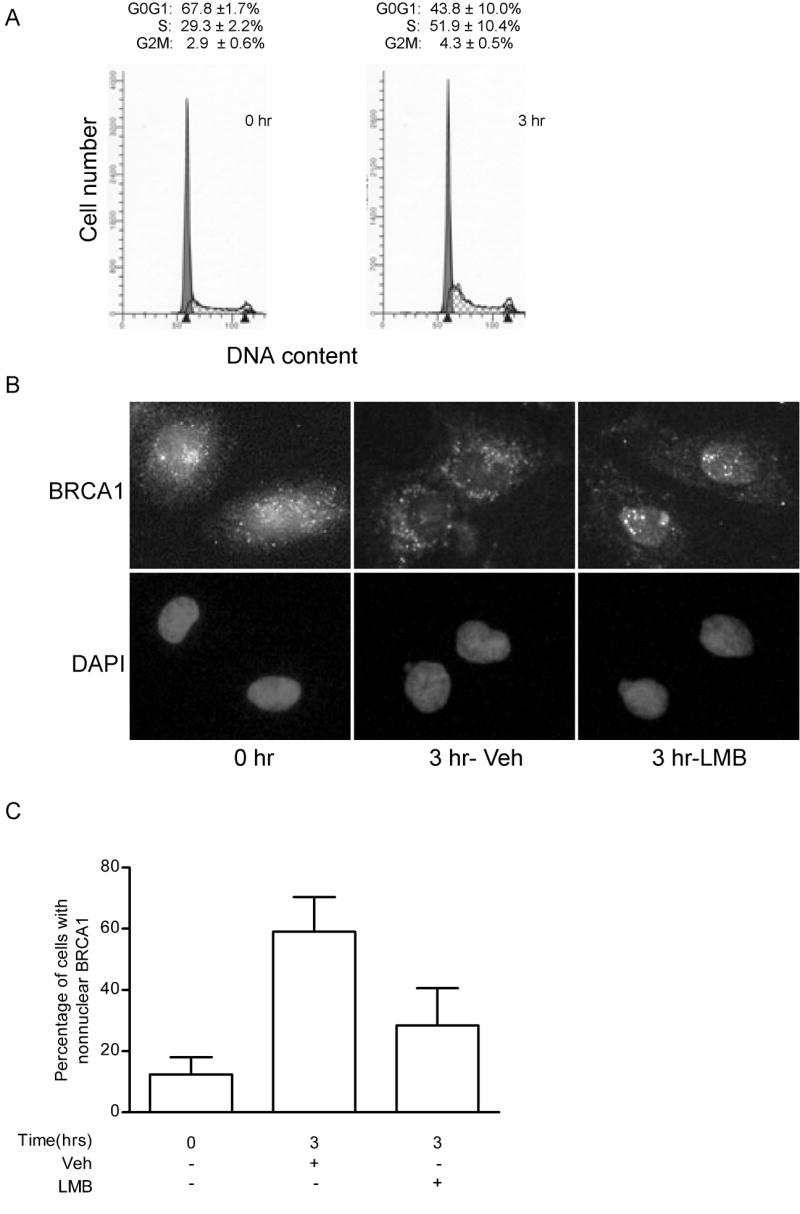
MCF-12A cells were enriched for G0/G1 by treatment with 0.5 mM mimosine for 24 hours and released for 3 hours. (A) Representative histograms of the cell cycle profile in MCF-12A cells. Numbers are the mean ± s.e. of percentage of cells in each phase of the cell cycle (n=3). (B) Immunocytochemical analysis of BRCA1 in MCF-12A cells synchronized in G0/G1 and released for 0 and 3 hours in the presence or absence of 10 ng/ml leptomycin B (LMB). Cells were fixed in ethanol:acetic acid and BRCA1 content probed with Ab-1. Nuclei were detected by staining DNA with DAPI. Magnification, 400x. (C) Graphical representation of the percentage of cells with predominantly nonnuclear BRCA1. (mean ± s.d.; at least 5 fields per treatment were analyzed).
