

Abstract
We tested the hypothesis that a transcriptional enhancer (CE), previously identified to increase human renin expression in choriodecidual cells is required to mediate tissue-specific, cell-specific and regulated expression of human renin in transgenic mice. Recombineering was used to delete the CE upstream of the renin gene alone or in combination with the kidney enhancer (KE) in a large artificial chromosome construct containing the entire human renin gene and extensive flanking sequences. Deletion of the CE had no qualitative or quantitative effect on the tissue-specific expression of human renin, nor on the cellular localization of human renin in the kidney or placenta. Combined deletion of both the CE and KE caused a decrease in the level of renal renin expression consistent with the established role of the KE. We also considered the possibility that the CE, is a downstream enhancer of the KiSS1 gene which lies directly upstream of renin and is also expressed in the placenta. Deletion of the CE alone, or the CE and KE together, had no effect on the level of KiSS1 expression in the placenta. These data provide convincing evidence that the CE is silent in vivo, at least in the mouse. The absence of a phenotype caused by deletion of the CE is consistent with the observation that the sequence is not evolutionarily conserved.
Introduction
The complexity of renin gene regulation in rodents and humans is evidenced by the abundance of transcription factors now reported to be involved (reviewed in (17)). Regulatory elements controlling human renin gene expression have been identified in the proximal promoter region and in two distal enhancers. For example, in the proximal promoter no less than 9 transcription factors including HoxD10, PBX1b, PREP1, Ets1, notch, CREB, Sp1/Sp3, LXRα and PPARγ have been reported to bind and have either stimulatory or inhibitory effects on renin expression (16; 18; 20; 28; 30; 32). The most distal enhancer, termed the kidney enhancer (KE), is located at 2.6 Kb upstream of the mouse and 11 kb upstream of the human renin gene. Unlike the proximal promoter, the enhancer is an assemblage of extremely closely linked and overlapping binding sites for the transcription factors NF-Y, RARα, RXRα, EAR2, CREB, CREM, NFI, and Sp1/Sp3 (9; 14; 15; 22; 23). Some of these factors and their binding sites are required for high level activity of the enhancer while others antagonize enhancer activity. The enhancer may also be required to mediate some of the negative regulatory effects of cytokines (2; 9; 19). Further complicating the picture is the recent emergence of a trans-repression pathway whereby NF-κB (induced by cytokines) and vitamin-D receptor (induced by vitamin D analogs) interfere with transcription factors that bind to sites in the renin enhancer (31; 33). Although the kidney enhancer (termed the KE) robustly transactivates reporter genes in renin expressing As4.1 cells in culture, recent in vivo studies using knockout and transgenic mice reveal the sequence to be mainly important for the maintenance of baseline renin expression in kidney, and may be necessary to control the level of renin protein stored in renal JG cells in response to stimuli (1; 34). Enhancer-dependent effects may be more important in the transcriptional regulation of renin in the adrenal and salivary glands as its disruption nearly abolishes renin expression in those tissues (10).
A second enhancer was identified upstream of the human renin gene by Pinet and colleagues (5). This sequence, centered at about −5.5 kb, is located halfway between the KE and the proximal promoter. It was identified by transfection analysis in choriodecidual cells and therefore is herein defined as the chorionic enhancer (CE). Recently polymorphisms near this enhancer have been reported to influence the activity of the renin promoter (4). The primary purpose of this study was to test whether the chorionic enhancer is required to mediate the tissue- and cell-specific expression of renin in the kidney and placenta. Secondarily, based on the observation that the gene directly upstream of renin in mammalian genomes, KiSS1, is also highly expressed in the placenta, we tested whether the CE could potentially be a downstream enhancer of KiSS1.
