

Abstract
Central sensitization, increased sensitivity in spinal cord dorsal horn neurons after injuries, plays an essential role in the induction and maintenance of chronic pain. However, synaptic mechanisms underlying central sensitization are incompletely known. Growing evidence suggests that proinflammatory cytokines (PICs), such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNFα), are induced in the spinal cord under various injury conditions and contribute to pain hypersensitivity. Using patch-clamp recordings in lamina II neurons of isolated spinal cord slices, we compared the effects of IL-1β, IL-6, and TNFα on excitatory and inhibitory synaptic transmission. Whereas TNFα enhanced the frequency of spontaneous EPSCs (sEPSCs), IL-6 reduced the frequency of spontaneous IPSCs (sIPSCs). Notably, IL-1β both enhanced the frequency and amplitude of sEPSCs and reduced the frequency and amplitude of sIPSCs. Consistently, TNFα and IL-1β enhanced AMPA- or NMDA-induced currents, and IL-1β and IL-6 suppressed GABA- and glycine-induced currents. Furthermore, all the PICs increased cAMP response element-binding protein (CREB) phosphorylation in superficial dorsal horn neurons and produced heat hyperalgesia after spinal injection. Surprisingly, soluble IL-6 receptor (sIL-6R) produced initial decrease of sEPSCs, followed by increase of sEPSCs and CREB phosphorylation. Spinal injection of sIL-6R also induced heat hyperalgesia that was potentiated by coadministration with IL-6. Together, our data have demonstrated that PICs induce central sensitization and hyperalgesia via distinct and overlapping synaptic mechanisms in superficial dorsal horn neurons either by increasing excitatory synaptic transmission or by decreasing inhibitory synaptic transmission. PICs may further induce long-term synaptic plasticity through CREB-mediated gene transcription. Blockade of PIC signaling could be an effective way to suppress central sensitization and alleviate chronic pain.
Keywords: EPSC, IPSC, disinhibition, GABA, glycine, soluble IL-6 receptor, proinflammatory cytokines, PICs
Introduction
Peripheral inflammation is associated with pain hypersensitivity that is produced by the release of inflammatory mediators from immune cells and non-neuronal cells in the periphery. The proinflammatory cytokines (PICs) such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNFα) are an important group of inflammatory mediators and play an essential role in pain sensitization (Sorkin et al., 1997; Woolf et al., 1997; Sommer and Kress, 2004). The peripheral effects of these PICs on sensitizing nociceptors have been well documented. For example, PICs enhance the activity of TRPV1 (transient receptor potential subtype V1) (Nicol et al., 1997; Opree and Kress, 2000; Jin and Gereau, 2006), induce the expression of pronociceptive genes in dorsal root ganglion (DRG) neurons (Fehrenbacher et al., 2005; von Banchet et al., 2005), and further increase spontaneous activity in DRG neurons (Schafers et al., 2003).
Increasing evidence suggests that the PICs also enhance pain via central mechanisms. First, PICs are induced in the spinal cord, especially in glial cells (e.g., microglia and astrocytes), in different chronic pain conditions. Second, intrathecal injection of the PICs was shown to enhance pain. Third, spinal blockade of PIC signaling attenuates chronic pain (for review, see DeLeo and Yezierski, 2001; Watkins et al., 2001). However, little is known as to how PICs alter synaptic transmission and neuronal activity in the spinal cord. We used patch-clamp recording in dorsal horn neurons in isolated spinal cord slices to investigate whether PICs have similar or different effects on synaptic transmission in lamina II neurons in which nociceptive information is modulated and conveyed to projection neurons. In addition, spinal IL-1β was also shown to induce the transcription of pronociceptive genes (e.g., Cox-2) in the spinal cord (Samad et al., 2001). We further examined whether PICs can activate the transcription factor cAMP response element-binding protein (CREB), a critical mediator for the transcription of pronociceptive genes and long-term neuronal plasticity (Ji et al., 2003).
Increased synaptic transmission in dorsal horn neurons, central sensitization, has been strongly implicated in persistent pain development (Woolf and Salter, 2000; Ji et al., 2003). Central sensitization is caused by increased excitatory synaptic transmission and decreased inhibitory synaptic transmission. Loss of inhibitory synaptic transmission, or disinhibition, is emerging as a critical mechanism for neuropathic pain sensitization (Moore et al., 2002; Coull et al., 2005; Zeilhofer, 2005). We now show that PICs also contribute to modulating inhibitory synaptic transmission in the spinal cord.
