

Abstract
Using spacers between the C-termini of the long (LF) or short (SF) human prolactin receptors and luciferase/GFP such that BRET occurred minimally in intact versions of these receptors in the absence of ligand, we have monitored the BRET signal after deletion of regions of the extracellular domain (ECD). Deletion of S2 produced ligand-independent BRET for only those pairings normally occurring in the presence of ligand with the intact receptor. Deletion of the similarly-sized S1, or S1 plus S2, produced no ligand-independent or -dependent BRET. When deleted receptors were transfected into human breast (T47D) or prostate (DU145) cancer cells incubated in the absence of added PRL and presence of anti-PRL, expression of the ΔS2LF resulted in increased cell number, whereas expression of the intact receptor did not. When endogenous β-casein expression was examined in T47D cells, the ΔS2LF and ΔS2F1a both showed ligand-independent activation of transcription, again not duplicated by the intact receptor. Paired with evidence in the literature for pre-dimerization of PRLRs, these results demonstrate that altered ECD conformation, and not just a change in bulk, produces altered conformation of the intracellular signaling region of the receptors, supporting the concept that ligand binding to the ECD of intact pre-dimerized receptors could initiate signaling. In addition, the current work supports a dual proliferative and differentiative role for the LF receptor, but only a differentiative role for the SF1a receptor. Naturally-occurring ΔS2 PRLRs were also found in normal and cancerous human cells. This additionally suggests a heretofore unappreciated ligand-independent role for PRLRs.
Keywords: human prolactin receptor, naturally-occurring variant, long and short forms, constitutive activity, cell proliferation, β-casein
Prolactin (PRL) is a pituitary polypeptide hormone characterized by multiple biological actions including growth control in the prostate, and stimulation of development and milk protein gene expression in the mammary gland (1-3). The effects of PRL are mediated by interaction with specific receptors located on the plasma membrane of many target tissues (4,5). PRL receptors (PRLR) belong to the superfamily of hematopoietic cytokine receptors, which are devoid of intrinsic catalytic activity (6). Rather, family members associate with tyrosine kinases that are activated upon binding of the ligand, which in the case of the PRLR, results in one ligand-two receptor ternary complexes (7). Activation requires close proximity of the intracellular domains (7) and can be measured by bioluminescence resonance energy transfer (BRET), as described previously (8,9). Receptors in this family have an extracellular ligand-binding domain, a single hydrophobic transmembrane domain, and an intracellular domain (10). The extracellular domain (ECD) of the prolactin receptor (PRLR) (and other members of this cytokine receptor family) consists of two subdomains of approximately 100 amino acids each, S1 and S2, both of which are involved in ligand binding (10,11). Increasing evidence shows that the two subdomains of the ECD function in very different ways. Gourdou and coworkers (12) examined the properties of mutant forms of the rabbit long form PRLR, in which the S1 or S2 subdomains were deleted, and found deletion of S2 made the receptor constitutively active when using milk protein gene expression as the measure of bioactivity. Lee et al (13) also showed that deletion of most of the ECD (but not specifically the S2 region) of the human long PRLR led to constitutive activation and ligand-independent proliferation of cells. These investigators produced such mutant forms to analyze regions of the ECD involved in ligand binding and the process of receptor dimerization.
More recent work on the PRLR, however, has shown that the receptors are predimerized (14-16), a conclusion previously drawn for several other members of this receptor superfamily (17,18). It is thus suggested that the binding of ligand initiates signal transduction by causing a change in the conformation of the preformed receptor pair. In the current study, we have used the generation of a BRET signal to examine whether a change in conformation of the ECD could in fact result in a change in conformation of the intracellular, signaling region. Binding of PRL to the PRLRs involves parts of both the S1 and S2 regions of the ECD (7,10,11,19). We therefore deleted either the S1 or similarly-sized S2 region of the ECD (or both). In addition, we used the LF and both SFs (SF1a and SF1b) of the human PRLR to take advantage of our previous determination that while all homo- and most hetero-pairs of these receptors form a signaling complex with PRL (8), SF1a and SF1b heteropairs do not.
The current study demonstrates that removal of the S2 extracellular subdomain changes the conformation of the intracellular signaling region of the LF and both SF receptors, thereby supporting the concept that the conformation of the ECD can affect the conformation of the intracellular domain of these single transmembrane receptors, and hence that ligand binding could initiate signaling by this means in predimerized receptors. In addition, the current work supports a dual proliferative and differentiative role for the LF receptor, but only a differentiative role for the SF1a receptor. Moreover, the finding that the ΔS2 versions are produced naturally in human prostate and endothelial cells suggests a heretofore unappreciated ligand-independent role for ΔS2 PRLRs in these tissues.
EXPERIMENTAL PROCEDURES
Materials
The codon-humanized pGFP2-N1 (h) and pRluc-N1 (h) vectors, containing multiple cloning sites upstream of the GFP2 gene or Rluc gene, and the luciferase substrate, DeepBlueC, were purchased from Perkin Elmer (Wellesley, MA). Coelenterazine h, another substrate for Rluc used for the spectral studies and receptor binding assays was purchased from Assay Designs, Inc. (Ann Arbor, MI). The PRLR cDNAs, encoding the LF, SF1a and SF1b were originally kindly provided by Dr B.K. Vonderhaar (NCI, Bethesda, MD) and were cloned into the GFP and Rluc vectors as described previously (8). The cell proliferation assay reagents, MTS ((3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) and PMS (phenazine methosulfate) were products of Promega (Madison, WI, USA). Reverse transcriptase and pfu polymerase were purchased from Stratagene (La Jolla, CA, USA). The transfection reagent, lipofectamine 2000, was from Invitrogen (Carlsbad, CA). Unmodified PRL was purified from E. Coli (BL21) as described previously (20).
Generation of eukaryotic expression vectors for PRLR mutants tagged with GFP2 or Rluc
The cDNA for the human PRLR in a mammalian expression vector has been described previously (8). Mutants were established by PCR splicing. As shown in Fig.1, to create ΔS1 or ΔS2 forms of the PRLRs, 101 amino acid (303 bp nucleotide) were deleted (amino acid 3−103 in S1 or 106−206 in S2). Furthermore, to create ΔS1S2, 204 amino acid (3−206) were deleted. Four primers (Table 1) were required for each construct, two flanking primers (primers 2 and 3) and two hybrid primers (primers 1 and 4). The two flanking primers were based on the sequence immediately before and after the part to be deleted, and primers 2 and 3 were complementary to each other. In this study, 15 bases were designed on each side of the sequence to be deleted, and combined with a 30 base primer in the next PCR reaction to generate fragments with deletion of the S1, S2 sequence, or both.
Fig 1. Diagram illustrating the different versions of the PRLR used.
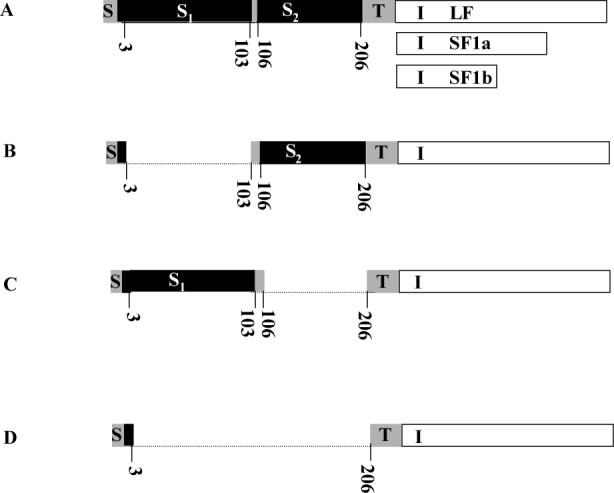
A, intact human LF PRLR with the shorter intracellular regions of SF1a and SF1b illustrated. The transmembrane and ECD regions are the same for all three; B, S1- deleted; C, S2-deleted and D, S1- and S2-deleted The dotted line illustrates the part deleted and the numbers given are the amino acids in the mature sequence. S, signal peptide; S1, subdomain 1; S2, subdomain 2, of the extracellular domain; T, transmembrane; I, intracellular part of long form (LF), short form (SF) 1a and SF1b.
Table 1.