Materials and Methods
Generation of CE-deficient and CEKE double enhancer-deficient PAC 160 transgenic mice
PAC160 (Genome System control number 4917) was used as the template for deletion of the CE in a manner similar to that previously reported by us (13; 34). Briefly, 550bp upstream (CE-L) and downstream (CE-R) homologies surrounding the CE were PCR cloned and inserted between χ-sites sequence in the vector pRM4-N (8). The CE is defined as sequences from −5777 to −5552 upstream of the hREN gene as previously described (5). The chloramphenicol acetyltransferase (CAM) gene flanked by heterotopic lox2272 was also PCR amplified and cloned. Lox2272 does not recombine with either loxP or Lox511 but mediates efficient recombination with other lox2272 sites. The lox2272-CAM-lox2272 fragment was then inserted between CE-L and CE-R in pRM4-N to generate the final targeting construct which was electroporated in a linearized form into E. coli MC1061 containing PAC160. Homologous recombinants were selected by CAM and kanamycin resistance and ampicillin sensitivity. This results in deletion of the CE and replacement with the Lox2272 flanked CAM gene. The CE deleted PAC160 was purified and retransformed into E. coli strain BS591 which contains strong Cre-recombinase enzyme activity (a kind gift of Brian Sauer). The resultant PAC160ΔCE replaces the CE sequence with a single lox2272 site. To make the double mutant, the CE targeting vector was transformed into MC1061 cells carrying PAC160ΔKE and then carried through the same steps. PAC160ΔKE was reported previously (34). The resultant vector PAC160ΔCEKE replaces the KE sequence with a lox511 site and the CE with a lox2272 site. The resultant purified PAC160ΔCE and PAC160ΔCEKE constructs were extensively evaluated by PCR, Southern blot and sequencing to ensure that the mutations were made faithfully and specifically and that there were no gross deletions elsewhere in the construct.
PAC160ΔCE and PAC160ΔCEKE DNA was sent to the University of Iowa Transgenic Animal Facility for microinjection to generate transgenic mice. Three transgenic founders of each construct were obtained. All mice were fed with standard mouse chow and water ad libitum. Care and use of mice met the standard set by National Institutes of health and all protocols were approved by University Animal Care and Use Committee at the University of Iowa. Captopril was dissolved in drinking water (0.5mg/ml) and administered to mice for 10 days. Drinking water was administered to control mice as the vehicle. Timed breedings were performed by inspection for vaginal plugs daily. Placentas were recovered at 18.5 days of gestation.
Molecular Analysis of PAC160ΔCE and PAC160ΔCEKE Transgenic Mice
We first confirmed that the CE was indeed deleted in the PAC160 constructs integrated into the mouse genome. Southern Blots were performed as previously described (34) using the following probes: 1) hREN cDNA probe, 2) CE probe; 3) DNA 5' of the CE (CE-L), 4) DNA 5' of the KE (KE-L), and 5) whole PAC160 DNA.
RNA was isolated using Tri-Reagent (Molecular Research Center Inc. Cincinnati, OH), and 50μg of RNA was used in RPA. RNase protect assays (RPA) designed to examine expression of GOLT1A, hREN, ETNK2 were performed using probes as previously described (34). Two new KiSS-1 probes, KiSS1A and KiSS1B were generated by RT-PCR of RNA isolated from human placenta RNA (Clontech) using the following primer sets: KiSS1AGGGAGAAGGACCTGCCGAACTACAACTGGAACT and CGCCCCCGCCCCGCATGCTCTGACT; KiSS1B- CAGCCAGGTGGTCTCGTCA and GTTCCAGTTGTAGTTCGGCAGGTC. Both PCR fragments were inserted into PCT4.TOPO (Invitrogen). KiSS1A-PCR4 was digested with Not1 and T3 RNA polymerase (Stratagene) was used to label the probe protecting 167 nucleotides of KiSS1 mRNA, whereas KiSS1B-PCR4 was digested with Xba1 and T7 RNA polymerase (Stratagene) was used to label the probe protecting 265 nucleotides of KiSS1 mRNA. Quantification of RPA results were performed with PhosphorImager and ImageQuant software (GE healthcare).