Materials and Methods
Animals and drugs.
Adult male Sprague Dawley rats were used according to a protocol approved by the Standing Committee for Animals at Harvard Medical School. We purchased the rat recombinant IL-1β, IL-6, and TNFα, and human soluble IL-6 receptor (sIL-6R) from R&D Systems (Minneapolis, MN). AMPA, NMDA, GABA, and glycine were obtained from Sigma-Aldrich (St. Louis, MO). All of the cytokines were prepared as 1000× stock solution in PBS and finally used at the concentration of 10 ng/ml (i.e., 0.58, 0.45, and 0.59 nm for IL-1β, IL-6, and TNFα, respectively).
Spinal slice preparation.
As we described previously (Baba et al., 2003; Kawasaki et al., 2004), a portion of the lumbar spinal cord (L4–L5) was removed from adult rats (200 g) under urethane anesthesia (1.5–2.0 g/kg, i.p.) and kept in preoxygenated ice-cold Krebs' solution. The spinal segment was glued to the bottom of the microslicer stage. Transverse slices (600 μm) were cut on a vibrating microslicer. The slices were continuously perfused with Krebs' solution (10 ml/min) and then saturated with 95% O2 and 5% CO2 for 1–3 h before experiment. The perfusion solution was heated to 36 ± 1°C via a temperature controller (Warner Instruments, Hamden, CT). The Krebs' solution contained the following (in mm): 117 NaCl, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose. All the drugs (1000×) were added to the perfusion system.
Patch-clamp recordings in spinal slices.
The whole-cell patch-clamp recordings were made from lamina II neurons in voltage-clamp mode (Baba et al., 2003). Under a dissecting microscope, the substantia gelatinosa (SG) (lamina II) is clearly visible as a relatively translucent band across the dorsal horn. However, the shapes of individual SG neurons cannot be visualized under this condition, and thus giga-ohm sealing was performed blindly. Patch pipettes were fabricated from thin-walled, borosilicate, glass-capillary tubing (1.5 mm outer diameter; World Precision Instruments, Sarasota, FL). After establishing the whole-cell configuration, neurons held their holding potentials at −70 and 0 mV for recording spontaneous EPSCs (sEPSCs) and spontaneous IPSCs (sIPSCs), respectively. The resistance of a typical patch pipette is 5–10 MΩ when filled with the internal solution that contains the following (in mm): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, and 5 ATP-Mg for sEPSC recording; 110 Cs2SO4, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, 5 tetraethylammonium, and 5 ATP-Mg for sIPSC recording. Membrane currents were amplified with an Axopatch 200A amplifier (Molecular Devices, Sunnyvale, CA) in voltage-clamp mode. Signals were filtered at 2 kHz and digitized at 5 kHz. Data were stored with a personal computer using pCLAMP 6 software and analyzed with Axograph 4.0 (Molecular Devices). Cells were perfused with Krebs' solution during baseline recording; the perfusate was then shifted to drug-containing Krebs' solution for additional recording.
Immunohistochemistry.
Thirty minutes after cytokine stimulation, all the slices were fixed with 4% paraformaldehyde for 1 h at room temperature. Transverse spinal sections (15 μm) were cut in a cryostat and processed for immunostaining. All the sections were blocked with 2% goat serum in 0.3% Triton for 1 h and incubated overnight at 4°C with anti-phosphoCREB antibody (anti-rabbit, 1:1000; Cell Signaling Technology, Danvers, MA). The sections were then incubated for 1 h at room temperature with cyanine 3 (Cy3)-conjugated secondary antibody (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA). Some spinal sections were used for phosphorylated cAMP response element-binding protein (pCREB)/neuronal-specific nuclear protein (NeuN) double staining by incubating with a mixture of polyclonal pCREB antibody (1:1000) and monoclonal NeuN antibody (anti-mouse, 1:5000; Millipore Bioscience Research Reagents, Temecula, CA) followed by a mixture of Cy3 and FITC-conjugated secondary antibodies (Kawasaki et al., 2004). The immunostained sections were examined with a Nikon (Tokyo, Japan) fluorescence microscope, and images were captured with a CCD Spot camera (Diagnostic Instruments, Sterling Heights, MI). To quantify pCREB signaling, we counted the number of pCREB-positive neurons in laminas I–II from six randomly picked spinal sections (Kawasaki et al., 2004).