Flanking (2,3) and hybrid (1,4) primers used in PCR splicing(a)
| Construct | flanking primers | hybrid primers | ||
|---|---|---|---|---|
| ΔS1-LF | 2 | 5′-ctccaaaggagggtctaactgtccattcag-3′ | 1 | 5′-gacacgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-ctgaatggacagttagaccctcctttggag-3′ | 4 | 5′-aacggtaccagtgaaaggagtgtgt-3′ | |
| ΔS1-SF1a | 2 | 5′-ctccaaaggagggtctaactgtccattcag-3′ | 1 | 5′-gacacgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-ctgaatggacagttagaccctcctttggag-3′ | 4 | 5′- aacggtaccactggactgtggtcaa-3′ | |
| ΔS1-SF1b | 2 | 5′-ctccaaaggagggtctaactgtccattcag-3′ | 1 | 5′-gacacgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-ctgaatggacagttagaccctcctttggag-3′ | 4 | 5′- aacggtaccaaggggtcacctccaa-3′ | |
| ΔS2-LF | 2 | 5′-tgtatcattcatggtagggtctggctgaac-3′ | 1 | 5′-gac acgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-gttcagccagaccctaccatgaatgataca-3′ | 4 | 5′-aacggtaccagtgaaaggagtgtgt-3′ | |
| ΔS2-SF1a | 2 | 5′-tgtatcattcatggtagggtctggctgaac-3′ | 1 | 5′-gac acgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-gttcagccagaccctaccatgaatgataca-3′ | 4 | 5′- aacggtaccactggactgtggtcaa-3′ | |
| ΔS2-SF1b | 2 | 5′-tgtatcattcatggtagggtctggctgaac-3′ | 1 | 5′-gac acgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-gttcagccagaccctaccatgaatgataca-3′ | 4 | 5′- aacggtaccaaggggtcacctccaa-3′ | |
| ΔS1S2-LF | 2 | 5′-tgtatcattcatggttaactgtccattcag-3′ | 1 | 5′-gacacgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-ctgaatggacagttaaccatgaatgataca-3′ | 4 | 5′-aacggtaccagtgaaaggagtgtgt-3′ | |
| ΔS1S2-SF1a | 2 | 5′-tgtatcattcatggttaactgtccattcag-3′ | 1 | 5′-gacacgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-ctgaatggacagttaaccatgaatgataca-3′ | 4 | 5′- aacggtaccactggactgtggtcaa-3′ | |
| ΔS1S2-SF1b | 2 | 5′-tgtatcattcatggttaactgtccattcag-3′ | 1 | 5′-gacacgcgtaccatgaaggaaaatgtg-3′ |
| 3 | 5′-ctgaatggacagttaaccatgaatgataca-3′ | 4 | 5′- aacggtaccaaggggtcacctccaa-3′ |
Note: The nucleotides in bold face are restriction sites designed to ligate into eukaryotic expression vector GFP2N1 or Rluc N1.
Additionally, to generate these deleted PRLRs tagged with Rluc or GFP2 at the carboxy-terminus, forming ΔS1 PRLR-GFP2, ΔS2 PRLR-GFP2, ΔS1S2 PRLR-GFP2, and ΔS1 PRLR-Rluc, ΔS2 PRLR-Rluc, ΔS1S2 PRLR-Rluc, the extra DNA base pairs corresponding to Mlu I (ACGCGT) and Kpn I (GGTACC) were designed to be upstream of the initiation codon on the forward primer 1 and to replace the stop codon on the reverse primer 4 (the nucleotide in bold face in table 1). An extra triplet ACC was also designed to be immediately prior to the initiator codon, ATG, to facilitate and therefore increase the translation rate in transfected mammalian cells. The high fidelity proof-reading DNA polymerase (pfu polymerase) from Stratagene was used. The original plasmids, pEF6-LF, pEF4-SF1a and pCR2.1-SF1b, which contain the entire coding sequence for LF (1866 bp), SF1a (1128 bp) and SF1b (864 bp), respectively, were used as template in the first stage PCR.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) containing high glucose, 1 mM sodium pyruvate and pyridoxine hydrochloride, 2 mM L-glutamine, supplemented with 10% fetal bovine serum, 100 units /ml penicillin and 0.7 μM streptomycin. The cells were seeded at a density of 5 × 105 cells per well of a six-well (or as indicated) plate. Transient transfections were performed the following day when the cells were 90%−95% confluent using Lipofectamine™ 2000 (Invitrogen) in accordance with the protocol provided by the vendor. Briefly, 3.5−4.5 μg DNA was used per well (depending on the experiment). The DNA was initially incubated in 250 μl of Opti-MEM®I (Invitrogen,) (without serum and antibiotics) and mixed gently. A 1:25 dilution of Lipofectamine™ 2000 in 250 μl of the same medium was then prepared. After a 5-min incubation at room temperature, the diluted DNA and Lipofectamine™ 2000 solutions were mixed and incubated at room temperature for 20 min to allow the DNA-Lipofectamine™2000 complexes to form. The complexes were then added to the cells (in medium with serum but without antibiotics) and mixed by rocking the plate back and forth. After a 48h incubation of the cells at 37°C in a CO2 incubator, the cells were subjected to BRET analysis as described previously (8).
Confocal Imaging
Confocal microscopy (using a Zeiss 510, Germany) was applied to check cellular expression and localization of GFP2-tagged PRLRs. HEK293 Cells were plated at a density of 5 × 105 cells/35mm well on polylysine-coated coverslips (Φ=12 mm) and cultured in DMEM as described above. One day following plating, when the cells reached about 90% confluency, the cells were transfected with 0.8 μg DNA/35mm well using lipofectamine 2000 as described above. Forty eight hours after transfection, microscopic observation was performed.
Living cell-based binding and uptake study
In order to investigate the binding/uptake properties of the deleted receptors in living cells, we developed a luminescence based method. Briefly, the cDNA coding sequence of PRL and Gaussia-luciferase (Nanolight technologies, Pinetop, AZ), a luciferase of 17kD, were amplified and fused from the original plasmid PT7-SCII-prl and pcDNA-3.1Gluc based on a PCR approach. A spacer coding for a 14 amino acid linker in the fusion protein was also included. The sequence of the spacer was GSRYRGPGLPPVAT. The whole fusion fragment cDNA (prl-Gluc) was then sub-cloned into a mammalian expression vector, pcDNA3.1 which additionally codes for the signal sequence. Finally, transfection of pcDNA3.1-prl(Gluc) into 293 cells was carried out and conditioned medium (using horse serum so that the conditioned medium could be assayed in the Nb2 bioassay) containing PRL-Gluc was collected. The PRL bioactivity of the fusion protein was determined by Nb2 assay, as previously described (20,21). A dilution of conditioned medium equivalent to 50 ng/ml of unmodified PRL in the same bioassay was chosen to compare binding and uptake abilities at physiological concentration. To assess binding/uptake, cells were incubated in conditioned medium (diluted with fresh culture medium containing serum to achieve the correct concentration) for 3 h in the absence and presence of a 50 fold excess of unmodified PRL. Binding/uptake in the presence of excess PRL was considered non-specific. A separate, non-specific binding/uptake control was run for each receptor type. The cells were then washed three times with PBS before incubation in BRET2 buffer (Dulbecco's PBS containing 0.9 mM CaCl2, 0.5 mM MgCl2·6H2O and 5.5 mM D-glucose) for 5 min and measurement of luminescence intensity.
RNA isolation, RTPCR and Western blot
Forty eight hours after transfection with intact or deleted PRLR, the T47D cells were washed three times with Dulbecco's PBS, and RNA extractions were performed using TRIzol reagent (Invitrogen) according to the protocol suggested by the vendor. Total RNA was used for first-strand synthesis of cDNA using reverse transcriptase (Stratagene, La Jolla, CA, USA). Briefly, 5μg total RNA, 3μl random primer (Epicentre, Madison, WI, USA) in nuclease-free water were incubated at 65°C for 5 min, slowly cooled to room temperature to allow the primers to anneal to RNA. Then, 5μl 5 × RT buffer, 1μl RNase inhibitor, 2μl 100 mM dNTP mix, 1μl reverse transcriptase (Stratagene), were added and mixed and incubated at 42°C for 1 h. For PCR, 1μl RT product was added to a 50μl reaction containing 5μl 10 × pfu polymerase buffer, 4μl dNTP (4mM), 2.5μl forward or reverse primer (10mM) and 1μl pfu polymerase. β-casein mRNA was detected using the following two primers: 5’-acactgtctacactaagggc-3’(forward) and 5’-tggctgagtcacagggtaga-3’(reverse), which yield a 303 bp fragment of the β-casein open reading frame. PCR parameters were as follows: 95°C for 1 min, 55°C for 50 sec, 72°C for 2 min, with a final extension period of 72°C for 10 min. β-casein mRNA is normally expressed at very low levels in these cells. Cycle titration ensured amplification in the linear range and a semi-quantitative result. All data were normalized to β-actin.
When detecting the presence of the ΔS2 receptor in prostate cancer and endothelial cells, PCR primers with the following sequences were utilized: 5’-atcatgatggtcaatgccacta-3’ (forward) and 5’-tccaacagatgagcatcaaatc-3’ (reverse). Amplifications were carried out for 35 cycles of 95°C for 30 sec, 62°C for 30 sec, and 72°C for 45 sec. A final extension at 72°C was performed after the last cycle for 5 min. For detection of the different isoforms of ΔS2 PRLR, the same forward primer was used, but isoform-specific reverse primers were utilized: 5’-tggggttcctcacacttttc-3’ (LF), 5’-gtaatgagaggcacccaacat-3’ (SF1a) and 5’-aggggtcacctccaacagat-3’ (SF1b). Cycle number differed (see figure legend). The final PCR product was resolved in a 1% agarose gel, stained with ethidium bromide and visualized under UV light. All bands obtained were sequenced to confirm their identity.