3.4 Nonnuclear localization of BRCA1 is calcium dependent
Because calcium signaling is involved in cell cycle progression from G1 to S phases, we queried whether calcium was also required for the movement of BRCA1 out of the nucleus during early S phase. MCF-7 cells were released from G0/G1 in the presence of increasing concentrations of BAPTA-AM (Fig. 6A), an intracellular calcium chelator. Treatment of cells with BAPTA showed a dose dependent decrease in the percentage of cells exhibiting nonnuclear accumulation of BRCA1 with a significant decrease occurring at 50 μM. These data implicate intracellular calcium in the nuclear exclusion of BRCA1 during the G1/S phase transition.
Fig. 6. Nonnuclear localization of BRCA1 is calcium-dependent.

(A) MCF-7 cells were synchronized by thymidine-mimosine. Cells were released from cell cycle arrest for 4 hours in the presence of increasing concentrations BAPTA-AM and immunostained for BRCA1. A dose response of changes in the percentage of cells with predominantly nonnuclear BRCA1 is shown. Points shown are mean ± s.e. of three experiments. (B) MCF-7 cells were treated with increasing doses of A23187 for 5 minutes prior to immunostaining for BRCA1. Graph depicts the dose response of changes in the percentage of cells with predominantly nonnuclear BRCA1. Points shown are mean ± s.d. of two experiments. (C) MCF-7 cells were treated with 10 μM A23187 for increasing lengths of time prior to immunostaining for BRCA1. Graph depicts the time course of changes in the percentage of cells with predominantly nonnuclear BRCA1 and shows the mean ± s.e. of three experiments. (D) MCF-7 cells were treated with increasing doses of thapsigargin for 4 hours minutes prior to immunostaining for BRCA1. Graph depicts the dose response of changes in the percentage of cells with predominantly nonnuclear BRCA1. Points shown are mean ± s.d. of two experiments. (E) MCF-7 cells were treated with 10 μM A23187 for 5 minutes. Media containing A23187 was removed and cells washed prior to immunostaining for BRCA1. Graph depicts the changes in the precentage of cells with nonnuclear BRCA1 at 0, 3, and 7 minutes after removal of A23187. Points shown are mean ± s.d. of two experiments.
If calcium were mediating the cell cycle dependent changes in BRCA1 localization, the administration of a calcium ionophore should mimic cell cycle changes. MCF-7 cells were treated with increasing doses of A23187 for 5 minutes prior to immunostaining for BRCA1. The results indicate a dose response effect of BRCA1 accumulation outside the nucleus in response to A23187 (Fig. 6B), with the greatest effect occurring at 10 μM. To examine the time course of the A23187 effect, MCF-7 cells were treated with 10 μM A23187 for a 10-minute time course prior to immunostaining for BRCA1. Treatment with the calcium ionophore resulted in a significant increase in the percentage of cells with BRCA1 localization predominantly outside the nucleus (from 30% to 70%) within 2 minutes of A23187 treatment (Fig. 6C). This further implicates calcium involvement in regulating BRCA1 localization. Like A23187, thapsigargin, which increases intracellular calcium, was also able to increase the percentage of cells with nonnuclear BRCA1 in a dose-dependent manner (Fig. 6D).
To assess whether the calcium-mediated change in BRCA1 localization was reversible, MCF-7 cells were treated with 10 μM A23187 for 5 minutes. After removal of the ionophore, cells were fixed immediately or 3 or 7 minutes later. Figure 6E demonstrates that within 3 minutes after the removal of A23187, the percentage of cells with nonnuclear BRCA1 had decreased from ~75% (in the presence of A23187) to ~57%. The effect was similar seven minutes after removal of A23187. These data indicate that the calcium-mediated change in BRCA1 localization is reversible.
3.5 Calcium-mediated change in BRCA1 localization depends on nuclear export
In order to demonstrate that the calcium-mediated change in BRCA1 localization was dependent on CRM-1, MCF-7 cells were treated with either vehicle or leptomycin B for 30 minutes prior to administering A23187, and then immunostained for BRCA1. Similar to results obtained in Fig. 6C, treatment of MCF-7 cells with the calcium ionophore resulted in a significant increase in the percentage of cells showing predominantly nonnuclear BRCA1. Inhibition of CRM-1-dependent nuclear export with leptomycin B blocked the ability of A23187 to alter BRCA1’s localization, resulting in only 37% of cells having a predominantly nonnuclear presence (Fig. 7A). Thus, calcium-dependent changes in BRCA1 localization are CRM-1-dependent.