Behavior.
The cytokines and vehicle (saline) were delivered to CSF space between L5 and L6 vertebrae via a spinal cord puncture, which is made by a 30 ga needle. Before puncture, the rats' heads were covered by a piece of cloth. Twenty microliters of solution were injected with a microsyringe. A successful spinal puncture was confirmed by a brisk tail-flick. Animals were put in plastic boxes and habituated to the testing environment before baseline testing. Rat paw withdrawal latency (PWL) was measured using Hargreaves' radiant heat test and adjusted to 9–11 s for baselines. After drug treatment, the PWL values were expressed as a percentage of baselines.
Statistics.
For electrophysiology, 5–13 neurons showing responses to a cytokine (5% above or below baselines) were included for each current analysis. PIC-pretreated and -treated groups were compared using paired t test. For immunostaining, four to five slices from separate rats were included for each group. Differences between groups were compared using ANOVA followed by Fisher's PLSD post hoc analysis or using t test. Data were expressed as mean ± SEM. The criterion for statistical significance was p < 0.05.
Results
Proinflammatory cytokines enhance excitatory synaptic transmission and potentiate AMPA- and NMDA-induced currents
We first recorded sEPSCs in lamina II neurons in isolated spinal slices. Perfusion of IL-1β and TNFα, but not IL-6 at the concentration of 10 ng/ml (2 min), induced a significant increase in the frequency of sEPSCs (Fig. 1a,b), suggesting a possible presynaptic mechanism of these PICs to enhance glutamate releases (Baba et al., 2003; Kohno et al., 2005). Notably, TNFα-produced frequency increase (71%, p < 0.01) was more robust than that of IL-1β (27%, p < 0.05). However, only a portion of recorded lamina II neurons responded to TNFα (8 of 14) and IL-1β (13 of 25). Importantly, most neurons that re-sponded to IL-1β also responded to TNFα (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In addition to frequency change, IL-1β, but not TNFα and IL-6, also increased the amplitude of sEPSCs (28%, p < 0.05) (Fig. 1a,c), suggesting additional postsynaptic regulation of IL-1β (Kohno et al., 2005).
Figure 1.

a–e, Potentiation of excitatory synaptic transmission by PICs. a, Patch-clamp recording of sEPSCs in lamina II neurons shows increase in the frequency and amplitude of sEPSCs after perfusion of IL-1β (10 ng/ml, 2 min). a1 and a2 are enlargements of the recording before and after IL-1β treatment, respectively. b, c, Ratio of the frequency (b) and amplitude (c) of sEPSCs after the treatment of IL-1β, TNFα, and IL-6 (10 ng/ml, 2 min). d, Effects of PICs on AMPA-induced currents recorded at holding potential of −70 mV. Top, A mild potentiation of AMPA (10 μm)-induced current by IL-1β. Bottom, Ratio of the amplitude of the AMPA-induced currents after treatment of IL-1β, TNFα, and IL-6 (10 ng/ml, 2 min). e, Effects of PICs on NMDA-induced currents recorded at holding potential of −50 mV. Top, Potentiation of NMDA (50 μm)-induced current by IL-1β. Bottom, Ratio of the amplitude of the NMDA-induced currents after treatment of IL-1β, TNFα, and IL-6 (10 ng/ml, 2 min). *p < 0.05 compared with pretreatment baseline (t test). Inside each column, the number of total recorded neurons and number of responding neurons are indicated.
Because excitatory synaptic transmission is mainly mediated by AMPA and NMDA receptors, we further examined the effects of the PICs on inward currents induced by AMPA (10 μm) and NMDA (50 μm) when holding the voltage at −70 and −50 mV, respectively. AMPA-induced current was significantly enhanced by TNFα (43%, p < 0.05) and moderately enhanced by IL-1β (28%, p > 0.05) (Fig. 1d). NMDA-induced current was also significantly increased by TNFα (65%, p < 0.05) and IL-1β (37%, p < 0.05) (Fig. 1e). In particular, TNFα enhanced NMDA currents in all the neurons (5 of 5), suggesting a powerful regulation of TNFα on NMDA currents (Fig. 1e).