For Western blot, whole cell lysates were collected and 500 μg of protein were precleared with 1 μg normal mouse IgG and 20 μl protein A/G agarose (Santa Cruz Biotechnology, Santa Cruz, CA). Pre-cleared lysates were then incubated with 1 μg mouse anti-PRLR ECD antibody (Zymed, Carlsbad, CA) at 4°C overnight. Immunocomplexes were captured with protein A/G agarose and the beads were washed 3 times before dissolution of attached proteins in SDS loading buffer. SDS gel separation, transfer, membrane blocking and incubation in anti-PRLR were as described previously (22), using an antibody dilution of 1:1000. Immunoprecipitation with normal mouse IgG served as the negative control.
Cell proliferation assay (MTS assay)
This assay was performed under stringent conditions previously described (21). Briefly, 42 mg of MTS reagent powder was dissolved in 21ml DPBS and filter-sterilized using a 0.2μm filter. To this was added 100μl PMS/ 2ml MTS solution under light-protected conditions. Twenty μl of the freshly combined MTS/PMS solution was added to each well of a 96-well assay plate containing 100μl of cells in culture medium without serum. The cells plus reagents were then incubated at 37 °C, 5% CO2 for 3 h, after which time the absorbance at 490 nm was recorded using a microplate spectrophotometer. The number of cells was in the linear range of the assay.
Fluorescence and BRET2 Measurements
Transfected cells were harvested within 48 h of transfection by washing with DPBS (three times), detaching in DPBS containing 2 mM EDTA (Ethylenediaminetetraacetic acid), pelleting by centrifugation and resuspension in BRET2 buffer (DPBS containing 0.9 mM CaCl2, 0.5 mM MgCl2·6H2O and 5.5 mM D-glucose) at a density of 2 × 106/ml. After this and before the BRET2 measurements, the cells were incubated at 37°C for 1h. For each measurement, 0.5 ml of the cell suspension was loaded into a 0.5-cm2 quartz cuvette. Fluorescence and bioluminescence spectral scanning were performed using a FluoroMax-2 spectrofluorometer. Bioluminescence scanning and BRET2 signal detection were carried out immediately after the addition of 5 μM of the cell permeant luciferase substrate, DeepBlueC, with a black card in the path of the excitation light beam. Data were collected with the slits set at 5 nm, datum points were collected at 5 nm intervals, and signal integration was for 0.5 s per datum point). Energy transfer was defined as the BRET ratio (Emission500nm-520nm-background500nm-520nm)/(Emission385nm-420 nm-background 385nm-420 nm). The signals obtained from non-transfected cells were considered background.
Statistical analysis
All experiments were repeated a minimum of three times and most were repeated five or six times. Numerical data are presented as the mean ± SEM and statistical significance was determined by ANOVA with posttests using Bonferroni corrections for multiple comparisons against a single control, where appropriate.
RESULTS
Construction of eukaryotic expression vectors for ECD-mutated PRLRs tagged with GFP2 or Renilla luciferase
A set of ECD-mutant receptors were constructed using a rapid splicing PCR approach. For each receptor, we produced three mutant forms: S1 deletion, S2 deletion or a form with both S1 and S2 deleted, named ΔS1, ΔS2 or ΔS1S2, respectively (Fig.1). Mutant sequences were confirmed by sequencing. To detect close approximation of the receptors, we used BRET and hence each mutant receptor was tagged with either Renilla luciferase (Rluc), which acts as an energy donor, or a variant of green fluorescence protein (GFP2), as an energy acceptor. The luciferase or GFP2 were separated from the C termini of the receptors by 13 or 12 amino acid linkers, respectively. Insertion of these linkers has been previously shown to prevent most BRET in the intact receptors in the absence of ligand (8 and figure herein). Eighteen plasmids were constructed: ΔS1LF-GFP2, ΔS2LF-GFP2, ΔS1S2LFGFP2, ΔS1SF1a-GFP2, ΔS2SF1a-GFP2, ΔS1S2SF1a-GFP2, ΔS1SF1b-GFP2, ΔS2SF1b-GFP2, ΔS1S2SF1b-GFP2, ΔS1LF-Rluc, ΔS2LF-Rluc, ΔS1S2LF-Rluc, ΔS1SF1a-Rluc, ΔS2SF1a-Rluc, ΔS1S2SF1a-Rluc, ΔS1SF1b-Rluc, ΔS2SF1b-Rluc, ΔS1S2SF1b-Rluc. Energy transfer occurs when the Rluc and GFP2 molecules come within ∼ 100 Å of one another and is initiated by the addition of the Rluc substrate, DeepBlueC (DBC), as described previously (8, 9).
In order to verify expression and comparable localization of the receptors, confocal fluorescence microscopy and spectrofluorimetry were performed on HEK 293 cells 48h post transfection with the GFP2 plasmids. As shown in Fig 2 (A), a proportion of each expressed transfected receptor translocated to the region of the plasma membrane after synthesis. Because the receptors are synthesized in the rough endoplasmic reticulum and travel through the Golgi on their way to the surface, regions of the cell consistent with these intracellular organelles were also highly fluorescent. Similar subcellular distributions were observed in human breast cancer T47D cells and prostate cancer DU145 cells (data not shown). Intracellular distribution was very similar to GFP2 tagged versions of the intact receptors previously analyzed (8).
Fig 2. Confocal images demonstrating expression, localization (A) and spectral properties (B) of the GFP2 and/or Rluc-tagged deleted human PRLRs.
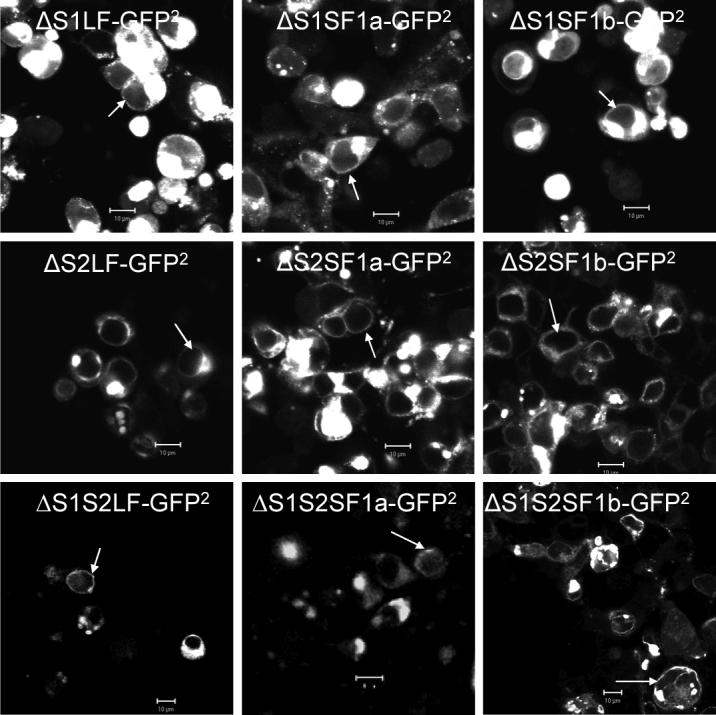
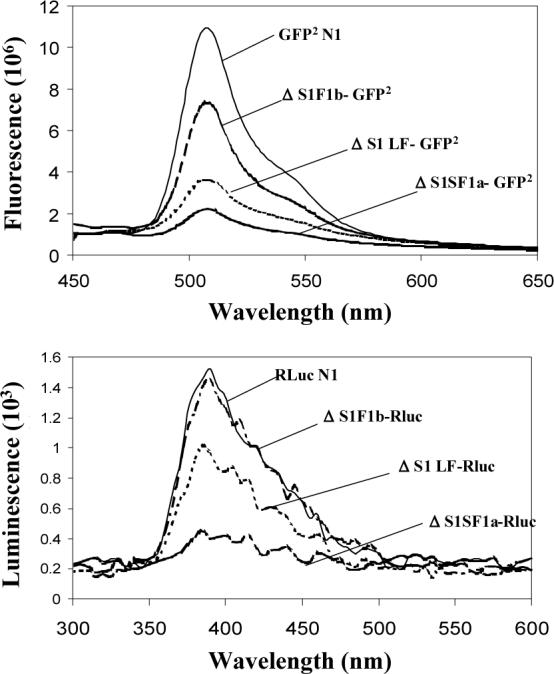
HEK 293 cells transiently transfected with plasmids containing cDNA coding for the receptors tagged with GFP2 were examined by confocal microscopy after 48h. bar,10μm; white arrowhead, localization of the tagged receptors to the region of the plasma membrane. For spectral scanning, GFP2 was excited at 405nm. Bioluminescence generated by Rluc was detected in the presence of substrate, DeepBlueC (5 μM), with blockade of the external excitation light source. Only the spectra for the ΔS1 versions are shown by way of example, but all spectra were unaltered by attachment to the various receptors. GFP2N1 is the GFP2 expressing vector without any receptor, and RLuc1N1 is the luciferase expressing vector without receptor. The relative expression of each receptor can be appreciated by comparing the amount of fluorescence or luciferase activity. Relative expression levels were the same for the intact and deleted versions of the receptor.