Immunofluorescence and in situ hybridization
Immunocytochemistry for mouse and human renin were performed as detailed previously (34). Female PAC160 WT, PAC160ΔCE or PAC160ΔCEKE were bred with male C57BL/6J nontransgenic mice (NT), and pregnant mice were sacrificed with CO2. The mice were perfused with PBS and 4% paraformaldehyde, and placenta were extracted and fixed overnight. Placenta were embedded in OCT and 5-10 μm sections were cut. In situ hybridization with DIG or FITC labeled hREN or mPL2 probes were performed according to protocols published (24). Both hREN and mouse placenta lactogen (mPL2) were cloned with Superscript III one-step RT-PCR kit and inserted into PCR TOPO II with dual promoter SP6 and T7 (Invitrogen). The total RNA used to amplify the hREN probe was from kidney of PAC160 WT transgenic mouse, whereas mPL2 was from murine 18.5 gd placenta. The RT-PCR primers were hREN: CCACCCCAAACCTTCAAAGTCG and TGCCCACAACCCCATCAAACTC; and primers for mPL2 were as previously published (26). Both SP6 and T7 RNA polymerases were used in vitro transcription to label sense (S) and antisense (AS) probe from the same phasmid. hREN was labeled with DIG, whereas mPL2 was labeled with FITC or DIG. Labeling and luminescent detection regents, including anti-FITC-AP or anti-DIG-AP antibody conjugates were from Roche.
Statistical analysis
Values were presented as mean ± SEM. Statistical significance was assessed by One Way ANOVA using the Bonferroni post hoc test. P < 0.05 was considered statistically significant.
Results
We previously reported that human renin expression in transgenic mice containing a large P1 artificial chromosome construct (PAC160) carrying the hREN and two upstream (GOLT1A and KiSS1) and one downstream (ETNK2) gene is tightly regulated in response to physiological cues (25). Deletion of the distal KE located at −11Kb resulted in a decrease in baseline expression but a preservation of cell-specific and regulated expression (34). In order to test the importance of the CE upstream of the human renin gene we employed a similar recombineering scheme to delete the CE in PAC160 and also in a PAC160 already lacking the KE (PAC160ΔKE). Figure 1 shows schematic maps of the resultant PAC160 constructs used to generate transgenic mice.
Figure 1. Generation of the PAC160 Transgenes.

Schematic representation of the PAC160 and mutant transgenes showing the hREN gene (blue), the kidney enhancer (KE, red) and its replacement by a lox511 site (red crosshatched arrow), the chorionic enhancer (CE, green) and its replacement by a lox2272 site (green crosshatched arrow), and neighboring genes (closed arrows). The direction of transcription is indicated by the direction of the arrows. The two terminal genes PEPP3 and Sox13 gene are truncated by the end of PAC160 as indicated by open boxes. The GOLT1A and KiSS1 genes lie upstream of hREN, whereas ETNK2 lies directly downstream. The LoxP site present in the PAC parent vector is indicated by the solid yellow arrowhead.
Initially, PCR was used to detect the presence of the modified PAC160 transgenes in the genome and to verify the presence of the upstream and downstream genes (data not shown). Southern blot analysis using probes detecting hREN (probe a), the CE itself (probe b), sequences adjacent to the CE (probe c), and sequences adjacent to the KE (probe d) were employed to ensure the integrity of the injected constructs (Figure 2A). This analysis confirmed the presence of hREN (Figure 2B probe a), demonstrated that the CE was deleted from the PAC160ΔCE and PAC160ΔCEKE but not from wildtype PAC160 (Figure 2B probes b and c), and that the KE was intact in PAC160ΔCE and wildtype PAC160 but not in PAC160ΔCEKE (Figure 2C).
Figure 2. Southern Blot of PAC160ΔKE Transgenic Mice.

A. Schematic representation of the region upstream of the hREN gene showing the location of the four probes (a-d) used in the Southern blots shown in B and C. B and C. Southern blots of genomic DNA isolated from mice carrying wildtype PAC160 (WT), lines carrying PAC160ΔCE, and lines carrying PAC160ΔCEKE transgenes. The numbers (8, 12, 16, 17,18) indicate the line numbers. The probes are indicated to the left of each blot. The DNA was digested with BamHI (B) and MscI (C). The blots in B initially probed with probe b was striped and re-probed with probe c.