Proinflammatory cytokines inhibit inhibitory synaptic transmission and suppress GABA- and glycine-induced currents
We next recorded sIPSCs in lamina II neurons by holding the voltage at 0 mV. All the PICs were perfused at the same concentration (10 ng/ml) and duration (2 min). Notably, most neurons (12 of 16) responded to IL-1β, and IL-1β produced a significant decrease of sIPSCs in both frequency (34%, p < 0.05) and amplitude (31%, p < 0.05) (Fig. 2a–c). IL-6 also inhibited the frequency of sIPSCs (22% decrease, p < 0.05). Of interest, most neurons that responded to IL-6 also responded to IL-1β (Fig. 2d; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, TNFα had no effect on sIPSC (Fig. 2b,c).
Figure 2.
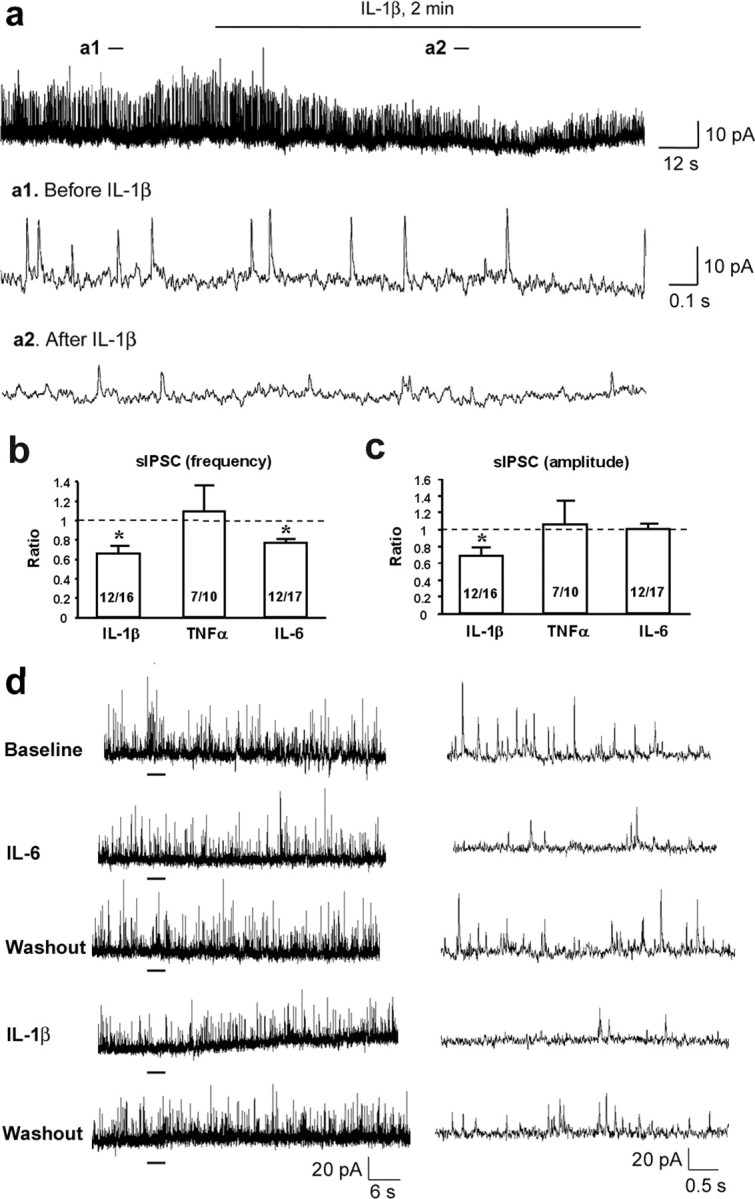
a–e, Suppression of inhibitory synaptic transmission by PICs. a, Patch-clamp recording of sIPSCs in lamina II neurons shows decrease in the frequency and amplitude of sIPSCs after perfusion of IL-1β (10 ng/ml, 2 min). The holding voltage is 0 mV. a1 and a2 are enlargements of the recording before and after IL-1β treatment. b, c, Ratio of the frequency (b) and amplitude (c) of sIPSCs after the treatment of IL-1β, TNFα, and IL-6 (10 ng/ml, 2 min). *p < 0.05 compared with pretreatment baseline (t test). Inside each column, number of total recorded neurons and number of responding neurons are indicated. d, A typical lamina II neuron shows inhibition of sIPSCs to both IL-6 and IL-1β treatment (10 ng/ml). Right, Enlarged traces of recordings that are underlined.