To verify normal fluorescence and bioluminescence spectral properties of the GFP2 and Rluc, when attached to all versions of the receptors, measurements were performed using a spectrofluorometer, as described previously for the intact receptors (8, 9). All GFP2- and Rluc-tagged receptors had normal spectral properties. The spectra for the ΔS1 forms are shown as an example (Fig. 2B). On the basis of amount of fluorescence and luciferase activity, one can appreciate that transfection with equal amounts of cDNA results in different degrees of expression of each receptor, with SF1b being most efficiently expressed, followed by the LF and SF1a, the same as previously reported for the intact receptors, either untagged or similarly tagged (8). The different degrees of expression were not a function of transfection efficiency since transfection efficiency was indistinguishable among constructs on any given occasion (data not shown). Also, all ECD versions of each receptor tested in the current experiments were expressed at the same level (i.e. intact, ΔS1, ΔS2 and ΔS1S2) and each had the described same relative expression (i.e. SF1b>LF>SF1a). In other words, only the intracellular region of the receptor appeared to influence the level of protein expression.
S1 or S2 deletion decreased binding and uptake of the ligand
To investigate the binding and uptake properties of the deleted receptors in living human cells (HEK 293) under physiological conditions with homologous ligand, we developed a luminescence-based approach in which a low molecular weight luciferase was attached to the C-terminus of human PRL (see materials and methods). This assay was conducted at 37°C to ensure normal interaction of the ligand with the ECD and normal movement of the receptors in the lipid bilayer. The PRL-Gluc construct was expressed in human cells (HEK 293), and conditioned medium (contained no FBS) was used as the source of ligand for binding. Biological activity of the Gluc-tagged human PRL was assessed by Nb2 PRL bioassay and compared to untagged human PRL, as shown in figure 3A. The PRL-Gluc showed a concentration-related ability to stimulate Nb2 cell proliferation which reached the same maximum as untagged PRL.
Fig 3. Biological activity of PRL-Gluc and the relative ability of the intact and deleted PRLRs on HEK293 cells to bind and internalize PRL-Gluc.
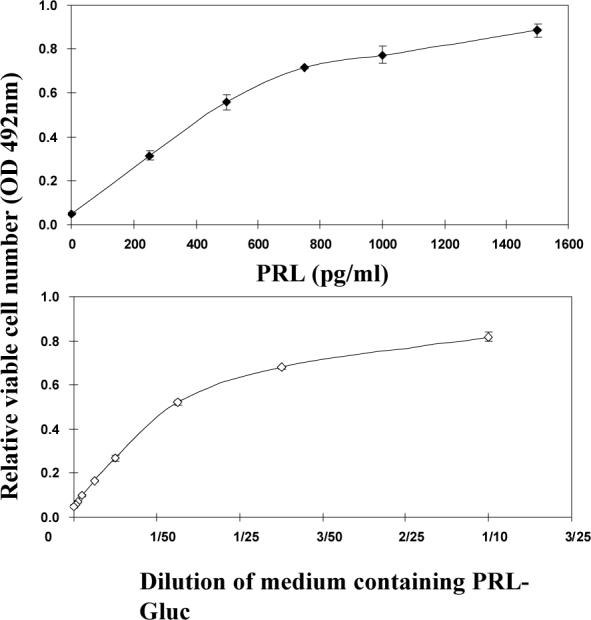
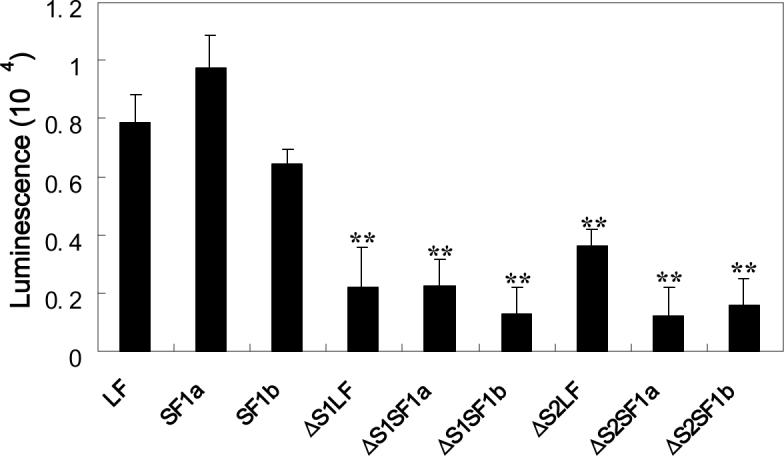
Panel A compares diluted conditioned medium with unmodified human PRL. PRL-Gluc was biologically active, reaching the same maximum as unmodified PRL. OD 492 nm is absorbance in the MTS assay and is representative of relative cell number. A dilution of conditioned medium with bioactivity equivalent to 50ng/ml unmodified PRL was used for the studies in panel B. After a 3h incubation at 37°C, the cells were washed three times and cell-associated luminescence was determined. Non-specific binding/uptake (binding/uptake in the presence of excess unmodified PRL for each receptor type) was subtracted from each result. ** significantly different from their intact counterpart with p<0.01.
For binding/uptake studies, 50 ng/ml PRL activity equivalents of PRL-Gluc were incubated with the cells for 3h at 37°C, the cells were then washed with PBS three times and the relative cell-associated luciferase activity was measured. Nonspecific binding/uptake (which was subtracted from the total and separately measured for each receptor construct) was assessed by binding/uptake in the presence of a 50 fold excess of unmodified PRL (2.5μg/mL). The results showed that deletion of the S1 or S2 subdomain markedly decreased binding/uptake for each of the receptors (Fig.3B). Although consistently higher, this level of binding/uptake was not statistically different from non-specific. The 3h incubation period was used in order to rigorously test for binding. Although this means that there was also uptake by the cells, the logic was that both binding and uptake are receptor-mediated processes and hence that the long incubation should amplify any specific binding.
Prolactin-independent BRET in homo- and hetero-pairs of ΔS2 receptors
To assess approximation of the intracellular regions of the deleted receptor forms, BRET was measured. In this technology, when the energy donor and acceptor protein come within 100 Å of each other, energy transfer from donor to acceptor occurs. The amount of energy transfer is given as the BRET ratio, defined under methods. In the absence of PRL, there was a small background BRET ratio for the intact homo-paired receptors (Fig. 4A). This was unchanged by S1 deletion. S2 deletion on the other hand increased the BRET ratio to a level more than 3 fold background with the intact receptors. Deletion of both S1 and S2 resulted in no significant change from the intact receptors (Fig. 4A). Only the ΔS2 versions were therefore tested for the receptor heteropairs (Fig. 4B). For ΔS2LF-ΔS2SF1a, the BRET ratio was 4 fold that of the intact receptor. For ΔS2LF-ΔS2 SF1b, the BRET ratio was double the intact receptor. Addition of PRL to ΔS1, ΔS2 and ΔS1S2 homopairs did not change the BRET ratios of these deleted mutants (Fig 4C), thereby illustrating a lack of significant effect of any potential residual PRL binding in these receptors. Although described sequentially as knowledge developed, the data presented in panels A-C of figure 4 were obtained on the same occasion.
Fig 4. Bioluminescence resonance energy transfer with intact and variously deleted receptors and combinations of receptors in the absence (A and B) and presence (C) of PRL.
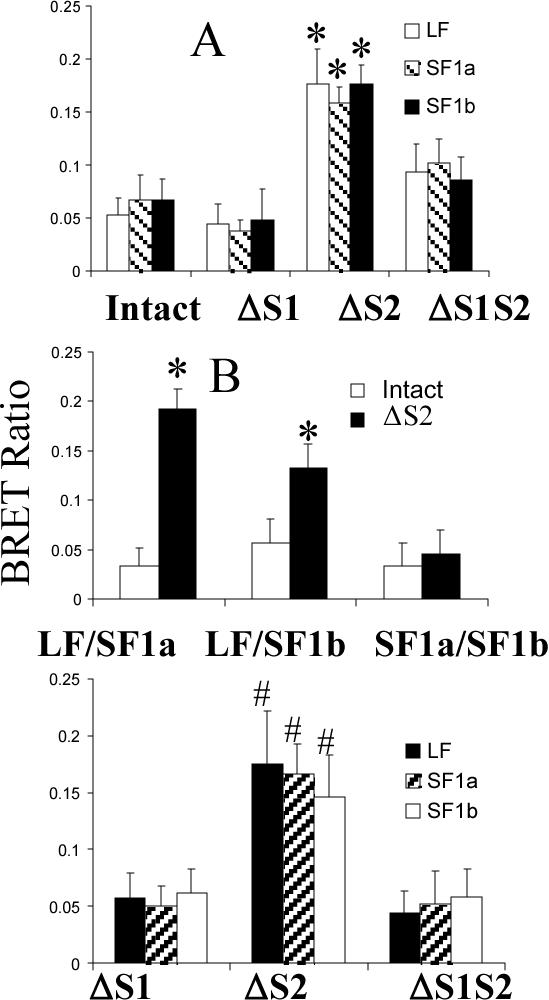
HEK 293 cells were transfected with equal quantities of GFP2- or luc-tagged receptors and 48h later were subjected to BRET analysis initiated by the addition of substrate. For panel B, the same result was obtained regardless of which receptor in the pair was tagged with GFP or luc. Results in all three panels are the mean ± SEM derived from experiments in which all variables from all panels were concurrently analyzed. Energy transfer is given as the BRET ratio (Emission500nm-520nm-background500nm-520nm)/(Emission385nm-420nm-background 385nm-420nm). BRET signals obtained from nontransfected cells were considered background in this equation. *, significantly different from the intact version with p<0.05; #, significantly different from ΔS1 and ΔS1S2 (p<0.05) and not different from ΔS2 in panel A.