Multiplex RNase protection assays were used to simultaneously assay for expression of hREN in addition to the upstream gene GOLT1A, the downstream gene ETNK2, and the endogenous 28S ribosomal RNA gene in wildtype PAC160, two lines of PAC160ΔCE and three lines of PAC160ΔCEKE mice (Figure 3). We previously reported that these probes accurately detect expression in transgenic mice but not in non-transgenic littermates (34). The results show a variable level of transgene expression compared with the endogenous 28S rRNA gene. In mice carrying the wildtype PAC160, hREN expression is evident in kidney and lung (uterus in females, data not shown), ETNK2 expression in kidney, liver and testes, and GOLT1A expression in brain, kidney, liver, and lung. Qualitatively, an identical pattern of tissue-specific pattern is seen for all three genes in mice carrying the altered PAC160 constructs. These data suggest that loss of the CE, either on its own or coupled with loss of the KE does not alter the overall pattern of tissue-specific expression of renin. Interestingly, in two lines of PAC160ΔCE mice, the ratio of hREN to ETNK2 in the kidney appear equal whereas in the PAC160ΔCEKE the relative expression of hREN is reduced relative to ETNK2. This is consistent with the loss of baseline expression when the KE is deleted (34). We next quantified the ratio of hREN to ETNK2 expression in the kidney of wildtype and mutant PAC160 transgenes (Figure 4). Although there was a significant decrease in hREN/ETNK2 expression in the kidney of PAC160 mice lacking the CE and KE (attributed to loss of the KE), there was no change in expression in mice only lacking the CE (Figure 4B). Moreover, induction of renal hREN expression in response to angiotensin converting enzyme inhibition was unaltered by deletion of either the CE alone or both the CE and KE (Figure 5).
Figure 3. Expression of the PAC160 Transgenes.

A. Representative multiplex RNase Protection Assays of hREN, GOLT1A, ETNK2 and 28S expression from total tissue RNA (50 μg) from male or female WT, PAC160ΔCE lines 14 and 16, PAC160ΔCEKE lines 8, 12 and 17 is shown. The location of the protected fragments is indicated. Tissues labels are: brain (B), heart (H), kidney (K), liver (L), lung (Lg), skeletal muscle (S), submandibular gland (Sg), testis (T) and uterus (U). Like PAC160CE and PAC160CEKE, hREN is expressed in the uterus of PAC160-WT mice (data not shown).
Figure 4. Influence of CE and CEKE Deletion on hREN Expression.

A. A representative RPA was performed with total kidney RNA from independent kidney samples from a representative line of wild type PAC160, PAC160ΔCEKE, and PAC160ΔCE mice. The position of the hREN, ETNK2, and Cyclophilin protected products is indicated. B. RPAs were quantified using the phosphorimager software and ratio of hREN/ETNK2 is shown. *, P<0.001 vs wildtype by ANOVA. The N is indicated within each bar. The RPAs in A were taken from a single large gel.
Figure 5. Induction of Renal hREN and mREN mRNA in Response to Captopril.

Total kidney mRNA was isolated from the indicated lines and analyzed by RNase protection assay (RPA) using probes specific for either mREN or hREN and normalized for expression of β-actin.
We performed immunohistochemistry using antisera directed specifically at hREN (Figure 6) or a combination of mREN-specific and hREN-specific antisera (Figure 7). Both studies reveal the absence of renal hREN protein in non-transgenic mice demonstrating the specificity of the hREN antisera. Juxtaglomerular cell-specific staining of hREN protein is evident in mice lacking the CE or both the CE and KE (Figure 6). Both hREN and mREN proteins are completely co-localized (Figure 7).
Figure 6. Immunostaining of Human Renin in Kidney.