Because inhibitory synaptic transmission is mediated by GABA and glycine receptors, we further examined the effects of the PICs on GABA- and glycine-induced outward currents by holding the voltage at 0 mV. GABA-induced current was significantly reduced by IL-1β (26%, p < 0.05) and IL-6 (20%, p < 0.05), but not by TNFα (Fig. 3a–d). Furthermore, glycine-induced current was also markedly suppressed by IL-1β (44% decrease, p < 0.05) and IL-6 (13%, p < 0.05), but not by TNFα (Fig. 3a–d).
Figure 3.

a–d, Suppression of GABA- and glycine-induced currents by PICs. a, b, Inhibition of GABA- (1 mm; a) and glycine- (1 mm; b) induced currents by IL-1β perfusion (10 ng/ml, 2 min). Both currents were recorded at holding potential of 0 mV. c, d, Ratio of the amplitude of the GABA- (c) and glycine- (d) induced currents after the treatment of IL-1β, IL-6, and TNFα (10 ng/ml, 2 min). *p < 0.05 compared with pretreatment baseline (t test). Number of total recorded neurons and number of responding neurons are shown inside each column. For TNFα treatment, only number of recorded neurons is shown, because all neurons failed to respond to TNFα.
Proinflammatory cytokines induce CREB phosphorylation and heat hyperalgesia
Phosphorylation of CREB on Serine 133 is essential for CREB-mediated gene transcription (Ji et al., 2002). To investigate whether PICs can induce CREB phosphorylation in the dorsal horn, we incubated spinal cord slices for 30 min with three PICs. As shown previously (Kawasaki et al., 2004), there was a basal pCREB expression in the superficial dorsal horn (Fig. 4a). However, all three PICs induced robust increase in pCREB levels. The increases induced by these PICs were comparable (Fig. 4b–e). Double staining indicated that all pCREB-labeled cells also expressed NeuN (Fig. 4f), suggesting that pCREB is only induced in dorsal horn neurons by PICs.
Figure 4.
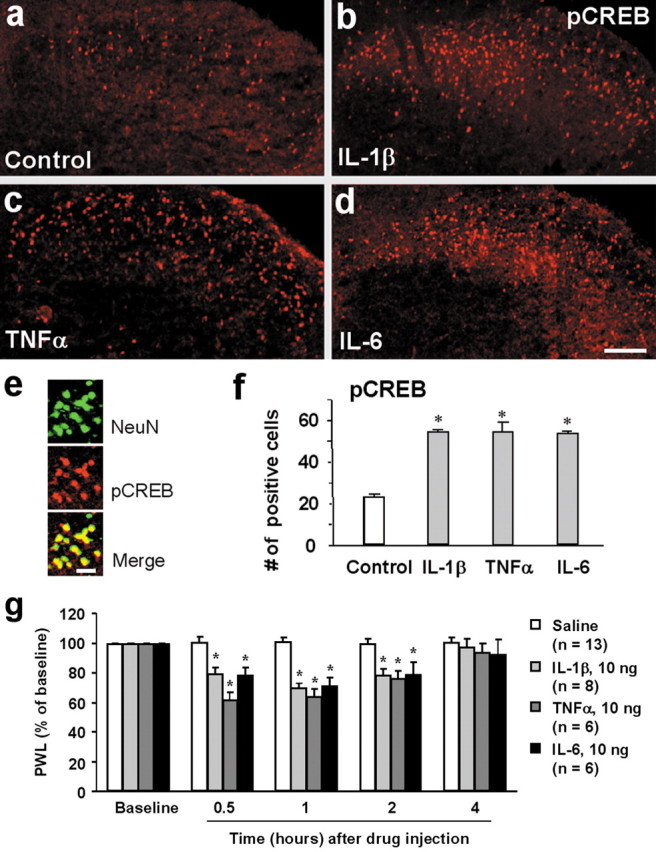
a–f, Induction of CREB phosphorylation and heat hyperalgesia by PICs. a–d, pCREB immunostaining in the superficial dorsal horn of nonstimulated control slice (a) and stimulated slices with IL-1β (b), TNFα (c), and IL-6 (d). Spinal cord slices were treated with the PICs for 30 min (10 ng/ml) and then fixed with 4% paraformaldehyde. Scale bar, 100 μm. e, Colocalization of pCREB (red) with neuronal marker NeuN (green) in the superficial dorsal horn after IL-1β treatment. Scale bar, 20 μm. f, Number of pCREB-positive nuclei of neurons in the superficial dorsal horn (laminas I–II) 30 min after PIC (10 ng/ml) treatment. *p < 0.05 compared with control (ANOVA); n = 4. g, Spinal injection of IL-1β, TNFα, and IL-6 (10 ng) induces heat hyperalgesia. *p < 0.05 compared with corresponding saline control.