Fig. 7. Calcium-mediated change in BRCA1 localization requires nuclear export.

(A) MCF-7 cells were treated with vehicle (methanol) or 10 ng/mL leptomycin B (LMB) for 30 minutes prior to treatment with either vehicle (DMSO) or A23187 for 5 minutes. Cells were immunostained for BRCA1. Graph depicts changes in percentage of cells displaying predominantly nonnuclear BRCA1. Graph shows the mean ± s.e. of 3 experiments in which at least 200 cells/concentration or time point were evaluated. (*, p<0.05, **, p<0.01). (B) MCF-7 cells were transfected with GFP-tagged BRCA1 constructs encoding either the wildtype BRCA1 or BRCA1 with its nuclear export sequences mutated. Pictures are representative of the predominant localization pattern of each expressed construct either in the presence of vehicle (DMSO) or A23187. Graph shows mean ± s.d. of two experiments.
The localization of BRCA1 in which both of its nuclear export sequences had been mutated was also assessed. MCF-7 cells were transfected with plasmids containing either GFP-tagged wildtype or mutant BRCA1 cDNAs and were treated with A23187. Figure 7B shows the localization of the tagged proteins. In cells expressing the wildtype GFP-tagged BRCA1, treatment with A23187 resulted in a two-fold increase in cells with nonnuclear GFP-BRCA1. This effect was not observed in cells expressing the export-defective GFP-tagged BRCA1.
3.6 Calcium-mediated change in BRCA1 localization occurs in several cell lines
To determine if the calcium-mediated change in BRCA1 localization was specific to MCF-7 cells or a more generalized occurrence, we evaluated BRCA1 localization in response to A23187 in MCF12A human breast cancer cells, HeLa human cervical cancer cells, T47D human breast cancer cells and nonmalignant human mammary epithelial cells (Fig. 8). In each cell line, A23187 was able to induce an increase in the percentage of cells with nonnuclear BRCA1. These results indicate that the ability of calcium to alter BRCA1 localization is not a consequence of cell line specificity.
Fig. 8. Calcium-mediated change in BRCA1 localization is not cell line specific.
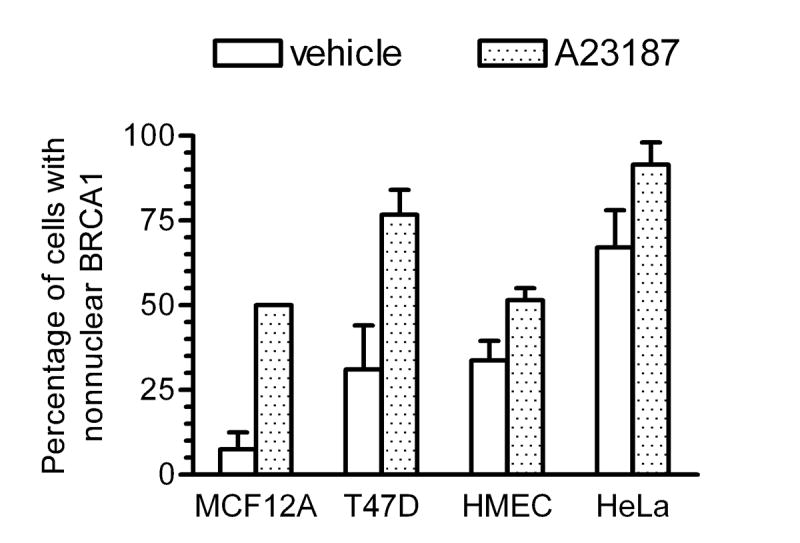
MCF12A, HeLa, T47D, and human mammary epithelial cells (HMEC) were treated with 10 μM A23187 or vehicle for 5 minutes prior to immunostaining for BRCA1. Graph depicts percentage of cells displaying predominantly nonnuclear BRCA1. Points shown are mean ± s.d. of two experiments.