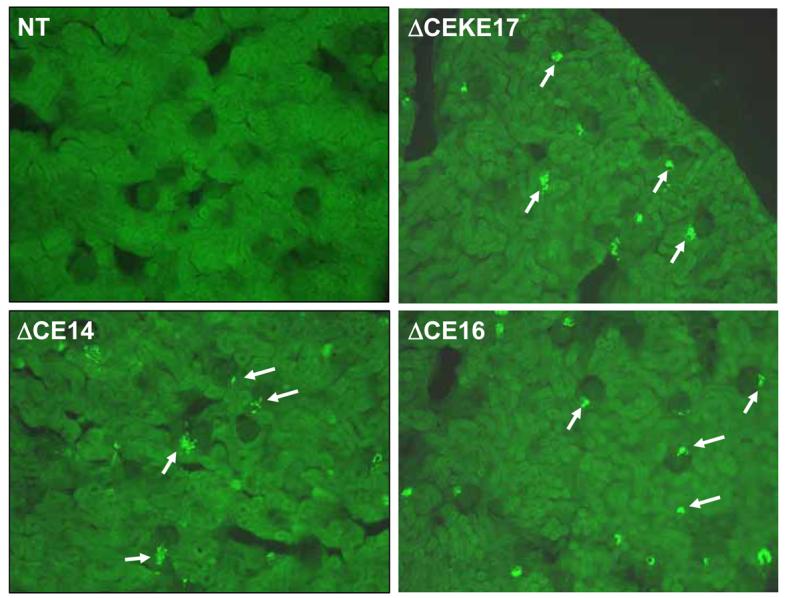
Representative immunofluorescent images of human renin staining in the kidney of non-transgenic (NT) and PAC160ΔCEKE17, PAC160ΔCE14 and PAC160ΔCE16. The arrows indicate human renin stained cells.
Figure 7. Co-localization of hREN and mREN in Kidney.
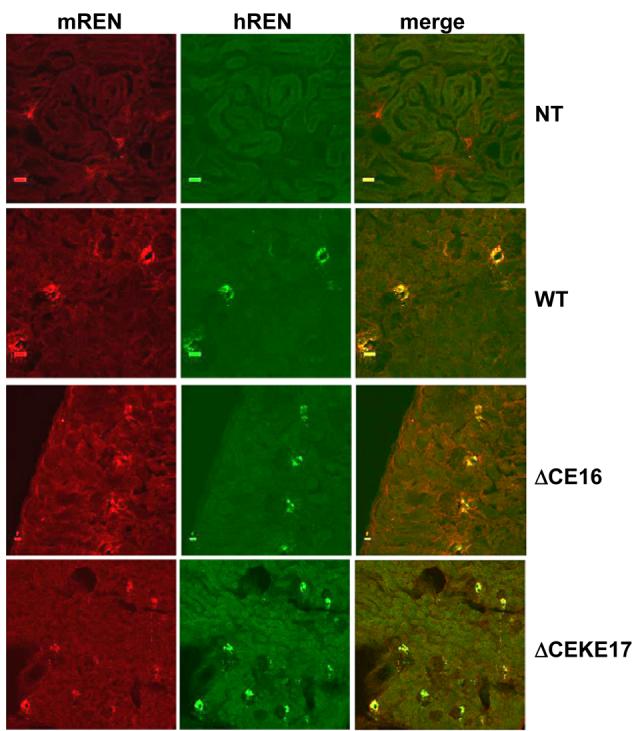
Representative immunofluorescent images of mREN (red) and hREN (green) staining in the kidney of non-transgenic (NT), PAC160 wildtype (WT), PAC160ΔCE16, and PAC160ΔCEKE17.
We next evaluated whether loss of the CE sequence causes a change in the expression of renin in the placenta. Timed breeding were performed resulting in a mix of 18.5 gd fetuses genotyped as either transgenic or non-transgenic. As shown in Figure 8A, there was essentially no effect on hREN expression in the placenta of mice lacking only the CE or lacking the CE in combination with the KE. The differences in the level of expression seen particularly in PACΔCEKE8 can be attributed to difference in transgene copy number which are lower in line 8 than in lines 12 or 14. Like renin, there was essentially no alteration in the expression of KiSS1 mRNA. The KiSS1 gene lies directly upstream of renin in the genome (Figure 1), is expressed in the placenta (7) and is present in PAC160 (Figure 8B). These data strongly suggest that the CE is neither required as an enhancer of renin nor a downstream enhancer of KiSS1 in the placenta.
Figure 8. Placental Expression of hREN and KiSS1.

Representative RNase Protection Assays of hREN (A) and KiSS1 (B) expression from total placental RNA (18.5 gd) from the indicated lines of WT, PAC160ΔCE, PAC160ΔCEKE mice are shown. The location of the protected fragments are indicated. The individual samples are derived from a mix of transgenic (expressing) and non-transgenic (non-expressing, i.e. lane 3) placentas.