To determine whether cytokine effects on spinal cord neurons are related to nociceptive behavior, we examined heat sensitivity by measuring PWL. Injection of all three PICs (10 ng) into spinal cord CSF induced marked heat hyperalgesia, as shown by a decrease in PWLs (Fig. 4g).
Effects of soluble IL-6 receptor on synaptic transmission, CREB phosphorylation, and pain behavior
Unlike IL-1β and TNFα, sIL-6R is required for IL-6 effects in neurons because membrane-bound IL-6 receptor may express at very low levels in neurons (März et al., 1999; Opree and Kress, 2000). Surprisingly, sIL-6R (10 ng/ml) produced a rapid inhibition of sEPSC frequency (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material) and an inhibition of AMPA- and NMDA-induced current (supplemental Fig. 4a,b, available at www.jneurosci.org as supplemental material). sIL-6R-induced inhibition of sEPSCs was prevented by coincubation of sIL-6R with IL-6 (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). Interestingly, after an initial inhibition (e.g., at 2 min), sEPSCs frequency was increased by sIL-6R at later time points (e.g., at 5 min) (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material).
sIL-6R also produced a moderate inhibition of sIPSC frequency (p = 0.138) (supplemental Fig. 3c, available at www.jneurosci.org as supplemental material). However, coapplication of sIL-6R and IL-6 produced significant inhibition of sIPSC frequency (p < 0.05) (supplemental Fig. 3c, available at www.jneurosci.org as supplemental material). Consistently, sIL-6R produced a moderate inhibition of GABA- and glycine-induced currents (supplemental Fig. 4b, available at www.jneurosci.org as supplemental material).
sIL-6R also increased CREB phosphorylation at 30 min after stimulation, which was further potentiated by coapplication with IL-6 (supplemental Fig. 5a, available at www.jneurosci.org as supplemental material). Finally, spinal injection of sIL-6R produced heat hyperalgesia, which was enhanced by coadministration with IL-6 (supplemental Fig. 5b, available at www.jneurosci.org as supplemental material).
Discussion
Accumulating evidence suggests that glial cells such as microglia and astrocytes in the spinal cord play an important role in persistent pain development. It is believed that PICs are predominantly induced in glial cells after inflammation and nerve injury and facilitate pain via neural–glial interaction (DeLeo and Yezierski, 2001; Watkins et al., 2001). Although spinal injection of IL-1β was shown to enhance C-fiber-evoked responses and windup in wide-dynamic-range dorsal horn neurons (Reeve et al., 2000), the role of PICs in regulating synaptic transmission and central sensitization is elusive. Our study has clearly demonstrated a powerful role of PICs in enhancing synaptic transmission and neuronal activity in the lamina II superficial dorsal horn neurons. Although we cannot be sure that the neurons we recorded are excitatory, it is generally believed that most neurons recorded in the lamina II of transverse spinal cord slices are excitatory and pronociceptive (Yang et al., 1998; Moore et al., 2002; Baba et al., 2003; Kohno et al., 2005). For example, lamina II neurons consistently show increased EPSCs after capsaicin incubation (Yang et al., 1998), but decreased EPSCs after opioid treatment (Kohno et al., 2005) and decreased GABA currents under neuropathic pain states (Moore et al., 2002). In particular, a recent study has shown that excitatory interneurons dominate in lamina II (Santos et al., 2007). Importantly, these neurons show consistent responses to PICs, and their electrophysiological responses to PICs are in parallel with PIC-induced pain behavior after spinal injection.