3.7 BRCA2 nuclear localization is independent of calcium
We next assessed whether the localization of BRCA2 was sensitive to increases in intracellular calcium. MCF-7 cells were treated with 10 μM A23187 for 5 minutes, after which the cells were immunostained for BRCA1 or BRCA2 (Fig. 9). In cells exposed to vehicle, BRCA2 localized exclusively within the nucleus. There was no difference in its localization pattern in cells exposed to A23187. In cells plated in duplicate wells, BRCA1 localization changed from predominantly nuclear to predominantly nonnuclear when exposed to A23187. These results indicate that the nuclear localization of BRCA2 is not regulated by increases in intracellular calcium.
Fig. 9. BRCA2 localization is not calcium-dependent.

MCF-7 cells were treated with vehicle or 10 μM A23187 for 5 min prior to fixation in paraformaldehyde and immunostaining for BRCA1 or BRCA2. Pictures represent results obtained in two experiments.
3.8 Calcium-calmodulin dependent kinase II is involved in regulating BRCA1 localization
Calmodulin is a key regulator of calcium signaling. It binds calcium to enable it to activate calmodulin-dependent protein kinases. To examine whether calmodulin was involved in the calcium-mediated change in BRCA1 localization, MCF-7 cells were treated with W-7 (30 μM) for 2 hours prior to the addition of A23187. This resulted in the inability of A23187 to increase the percentage of cells with nonnuclear BRCA1 (Fig. 10). We also examined the role of calmodulin-dependent protein kinase II in this process. MCF-7 cells were pre-incubated with KN-62, a specific inhibitor of calmodulin-dependent protein kinase II, before the addition of A23187. This, too, attenuated the calcium-mediated response. These data indicate that the calcium-mediated change in BRCA1 localization is meditated through calmodulin-dependent protein kinase II.
Fig 10. Calmodulin-dependent kinase II regulates calcium-mediated BRCA1 localization change.
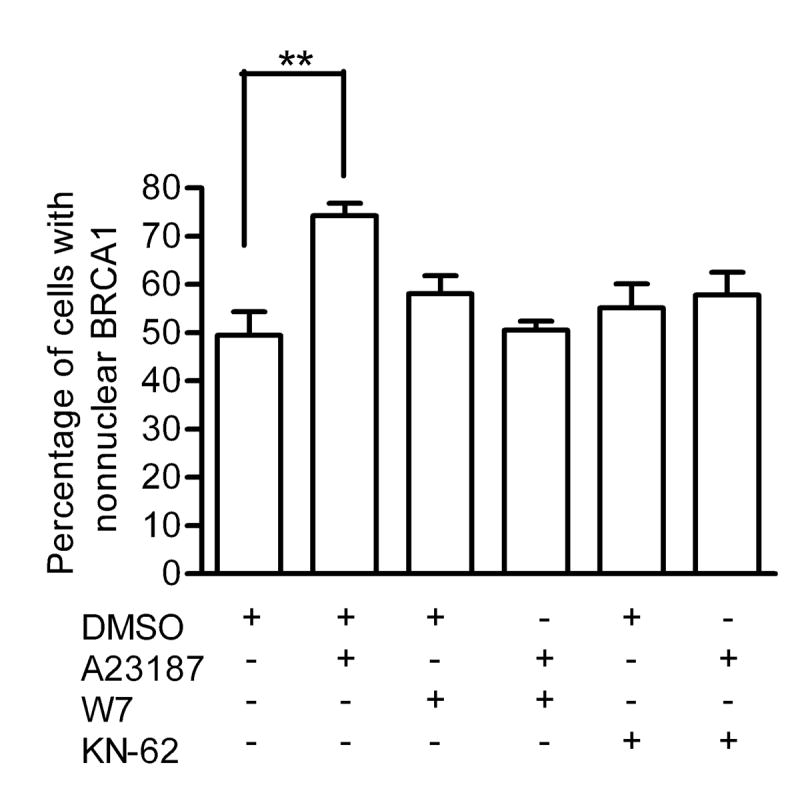
MCF-7 cells were pretreated with 30 μM W-7 or 10 μM KN-62 for two hours prior to the administration of 10 μM A23187 for 5 minutes. Cells were immunostained for BRCA1. Graph depicts percentage of cells displaying predominantly nonnuclear BRCA1. At least 200 cells per treatment were analyzed. Points shown are mean ± s.e. of three experiments (** p<0.01).