Finally, we performed in situ hybridization to assess if the CE is required to mediate cell-specific expression of hREN in the placenta. We first demonstrated that the hREN antisense probe faithfully detected hREN mRNA in juxtaglomerular cells of the kidney (Figure 9A-B). There was no hybridization signal detected when a control sense probe was used on PAC160 wildtype placenta (Figure 9C). A robust signal was detected with the antisense probe on placenta from PAC160 wildtype (Figure 9D), PAC160ΔCE (Figure 9F) and PAC160ΔCEKE (Figure 9E). Previous studies reported that renin is expressed in trophoblast giant cells of the labyrinth where it can be secreted into the maternal circulation (26), and the localization of the in situ hybridization signal is consistent with this. There was no difference in the cellular localization of hREN irrespective of the presence of the CE. To further characterize the location of renin expression in the placenta, we performed dual in situ hybridization design to detect both hREN and placental lactogen 2 (PL2) mRNA. PL2 expression has been reported in the trophoblast cells of the placental labyrinth of mice (24; 26). As expected, only PL2 mRNA was detected in the non-transgenic placenta (Figure 10D). In the transgenic placenta, hREN-expressing and PL2-expressing cells were found in adjacent cells and in co-localized cells around canals and sinusoids. The pattern of adjacent and co-localization of renin and PL2 expressing cells was identical in transgenic mice carrying the wildtype and CE-deleted PAC160 constructs (Figure 10A-C).
Figure 9. In Situ Hybridization of Human Renin in Kidney and Placenta.
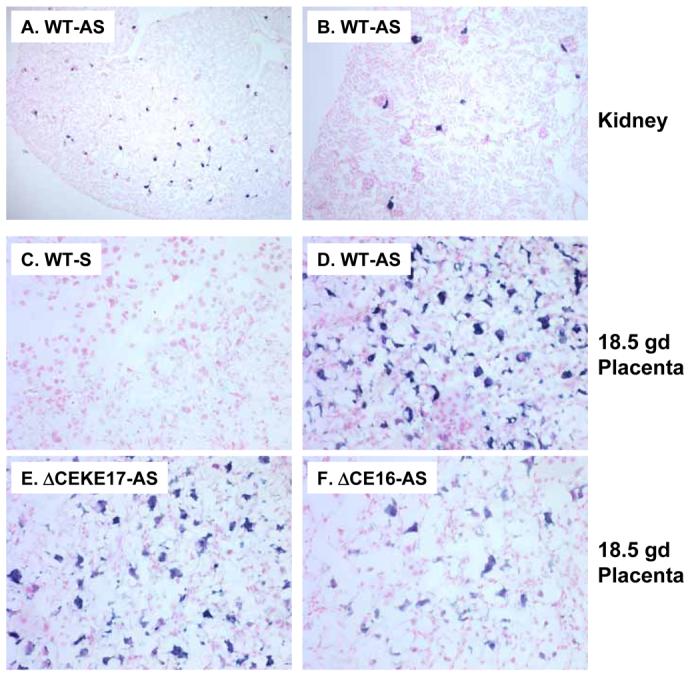
Representative in situ hybridization images of human renin mRNA in the kidney (A,B) and placenta (C-F) of PAC160 wildtype (WT), PAC160ΔCEKE, and PAC160ΔCE. AS, antisense probe; S, sense probe. The image in panel B is a higher magnification from the same slide as panel A.
Figure 10. In Situ Hybridization of Human Renin and PL2 in Placenta.