Although these PICs appear to have similar mechanisms in regulating the sensitivity of primary sensory neurons (Opree and Kress, 2000), they have different mechanisms in modulating synaptic activities in lamina II dorsal horn neurons: TNFα and IL-6 modulate excitatory and inhibitory synaptic transmission, respectively, whereas IL-1β controls both excitatory and inhibitory synaptic transmission. Our data have shown that TNFα induced a dramatic increase (71%) of sEPSC frequency, suggesting a potent role of this cytokine in inducing glutamate release from central terminals of primary afferents. TNFα also enhanced AMPA-induced current in dorsal horn neurons, in agreement with a previous report from hippocampal neurons (Stellwagen et al., 2005). Our data also demonstrated an enhancement of NMDA current by TNFα and IL-1β, in parallel with reports showing that IL-1β increases NMDA receptor phosphorylation in the trigeminal nucleus (Guo et al., 2007) and NMDA-induced Ca2+ influx in hippocampal neurons (Viviani et al., 2003). In particular, IL-1β exhibited a powerful influence on inhibitory neurotransmission by suppressing sIPSC and GABA- and glycine-induced currents in most recorded neurons.
Although there is controversy regarding the role of IL-6 in pain regulation (Flatters et al., 2003), our data support a pronociceptive role of IL-6 by suppressing inhibitory neurotransmission, promoting CREB phosphorylation, and inducing heat hyperalgesia. It is likely that low levels of sIL-6R could be present in extracellular spaces of spinal slices to facilitate IL-6 effects. Surprisingly, sIL-6R alone (with a relatively high dose) had complex effects on synaptic transmission: it initially inhibited sEPSCs and then enhanced sEPSCs at late times. However, the overall effect of sIL-6R is pronociceptive, because it induced pCREB at 30 min and heat hyperalgesia at 30–120 min, and both are potentiated by coapplication with IL-6.
Increasing evidence indicates that nerve injury disrupts a balance between excitatory and inhibitory synaptic activities in dorsal horn neurons, leading to the development of neuropathic pain. Nerve injury not only enhances excitatory synaptic transmission, initiated by the activation of spinal NMDA receptors, but also induces a loss of inhibitory synaptic transmission (disinhibition) in dorsal horn neurons (Woolf and Salter, 2000). Several mechanisms may underlie the disinhibition. First, inhibitory neurons in the lamina II may undergo apoptosis after nerve injury, leading to a loss of GABA currents (Moore et al., 2002) (but see Polgar et al., 2005). Second, nerve injury produces a BDNF-mediated change of chloride reversal potential in lamina I neurons so that GABA causes excitation (Coull et al., 2005). Third, PGE2 production via COX-1/2 suppresses glycine current in superficial dorsal horn neurons (Zeilhofer, 2005). We now show another mechanism of disinhibition induced by cytokines. Because PICs can suppress both GABA and glycine currents, their roles in disinhibition can be powerful.
Despite distinct synaptic and neuronal mechanisms, all the PICs positively regulate central sensitization by enhancing excitatory neurotransmission and suppressing inhibitory neurotransmission. Additionally, all three PICs induced phosphorylation of the transcription factor CREB, which is essential for the maintenance of long-term neural plasticity in dorsal horn neurons (Ji et al., 2003). CREB may maintain persistent pain sensitization by inducing the transcription of pronociceptive genes such as neurokinin-1 (Ji et al., 2002) and Cox-2 (Samad et al., 2001; Ji et al., 2003).