Effect of the deleted PRLRs on breast/prostate cancer cell growth and endogenous β-casein gene transcription
To assess the biological result of the deletions, receptors were transiently transfected into human breast (T47D) or prostate (DU145) cancer cells. Once again, transfection efficiency of each ECD form of the receptor was the same, but expression levels of the protein were different with the same relative expression levels as for the HEK293 cells. Forty eight hours post-transfection, the cells were cultured for a further 24h period in fresh medium in the continued absence of added PRL. Cell number was then assessed by MTS assay in serum free medium (21), or RNA was extracted. As shown in Figs. 5A and B, which show relative viable cell number, only the ΔS2LF had a significant effect and this was to increase cell number in both human breast cancer cells (A) and human prostate cancer cells (B). The presented data are only normalized to the control transfected cells so that the degree of change can be appreciated and are not adjusted for transfection efficiency in the culture. Adjustment for transfection efficiency would approximately triple the response. Addition of an antibody to human PRL did not change this overall result (data not shown), although cell number in all incubations went down. The reduced cell number demonstrates that the antibody was effective at binding and neutralizing autocrine PRL. The antibody used was a chicken anti-human PRL raised in the laboratory and demonstrated at the same concentration (1μg/ml) to completely block human PRL-stimulated Nb2 cell proliferation during a 3 day incubation (data not shown). The cell proliferation result was therefore unlikely due to autocrine PRL and residual binding to the ΔS2LF. To test the possibility that overcrowding of receptors (due to overexpression) could produce proliferation, intact versions of the receptors were also used. No significant effect on cell proliferation (versus controls similarly incubated) was observed with the intact receptors (which were expressed at very similar levels) in the absence of added ligand and presence of anti-PRL. This control also negates the possibility that autocrine PRL may have interacted with the ΔS2 receptors in an intracellular compartment not accessed by anti-PRL.
Fig 5. Effect of intact and deleted PRLRs on the growth of human breast cancer T47D cells (A) and human prostate cancer DU145 cells (B) in the absence of added PRL.
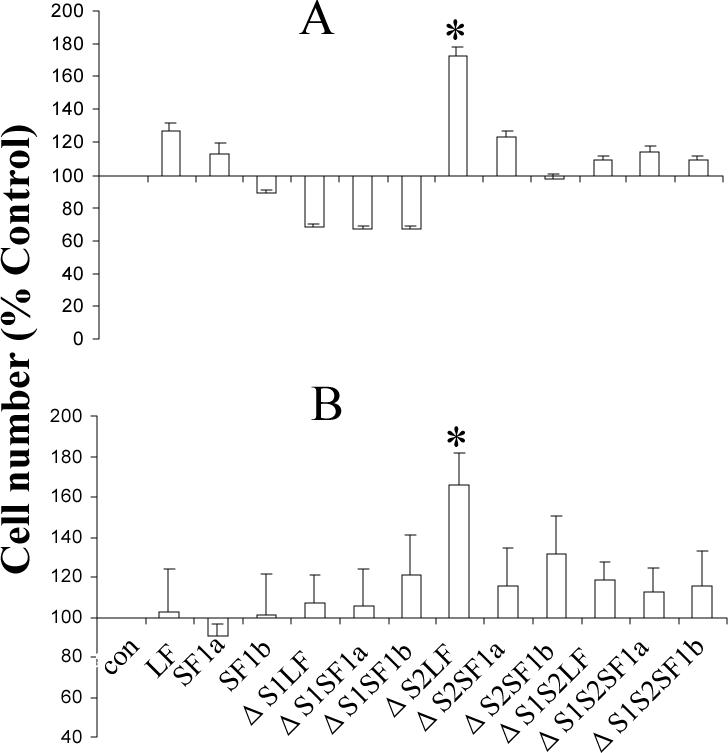
Receptor constructs were transfected into either T47D or DU145 cells. Forty eight hours post transfection, the medium with serum was refreshed. After a further 24h, the medium was changed to serum-free DMEM to conduct the MTS assay. Data are expressed as a percent of the control to illustrate the change induced. The data are not corrected for either transfection efficiency or expression efficiency for reasons discussed in the text. The same overall result was obtained when anti-PRL was added to the incubation, although cell number was decreased in each incubation indicative of an effect of the antibody on autocrine PRL. *, significantly different from the intact counterpart with p<0.05.
Also, in human breast cancer cells, but not in the DU145 cells, a consistent reduction, but not statistically significant decrease, in relative cell number was observed with all three ΔS1 receptors (ΔS1LF, ΔS1SF1a and ΔS1SF1b).
When endogenous β-casein gene expression was analyzed, a significant increase in expression occurred in ΔS2LF and ΔS2SF1a transfected cells (Fig.6). Once again, although normalized to the control transfected cells, these data have not been normalized for transfection efficiency. This result was also not altered by the addition of anti-human PRL to the medium (data not shown). Since the ΔS2SF1a generated a response that was indistinguishable from that of the ΔS2LF in terms of β-casein gene expression, it seems unlikely that the lack of effect of the ΔS2SF1a on cell proliferation was due to the lower level of expression of this form. Clearly, since the ΔS2SF1b was expressed the most efficiently, the level of expression is not the cause for an absence of significant effect of this form.
Fig 6. Effect of the ΔS1 and ΔS2 receptors on endogenous β-casein gene expression in T47D cells.
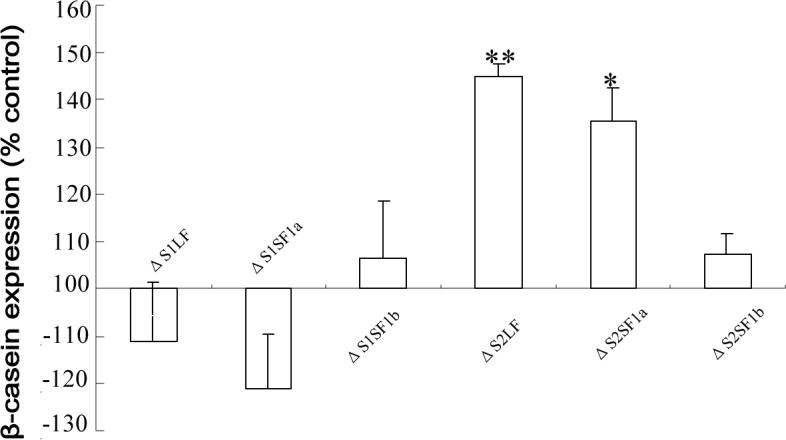
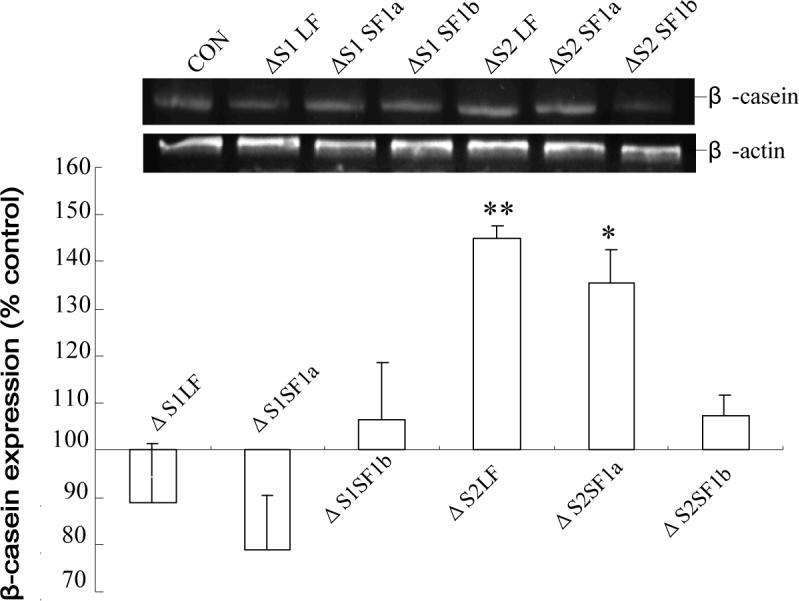
Forty eight hours post transfection, the cells were subjected to a further 24 h incubation in the absence of added PRL and then RT-PCR for β-casein was performed. Data are expressed relative to expression in non-transfected cells (which was the same as that in cells expressing the doubly deleted S1S2 receptor) to illustrate the changes observed. The data are not corrected for either transfection or expression efficiency. The same overall result was obtained in the presence of anti-PRL. *, significantly different from both the non-transfected and ΔS1S2-transfected cells with p<0.05.