4. Discussion
In this report, we demonstrate that nuclear export of BRCA1 can be a regulated process. Although the nuclear-cytoplasmic distribution of BRCA1 has been previously demonstrated to shift during S phase of the cell cycle in response to DNA damage, we have focused on changes which occurred only during the early part of the S phase and in the absence of any DNA damaging agents. Thus, we have been able to document several novel findings. First, the nonnuclear levels of BRCA1 transiently increase during the initial part of S phase. By late S, however, BRCA1 resumes being a predominantly nuclear protein. Secondly, as the nuclear-nonnuclear shift can be blocked, at least in part, by leptomycin B, the early S phase redistribution of BRCA1 requires CRM-1-mediated nuclear export. Thirdly, BAPTA-AM blocks, whereas A23187 and thapsigargin mimic the early S phase changes in BRCA1 distribution, indicating that calcium is involved in mediating this shift. Collectively, these data are consistent with calcium-dependent, cell cycle-mediated nuclear export of BRCA1.
After BRCA1 was cloned, several structural motifs, such as the carboxy-terminal transactivation domain and the BRCT sequences, were identified. This led to the theory that BRCA1 was a nuclear protein with no substantial cytoplasmic presence. This postulate was supported by the identification of two nuclear localization sequences within the protein [13]. However, in 2000, Rodriguez and Henderson [14] reported the presence of a nuclear export sequence within BRCA1, and this suggested that BRCA1 could also localize to the cytoplasm. The identification of a second nuclear export sequence [15] supported the nuclear-cytoplasmic shuttling of BRCA1.
The association of BRCA1 with the centrosome during mitosis was the first report of a cell cycle-dependent change in BRCA1 nuclear-nonnuclear distribution [1]. Subsequently, Okada and Ouchi [12] reported that in response to ultraviolet or ionizing radiation of S phase cells, BRCA1 becomes phosphorylated on serine 988 and its localization changes from predominantly nuclear to perinuclear. Data reported here demonstrate a similar change in BRCA1 localization from the nucleus to a nonnuclear location. In contrast to the previous report, however, we observed the localization change in the absence of DNA damage. The localization change is a transient event which occurs during the early stages of S phase and is dependent on CRM-1 mediated nuclear export.
Signaling molecules involved in regulating the nuclear export of BRCA1 have not been previously reported. Our data indicate that the early S phase nuclear export of BRCA1 depends on calcium, as demonstrated by the use of the intracellular calcium chelator BAPTA-AM and by use of the calcium ionophore, A23187. Intracellular calcium levels increase at the G1/S transition in mammalian cells, and this may serve as a stimulus to promote the early S phase relocalization of BRCA1 from the nucleus to the cytosol. Previous reports have demonstrated a change in BRCA1 phosphorylation when either its subnuclear or nuclear-cytoplasmic localization is altered. Thus, it may be that calcium is modulating the phosphorylation status of BRCA1 through calmodulin-dependent protein kinase II. This may be direct phosphorylation of BRCA1 by the kinase or it may be indirect through another kinase activated by calmodulin-dependent protein kinase II.
Although calcium is required for mammalian cell proliferation, BRCA1 is generally considered to negatively regulate cell cycle progression. BRCA1 induces the expression of the cdk inhibitor, p21Waf1/Cip1, which binds to cdk2 complexes to inhibit the kinase activity and cell cycle progression [3]. Therefore, it is possible that in order for normal cellular proliferation to occur, the transcriptional activity of BRCA1 needs to be transiently suppressed. One mechanism of doing this is to remove it from its site of action, the nucleus. Our data suggest that calcium is required for this to occur.
5. Conclusion
In conclusion, we have demonstrated that the subcellular localization of BRCA1 is cell cycle-modulated in the absence of DNA damage. We have also demonstrated that BRCA1 localization is subject to regulation by a calcium-dependent mechanism. Although currently not understood, elucidation of the molecular pathway(s) involved in this process will be vital to further understanding BRCA1 localization dynamics and function. Ultimately, knowing the basis for the calcium dependence of BRCA1 redistribution may lead to the identification of additional targets for manipulation therapeutically to prevent cancer progression by maintaining BRCA1 within the nucleus.