In conclusion, we have demonstrated novel synaptic/neuronal mechanisms in the superficial dorsal horn by which centrally produced PICs in injury and disease conditions can induce central sensitization and pain hypersensitivity. Specifically, TNFα and IL-6 regulate excitatory and inhibitory neurotransmission, respectively, and IL-1β regulates both excitatory and inhibitory neurotransmission. Also, these cytokines may maintain persistent pain by inducing transcriptional changes. Thus, blocking the actions of PICs in the spinal cord should provide effective relief of chronic pain.
Footnotes
The work was supported by National Institutes of Health Grants DE17794, NS 54932, and TW7180 and National Science Fund of China (NSFC) Grant 30528019. L.Z. was supported by a fellowship from East China Normal University and by NSFC Grant 30600171. J.-K.C. was supported by a John J. Bonica Trainee Fellowship from the International Association for the Study of Pain and by a fellowship from Mackay Memorial Hospital (Taipei, Taiwan).
References
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Fehrenbacher JC, Burkey TH, Nicol GD, Vasko MR. Tumor necrosis factor alpha and interleukin-1beta stimulate the expression of cyclooxygenase II but do not alter prostaglandin E2 receptor mRNA levels in cultured dorsal root ganglia cells. Pain. 2005;113:113–122. doi: 10.1016/j.pain.2004.09.031. [DOI] [PubMed] [Google Scholar]
- Flatters SJ, Fox AJ, Dickenson AH. Spinal interleukin-6 (IL-6) inhibits nociceptive transmission following neuropathy. Brain Res. 2003;984:54–62. doi: 10.1016/s0006-8993(03)03092-0. [DOI] [PubMed] [Google Scholar]
- Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, Wei F, Dubner R, Ren K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27:6006–6018. doi: 10.1523/JNEUROSCI.0176-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der MC, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Ji RR, Ito N, Allchorne AJ, Befort K, Karchewski LA, Woolf CJ. Peripheral axonal injury results in reduced mu opioid receptor pre- and post-synaptic action in the spinal cord. Pain. 2005;117:77–87. doi: 10.1016/j.pain.2005.05.035. [DOI] [PubMed] [Google Scholar]
- März P, Otten U, Rose-John S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors. Eur J Neurosci. 1999;11:2995–3004. doi: 10.1046/j.1460-9568.1999.00755.x. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol GD, Lopshire JC, Pafford CM. Tumor necrosis factor enhances the capsaicin sensitivity of rat sensory neurons. J Neurosci. 1997;17:975–982. doi: 10.1523/JNEUROSCI.17-03-00975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opree A, Kress M. Involvement of the proinflammatory cytokines tumor necrosis factor-α, IL-1 β, and IL-6 but not IL-8 in the development of heat hyperalgesia: effects on heat-evoked calcitonin gene-related peptide release from rat skin. J Neurosci. 2000;20:6289–6293. doi: 10.1523/JNEUROSCI.20-16-06289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar E, Hughes DI, Arham AZ, Todd AJ. Loss of neurons from laminas I-III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. J Neurosci. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain. 2000;4:247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Santos SF, Rebelo S, Derkach VA, Safronov BV. Excitatory interneurons dominate sensory processing in the spinal substantia gelatinosa of rat. J Physiol (Lond) 2007;581:241–254. doi: 10.1113/jphysiol.2006.126912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Lee DH, Brors D, Yaksh TL, Sorkin LS. Increased sensitivity of injured and adjacent uninjured rat primary sensory neurons to exogenous tumor necrosis factor-α after spinal nerve ligation. J Neurosci. 2003;23:3028–3038. doi: 10.1523/JNEUROSCI.23-07-03028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Xiao WH, Wagner R, Myers RR. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255–262. doi: 10.1016/s0306-4522(97)00147-4. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci. 2003;23:8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Banchet GS, Kiehl M, Schaible HG. Acute and long-term effects of IL-6 on cultured dorsal root ganglion neurones from adult rat. J Neurochem. 2005;94:238–248. doi: 10.1111/j.1471-4159.2005.03185.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Allchorne A, Safieh-Garabedian B, Poole S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol. 1997;121:417–424. doi: 10.1038/sj.bjp.0701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Yoshimura M. Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett. 1998;255:135–138. doi: 10.1016/s0304-3940(98)00730-7. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU. The glycinergic control of spinal pain processing. Cell Mol Life Sci. 2005;62:2027–2035. doi: 10.1007/s00018-005-5107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]