Expression of naturally occurring ΔS2 PRLRs in human cells
Given the constitutive activity of the ΔS2 PRLRs, we examined the possibility that some form of this variant occurred naturally in human cells. RT-PCR results showed that in addition to the intact receptors, an amplicon approximately 300 bp shorter than the intact receptor was detected in human prostate cancer cells, LNCaP, DU145, PC-3, and human microvascular endothelial cells, HmVEC-1 (Fig. 7A). In DU145 cells, this amplicon was present in larger amounts than the intact receptor. DNA sequence analysis revealed a 303 bp deletion, which generated a transcript lacking the S2 region (amino acids 106−206), and which serendipitously exactly matched the one we had constructed. Next, we examined whether the LF, SF1a and SF1b, each have a ΔS2 variant. Isoform-specific reverse primers were utilized in RT-PCR analysis. Figure 7B shows PC-3 cells as an example and illustrates that all three ΔS2 PRLR isoforms, as well as the intact receptors, can be detected. Once again, sequencing verified the nature of the band. It should be noted when looking at these results that different numbers of cycles were used for each form (see figure legend). Previous analysis of DU145 and PC3 cells had not detected any intact SF1a (22), but a very small amount of intact and more ΔS2SF1a was detected in the current study, perhaps due to the use of different primers (see methods). To ensure that the mRNA was translated into protein, Western blot analysis was performed. Figure 7C demonstrates an anti-PRLR positive band in LNCaP, PC3 and T47D cells that runs at the same mol wt (filled arrow) as ΔS2SF1b in stably transfected T47D cells (TΔS2). Because this methodology is not capable of distinguishing between ΔS1 and ΔS2, the possibility that this could be a ΔS1 form was examined by performing RT-PCR with primers specific for such forms. No ΔS1 forms were detected in these cells, thereby supporting the idea that the positive band on the Western was indeed the ΔS2SF1b. The band for a LF is also indicated by the open arrow since this serves as a positive control for the immunoprecipitation and Western. Although the antibody used for both immunoprecipitation and Western recognizes the remaining ECD with the ΔS2 versions, there is no means to quantify the relative affinity of this antibody versus the intact ECD and so one cannot draw conclusions as to the relative amounts of each form of the receptor expressed at the protein level. Making the assumption that the mRNA is translated in proportion to its presence, however, it appears that the ΔS2 versions could play a significant role in PRLR function in these cells.
Fig 7. Natural expression of ΔS2 PRLRs.
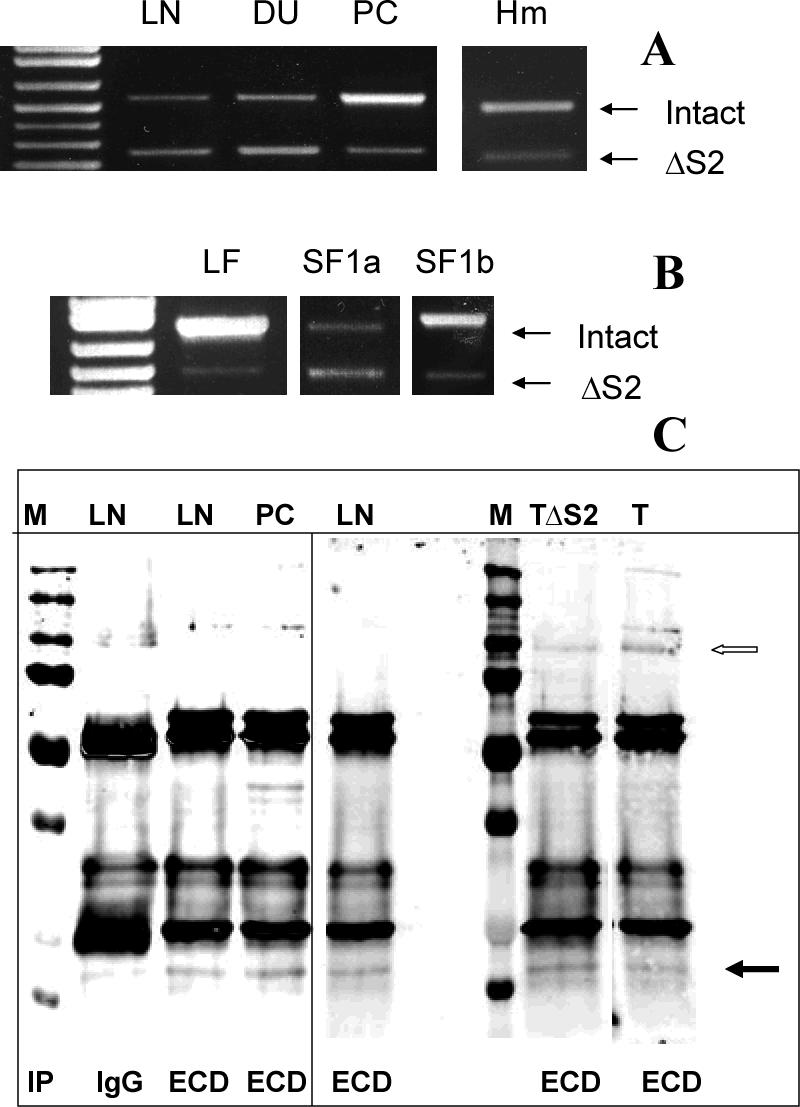
(A) Amplicons resulting from the expression of both intact and ΔS2 PRLR were observed in human LNCaP (LN), DU145 (DU), PC3 (PC) and microvessel human endothelial (Hm) cells and their identities were confirmed by sequencing. (B) Use of form-specific reverse primers demonstrated the presence of a ΔS2 variant of each receptor in all cells, and PC3 cells are illustrated by way of example. Again, the identities of the amplicons were verified by sequencing. Note that different numbers of cycles were required to demonstrate this for each receptor isoform and so the panels cannot be compared in terms of relative abundance (LF and SF1a, 35 cycles; SF1b, 40 cycles). (C) After immunoprecipitation, ΔS2 versions of SF1b are illustrated (closed arrow) as detected by Western blot (using the same anti-ECD antibody) in PC3, LNCaP and T47D (T) cells. Also illustrated is co-migration of immunoprecipitated ΔS2SF1b stably expressed under the control of tetracycline in T47D cells (TΔS2). M, molecular mass markers; IP, immunoprecipitated with control isotype-matched antibody (IgG) or antibody against the extracellular domain (ECD); open arrow marks a LF of the receptor. The vertical line separates samples run on different gels. The sample from normal T47D cells (T) was run on the same gel, but the lane was cut and pasted to be next to the TΔS2 cells to eliminate distracting additional experiments.
DISCUSSION
To ask the kinds of questions we wanted in this and previous studies (8,9), a spacer was placed between the C-termini of the receptors and the Rluc or GFP2 such that minimal BRET occurred in the intact receptors in the absence of ligand. A conformational change in the intracellular regions of the receptors therefore has to occur in order to generate a BRET signal. A BRET signal occurs when the donor and acceptor molecules are within 100 Å of each other, such as is necessary to generate transphosphorylation of the receptor-associated kinases (23).This methodology is in contrast to work from the Dufau lab (15) examining PRLRs where no spacer was reported, resulting in constitutive BRET in intact receptors in the absence of ligand. Our results therefore in no way contradict the notion of receptor pre-dimerization.
The nature of the conformational change resulting in closer approximation of the intracellular regions seems likely to involve rotation of one receptor in relation to the other. We cartooned this for the PRLR in a review in 2005 (24) based in large part on a pioneering study by Seubert et al. on the erythropoietin receptor (25). More recently, work from the Water's lab on the growth hormone receptor (GHR) (17) supports this concept. In the current study, deletion of the S2 domain of all three receptors resulted in close approximation of the intracellular domains and therefore constitutive BRET for all homopairs and LF-SF heteropairs. Thus, one might suggest that removal of the S2 region allowed a conformation closer to the ligand-bound state, and hence constitutive activation.
Heteropairs between the two ΔS2 SFs did not produce constitutive BRET, a finding in keeping with our previous publication which found no ligand-induced BRET after this pairing using the intact receptors (8). Thus, removal of the S2 region only facilitates the correct intracellular conformational change with normal receptor pairs and does not allow abnormal pairing. These results also suggest that the S2 region serves a similar function in all three forms of the human PRLR. That function appears to be to hold the intracellular region of the dimerized receptor in a conformation which does not allow signaling until PRL binds.
When BRET was examined using the similarly-sized ΔS1 receptors or the more completely deleted ΔS1S2 receptors, there was no significant difference from the controls. This means that the generation of a BRET signal is not the result of reduced bulk of the extracellular region of the receptor, but within the parameters tested in the current study is specific to the changes induced by removal of the S2 region. Since the entire membrane proximal region of S2 was removed, we can also conclude that adoption of an active signaling conformation of the intracellular domain does not require this part, but rather that some part of this subdomain of the intact receptor inhibits the adoption of the active signaling conformation by the intracellular domain, an inhibition that is relieved by the binding of PRL. This result is consistent with previous work by Gordou et al. using the LF of the rabbit receptor (12). Given that bulk of the extracellular region was not the significant parameter, we can conclude that it is conformation of the ECD that is important and further, that this constitutes evidence that a change in conformation of the ECD can result in a change in conformation of the intracellular signaling regions of the complex. In the T47D cells, there appeared to be a trend towards decreased cell number with the ΔS1 receptors. This was not reproduced in the DU145 cells. Since this effect was not statistically significant, it may only reflect a chance result. Alternatively, it may be an effect seen only in cells that normally express high levels of intact receptors. Analysis of this possibility will require further experimentation with adenoviral vectors to increase transfection efficiency.