Acknowledgments
We thank Dr. E. Motley and G. Chaudhuri for reagents, Dr. S. Eltom for HMEC cells, and Drs. T. Dermody and L. Limbird for helpful comments. Experiments/data analyses were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404 and the John F. Kennedy Center), the Flow Cytometry Special Resource Center (Veterans’ Administration Medical Center, Nashville, TN) and the DNA Core Facility (Meharry Medical College). We gratefully acknowledge the following support from the National Institutes of Health: NIH-NCRR supported Research Centers in Minority Institutions (RCMI) Grant 2G12RR03032, National Cancer Institute K01 CA89494 and U54 CA91408 (MET), National Institute of General Medical Studies T32 GM07347 (K.G-C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 2.Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta TJ. J Biol Chem. 2001;276:14537–14540. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 3.Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Zhang H, Wu GS, Licht J, Weber BL, El-Deiry WS. Nature. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 4.Moynahan ME, Chiu JW, Koller BH, Jasin M. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 5.Chapman MS, Verma IM. Nature. 1996;382:678–679. doi: 10.1038/382678a0. [DOI] [PubMed] [Google Scholar]
- 6.Jin S, Zhao H, Fan F, Blanck P, Fan W, Colchagie A, Fornace AJ, Jr, Zhan Q. Oncogene. 2000;19:4050–4057. doi: 10.1038/sj.onc.1203759. [DOI] [PubMed] [Google Scholar]
- 7.Ruffner H, Verma IM. Proc Natl Acad Sci USA. 1997;94:7138–7143. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudas JM, Li T, Nguyen H, Jensen D, Rauscher FJ, 3rd, Cowan KH. Cell Growth Differ. 1996;7:717–723. [PubMed] [Google Scholar]
- 9.Lotti LV, Ottini L, D’Amico G, Cama A, Belleudi F, Frati L, Torrisi MR, Mariani-Costantini R. Genes Chromosomes Cancer. 2002;35:193–203. doi: 10.1002/gcc.10105. [DOI] [PubMed] [Google Scholar]
- 10.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Ashley T, Livingston DM. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 11.Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 12.Okada S, Ouchi T. J Biol Chem. 2003;278:2015–2020. doi: 10.1074/jbc.M208685200. [DOI] [PubMed] [Google Scholar]
- 13.Chen CF, Li S, Chen Y, Chen PL, Sharp ZD, Lee WH. J Biol Chem. 1996;271:32863–32868. doi: 10.1074/jbc.271.51.32863. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez JA, Henderson BR. J Biol Chem. 2000;275:38589–38596. doi: 10.1074/jbc.M003851200. [DOI] [PubMed] [Google Scholar]
- 15.Thompson ME, Robinson-Benion CL, Holt JT. J Biol Chem. 2005;280:21854–21857. doi: 10.1074/jbc.M502676200. [DOI] [PubMed] [Google Scholar]
- 16.Kahl CR, Means AR. Endocr Rev. 2003;24:719–736. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- 17.Chawla S, Vanhoutte P, Arnold F, Huang C, Bading H. J Neurochem. 2003;85:151–159. doi: 10.1046/j.1471-4159.2003.01648.x. [DOI] [PubMed] [Google Scholar]
- 18.Bittinger MA, McWhinnie E, Meltzer J, Tourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M. Curr Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, McKeon F. Nature. 1999;398:256–260. doi: 10.1038/18473. [DOI] [PubMed] [Google Scholar]
- 20.Hinton CV, Fitzgerald LD, Thompson ME. Exp Cell Res. 2007;313:1735–1744. doi: 10.1016/j.yexcr.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzgerald LD, Bailey CK, Brandt SJ, Thompson ME. FEBS J. 2007;274:5137–5146. doi: 10.1111/j.1742-4658.2007.06033.x. [DOI] [PubMed] [Google Scholar]
- 22.Wilson CA, Ramos L, Villasenor MR, Anders KH, Press MF, Clarke K, Karlan B, Chen JJ, Scully R, Livingston D, Zuch RH, Kanter MH, Cohen S, Calzone FJ, Slamon DJ. Nature Gen. 1999;21:236–240. doi: 10.1038/6029. [DOI] [PubMed] [Google Scholar]