Because there are many unknowns about signal generation from the short receptors, we chose to monitor known end results of signaling, rather than activation of signaling molecules, in order to link the generation of a BRET signal to a biologically relevant effect such as cell proliferation and activation of β-casein gene expression.
Of the ΔS2 constitutively active forms, only ΔS2LF increased cell number over that seen with intact receptors in the absence of ligand. Use of the intact receptors was an important control because of the possibility that overexpression by itself could activate signaling. The magnitude of the increase was not large, primarily because these were transient transfection assays with about 30% transfection efficiency. The data presented were not normalized for transfection efficiency, but had they been they would have shown more than a doubling in the actual transfected cells. The same result was obtained with both T47D cells and DU145 cells which inherently express very different levels of PRLRs. From the current results, therefore, it is clear that signaling through the LF increases cell number. This function for the LF is in agreement with the results of Lee et al.(13) and work showing activation of cyclin D1 by the LF (26). Analysis of β-casein expression showed that constitutive activation of both the LF and SF1a increased expression, a result consistent with many previous studies for the LF and with a previous study from this laboratory in collaboration with the Vonderhaar group on the SF1a (8). Once again, the data presented were not normalized for transfection efficiency and hence the effect per transfected cell is greater than illustrated. It is also interesting to note that the effect of ΔS2SF1a is very similar to LF, despite the fact that ΔS2SF1a is expressed at a lower overall level in the cells. This may reflect a greater efficiency of SF1a receptors or may reflect an equal appropriate localization in the cell. Certainly, with the intact receptors, there was very similar cell-associated PRL-Gluc despite very different overall levels of expression. As described previously (8), there is no suggestion that activation of β-casein does not require Stat 5 since these cells have a high background Stat 5 activation to which activation of other signaling cascades can add.
Based on these findings, one might suggest that PRL promotes proliferation and differentiation in cells expressing mostly LF, and has a greater differentiative effect in cells with a significant expression of SF1a. Thus the deduced role for prolactin in cancer cell lines may be cell line dependent. The current experiments shed no light on the role of the SF1b receptor since constitutive activity of this receptor did not significantly alter either basal cell proliferation or β-casein expression during the duration of these transient transfection experiments.
In presenting the data, we have chosen not to normalize to either transfection efficiency or the level of expression, although we have reported their similarities or differences throughout. We opted not to normalize to transfection efficiency since this was not reflective of expression, but only the percentage of cells transfected. To additionally normalize to expression gave us two concerns. The first was that the data would then be triply normalized and the second was whether this was appropriate since although total expression of for example LF and SF1a were rather different, the amount of cell associated PRL-Gluc with both intact receptors was the same. This suggests that the total number of intact receptors on the plasma membrane was the same and that this might be the meaningful parameter to which to normalize. That said, however, it is unclear whether the ΔS2 receptors need a plasma membrane localization since they do not require ligand and, at least in other members of this cytokine family (27), Janus kinase 2 can associate with the receptor in the rough endoplasmic reticulum. With these unknowns, it seemed more appropriate to present the data in a more raw form and discuss the possible role of differential expression as we have.
The tertiary structure of the extracellular domain of the rat receptor, as determined by crystallization with placental lactogen, shows that the S1 and S2 regions form six ligand binding loops (L1-L6). Of these, L1, L2 and L3 are located in S1, and L5 and L6 are located in S2, while L4 is in the linker region between the domains (28). Since the ΔS1 and ΔS2 receptors retained some of the loops, it was not unreasonable to ask whether there was any residual ligand binding for these forms. Ligand binding was assessed using a new fusion protein developed in this laboratory in which the C-terminus of PRL is linked to Gaussia luciferase via a 14 amino acid linker. With this size linker and expression in eukaryotic cells, both the luciferase and PRL are biologically active. The luciferase makes the assay very sensitive, allowing us to look at binding under more physiological circumstances. Using an amount of the PRL-Gluc equivalent to 50 ng/ml unmodified PRL, we were able to show similar levels of cell-associated luciferase activity for each of the intact receptors. Removal of S1 or S2 reduced cell-associated luciferase activity to about 25% of that with the intact receptors, a result that was clearly statistically significant. Not significant, however, was the difference between residual luciferase activity and the subtracted non-specific activity (activity after binding in the presence of a 50 fold excess of unmodified PRL). HEK 293 cells do not produce PRL and hence the constitutive BRET seen with the ΔS2 receptors cannot be the result of PRL binding to the remaining portion of the receptor. When PRL was added to the system there was also no change in the amount of BRET. DU145 and T47D cells, on the other hand, produce autocrine PRL (29,30) and so the potential existed for this autocrine PRL to affect proliferation or β-casein expression through some residual binding. However, when antibody was added to the medium during these analyses there was no change in the overall result, although cell number in each incubation was decreased thereby demonstrating that the antibody bound and neutralized the autocrine PRL. Thus, even though others have reported residual binding for a LF devoid of most of the S1 region (31), our results show that this is insufficient to produce BRET or effects on cell proliferation or endogenous β-casein expression. However, in this cited article the very N terminal S1 residues were removed, whereas in our case, amino acids 1−3 and 103−106 remained in our ΔS1 constructs. Perhaps these amino acids before the S2 region decreased access for binding. Our ΔS1 and ΔS2 constructs were very similar to those of Gourdou et al (12) who found no residual binding for either. For the purposes of the current study, the most important issue, substantiated by the lack of effect of 1) intact receptors in the absence of added ligand and presence of anti-PRL, 2) added PRL on the BRET analysis, and 3) anti-PRL on the overall proliferation and β-casein result, was constitutive activity of the ΔS2 receptors.
Given the potential clinical importance of constitutively active receptors, we also asked whether there was any evidence for naturally-occurring constitutively active ΔS2 receptors in human cells. Analysis by RT-PCR showed the expression (at least at the level of mRNA) of ΔS2 forms in three prostate cancer cell lines and human microvascular endothelial cells. The high level of expression of other PRLR forms in T47D cells made definitive PCR demonstration of ΔS2 forms difficult and so these data are not shown. More detailed analysis in the prostate cancer cells showed that the ΔS2 version of all three receptor isoforms could be detected. Not only were these seen as PCR amplicons, but the bands were sequenced to confirm their identity. In addition, Western analysis of PC3 LNCaP and T47D cells showed an anti-PRLR positive band equivalent in size to transfected ΔS2SF1b. When the natural ΔS2 versions of all three receptors were sequenced, they serendipitously were found to have exactly the same sequence as those we had constructed. It therefore appears that human cells have the capability of splicing the receptor to produce a constitutively active form. Of importance is the number of cell types shown to express the ΔS2 forms of the receptor (LnCAP, PC3, DU145, T47D and HmVEC) since this makes the forms less likely to be an oddity of a single transformed cell line. The splice site is not a common one, but has similarities to other reported sites (32). This raises the possibility that the ΔS2 varieties play roles in the regulation of cell proliferation and differentiation.
We conclude that a change in the conformation of the extracellular domain can indeed cause a change in the conformation of the intracellular signaling region, thereby supporting the as yet unproven concept that ligand binding to the ECD of pre-dimerized receptors could have a similar effect. Using the constitutively active receptors, we also directly compared the functions of the LF and both SFs in the regulation of cell proliferation and β-casein expression and found that constitutive activation of the LF results in increased cell number, whereas constitutive activation of either the LF or SF1a results in increased β-casein gene expression. In addition, we showed that human cells express ΔS2 versions of the receptors, in some cells at levels greater than the intact versions at the mRNA level, suggesting a possible role in the regulation of proliferation and differentiation.
ACKNOWLEDGEMENTS
We are grateful to Dr. Barbara K.Vonderhaar for provision of the original receptor constructs and to Dr David Johnson for allowing us continued access to the spectrofluorimeter.
Abbreviations
- PRL
prolactin
- PRLR
prolactin receptor
- LF
long form
- SF
short form
- GFP
green fluorescent protein
- BRET
bioluminescence resonance energy transfer
- ECD
extracellular domain
- Rluc
Renilla luciferase
- Gluc
Gaussia luciferase
- MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- PMS
phenazine methosulfate
- ΔS1
receptor missing the S1 region of the extracellular domain
- ΔS2
receptor missing the S2 region of the extracellular domain
- ΔS1S2
receptor missing both the S1 and S2 regions of the extracellular domain
- DPBS
Dulbecco's phosphate buffered saline
- DMEM
Dulbecco's modified Eagle's medium
Footnotes
This work was supported by California Breast Cancer Research Program grant 10PB-0127, NIH grant DK61005, and by a grant from the Prostate Cancer Foundation. K-T Huang was a recipient of an individual predoctoral fellowship from the Department of Defense Breast Cancer Research Program - BC051103.
REFERENCES
- 1.Nevalainen MT, Valve EM, Ingleton PM, Nurmi M, Martikainen PM, Harkonen PL. Prolactin and prolactin receptors are expressed and functioning in human prostate. J. Clin. Invest. 1997;99:618–627. doi: 10.1172/JCI119204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neville MC, McFadden TB, Forsyth I. Hormonal regulation of mammary differentiation and milk secretion. J. Mammary Gland Biol. Neoplasia. 2002;7:49–66. doi: 10.1023/a:1015770423167. [DOI] [PubMed] [Google Scholar]
- 3.Goffin V, Binart N, Touraine P, Kelly PA. Prolactin: the new biology of an old hormone. Ann. Rev. Physiol. 2002;64:47–67. doi: 10.1146/annurev.physiol.64.081501.131049. [DOI] [PubMed] [Google Scholar]
- 4.Hu Z, Zhuang L, Maria L, Dufau ML. Multiple and Tissue-specific Promoter Control of Gonadal and Non-gonadal Prolactin Receptor Gene Expression. J. Biol. Chem. 1996;271:10242–10246. doi: 10.1074/jbc.271.17.10242. [DOI] [PubMed] [Google Scholar]
- 5.Nagano M, Kelly PA. Tissue distribution and regulation of rat prolactin receptor gene expression. Quantitative analysis by polymerase chain reaction. J. Biol. Chem. 1994;269:13337–13345. [PubMed] [Google Scholar]
- 6.Cosman D. The hematopoietin receptor superfamily. Cytokine. 1993;5:95–106. doi: 10.1016/1043-4666(93)90047-9. [DOI] [PubMed] [Google Scholar]
- 7.Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA. Prolactin (PRL) and its receptor: actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr. Rev. 1998;19:225–268. doi: 10.1210/edrv.19.3.0334. [DOI] [PubMed] [Google Scholar]
- 8.Tan D, Johnson DA, Wu W, Zeng L, Chen YH, Chen WY, Vonderhaar BK, Walker AM. Unmodified Prolactin (PRL) and S179D PRL-Initiated Bioluminescence Resonance Energy Transfer between Homo- and Hetero-Pairs of Long and Short Human PRL Receptors in Living Human Cells. Mol. Endocrinol. 2005;19:1291–1303. doi: 10.1210/me.2004-0304. [DOI] [PubMed] [Google Scholar]
- 9.Langenheim JF, Tan D, Walker AM, Chen WY. Two wrongs can make a right: dimers of prolactin and growth hormone receptor antagonists behave as agonists. Mol. Endocrinol. 2005;19:1291–1303. doi: 10.1210/me.2005-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goffin V, Kelly PA. The prolactin/growth hormone receptor family: structure/ function relationships. J. Mammary Gland Biol. Neoplasia. 1997;2:7–17. doi: 10.1023/a:1026313211704. [DOI] [PubMed] [Google Scholar]
- 11.Christinger HW, Elkins PA, Sandowski Y, Sakal E, Gertler A, Kossiakoff AA, de Vos AM. Crystallization of ovine placental lactogen in a 1:2 complex with the extracellular domain of the rat prolactin receptor. Acta. Crystallogr. D. Biol. Crystallogr. 1998;54:1408–1411. doi: 10.1107/s0907444998005794. [DOI] [PubMed] [Google Scholar]
- 12.Gourdou I, Gabou L, Paly J, A, Kermabon AY, Belair L, Djiane J. Development of a Constitutively Active Mutant Form of the Prolactin Receptor, a Member of the Cytokine Receptor Family. Mol. Endocrinol. 1996;10:45–56. doi: 10.1210/mend.10.1.8838144. [DOI] [PubMed] [Google Scholar]
- 13.Lee RC, Walters JA, Reyland ME, Anderson SM. Constitutive activation of the prolactin receptor results in the induction of growth factor-independent proliferation and constitutive activation of signaling molecules. J. Biol. Chem. 1999;274:10024–10034. doi: 10.1074/jbc.274.15.10024. [DOI] [PubMed] [Google Scholar]
- 14.Sakal E, Elberg G, Gertler A. Direct evidence that lactogenic hormones induce homodimerization of membrane-anchored prolactin receptor in intact Nb2−11C rat lymphoma cells. FEBS Lett. 1997;410:289–292. doi: 10.1016/s0014-5793(97)00581-4. [DOI] [PubMed] [Google Scholar]
- 15.Qazi AM, Tsai-Morris CH, Dufau ML. Ligand-independent homo- and heterodimerization of human prolactin receptor variants:inhibitory action of the short forms by heterodimerization. Mol. Endocrinol. 2006;20:1912–1923. doi: 10.1210/me.2005-0291. [DOI] [PubMed] [Google Scholar]
- 16.Gadd SL, Clevenger CV. Ligand-independent dimerization of the human prolactin receptor isoforms:functional implications. Mol. Endocrinol. 2006;20:2734–2746. doi: 10.1210/me.2006-0114. [DOI] [PubMed] [Google Scholar]
- 17.Brown RJ, Adams JJ, Pelekanos RA, Wan Y, McKinstry WJ, Palethorpe K, Seeber RM, Monks TA, Eidne KA, Parker MW, Waters MJ. Model for growth hormone receptor activation based on subunit rotation within a receptor dimer. Nat. Struct. Mol. Biol. 2005;12:814–821. doi: 10.1038/nsmb977. [DOI] [PubMed] [Google Scholar]
- 18.Constantinescu SN, Keren T, Socolovsky M, Nam H, Henis YI, Lodish HF. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc. Natl. Acad. Sci. 2001;98:4379–4384. doi: 10.1073/pnas.081069198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen TJ, Kuo CB, Tsai KF, Liu JW, Chen DY, Walker AM. Development of recombinant human prolactin receptor antagonists by molecular mimicry of the phosphorylated hormone. Endocrinology. 1998;139:609–616. doi: 10.1210/endo.139.2.5758. [DOI] [PubMed] [Google Scholar]
- 20.Huang KT, Chen YH, Walker AM. Inaccuracies in MTS assays: major distorting effects of medium, serum albumin, and fatty acids. BioTechniques. 2004;37:406–412. doi: 10.2144/04373ST05. [DOI] [PubMed] [Google Scholar]
- 21.Wu W, Ginsburg E, Vonderhaar BK, Walker AM. S179D prolactin increases vitamin D receptor and p21 through upregulation of short 1b prolactin receptor in human prostate cancer cells. Cancer Res. 2005;65:7509–7515. doi: 10.1158/0008-5472.CAN-04-3350. [DOI] [PubMed] [Google Scholar]
- 22.Rozakis-Adcock M, Kelly PA. Identification of ligand binding determinants of the prolactin receptor. J. Biol. Chem. 1992;267:7428–33. [PubMed] [Google Scholar]
- 23.Watson CJ, Burdon TG. Prolactin signal transduction mechanisms in the mammary gland: the role of the Jak/Stat pathway. Rev. of Reproduction. 1996;1:1–5. doi: 10.1530/ror.0.0010001. [DOI] [PubMed] [Google Scholar]
- 24.Walker AM. Prolactin receptor antagonists. Curr. Opin. Invest. Drugs. 2005;6:378–385. [PubMed] [Google Scholar]
- 25.Seubert N, Royer Y, Staerk J, Kubatzky KF, Moucadel V, Krishnakumar S, Smith SO, Constantinescu SN. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol. Cell. 2003;12:1239–1250. doi: 10.1016/s1097-2765(03)00389-7. [DOI] [PubMed] [Google Scholar]
- 26.Brockman JL, Schuler LA. Prolactin signals via Stat5 and Oct-1 to the proximal cyclin D1 promoter. Mol. Cell. Endocrinol. 2005;239:45–53. doi: 10.1016/j.mce.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang LJ, Constantinescu SN, Lodish HE. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8:1327–1338. doi: 10.1016/s1097-2765(01)00401-4. [DOI] [PubMed] [Google Scholar]
- 28.Elkins PA, Christinger HW, Sandowski Y, Sakal E, Gertler A, de Vos AM, Kossiakoff AA. Ternary complex between placental lactogen and the extracellular domain of the prolactin receptor. Nat. Struct. Biol. 2000;7:808–815. doi: 10.1038/79047. [DOI] [PubMed] [Google Scholar]
- 29.Xu X, Kreye E, Kuo CB, Walker AM. A molecular mimic of phosphorylated prolactin markedly reduced tumor incidence and size when DU145 human prostate cancer cells were grown in nude mice. Cancer Res. 2001;61:6098–6114. [PubMed] [Google Scholar]
- 30.Ginsburg E, Vonderhaar BK. Prolactin synthesis and secretion by human breast cancer cells. Cancer Res. 1995;55:2591–2595. [PubMed] [Google Scholar]
- 31.Kline JB, Rycyzyn MA, Clevenger CV. Characterization of a novel and functional human prolactin receptor isoform (delta S1PRLr) containing only one extracellular fibronectin-like domain. Mol. Endocrinol. 2002;16:2310–2322. doi: 10.1210/me.2001-0033. [DOI] [PubMed] [Google Scholar]
- 32.Jackson IJ. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res. 1991;19:3795–3798. doi: 10.1093/nar/19.14.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]