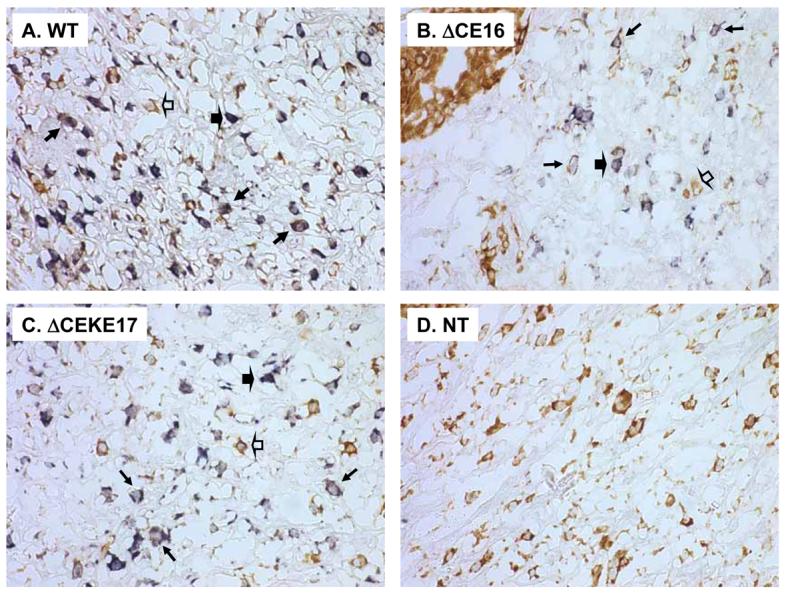
Representative dual in situ hybridization images of human renin mRNA (blue) and PL2 mRNA (brown) in the placenta of PAC160 wildtype (WT), PAC160ΔCEKE, PAC160ΔCE, and nontransgenic (NT) mice. Only PL2 expression is seen in the non-transgenic placenta. Open arrowhead denote PL2 expressing cells; Closed arrowhead denotes hREN expressing cell; regular arrows denotes cells co-expressing hREN and PL2.
Discussion
We show that the CE, a sequence previously reported to enhance transcriptional activity of human renin in choriodecidual cells is silent in vivo (5). This was accomplished by the generation of a precise and subtle deletion of the −5777 to −5552 CE sequence in a large P1 artificial chromosome (PAC) containing the entire human renin gene and nearly 150 kb of flanking sequences. We previously reported that expression of hREN in mice carrying this construct faithfully replicated the tissue-specific and cell-specific expression of the gene (as observed in humans)(25). Moreover, transcriptional activity of the hREN gene promoter responded to physiological cues known to regulate the renin gene including dietary sodium, ACE inhibition and angiotensin-II infusion. Deletion of the distal transcriptional enhancer, termed the kidney enhancer (KE) caused a reduction in baseline expression of hREN (34), a finding consistent with deletion of the highly homologous distal enhancer upstream of the endogenous mouse renin gene (1; 10).
There are a number of potential explanations for the absence of a phenotype in the PAC160ΔCE mice. The first is that the sequence is not physiologically important. Along these lines it is interesting to note that although the CE is conserved in the chimpanzee and Rhesus genomes, there is no obvious homology present upstream of either the mouse or rat renin genes. This lack of evolutionary conservation suggests that it is either unimportant or its importance is unique to humans and non-human primates. That polymorphisms in and around the CE have been reported to influence blood pressure and the blood pressure responses to renin-angiotensin system inhibition gives credence to this possibility (4; 11). Fuchs et al (4) first showed that the CE element could be used as decoy to lower activity of the renin promoter in choriodecidual cells.
They then identified two polymorphisms downstream of the 225 bp enhancer. One of the polymorphisms located at position −5312 when incorporated into a larger segment (−5870 to −5279) including the CE could modestly modulate transcriptional activity. Moore et al. (11) examined the same variant at position −5312 in a population of 387 white bank employees and in 259 white hypertensives. They found that carriers of the T allele, the allele with higher transcriptional activity in the Fuchs et al study, exhibited higher ambulatory blood pressure than CC homozygotes. Plasma renin activity did not differ in a different population of white hypertensives, although the blood pressure response to losartan was greater in −5312T carriers causing the authors to speculate that the −5312 allele may predict the anti-hypertensive effect of renin-angiotensin system blockers. Consequently, we cannot formally rule out the possibility that we may have obtained different results if a larger segment, including sequences downstream of the CE and including the −5312 region was deleted in our study.
We also considered the possibility that the CE sequence is not an upstream regulator of renin, but instead is a downstream regulator of the KiSS1 gene. The KiSS1 gene lies directly upstream of the renin locus, and like renin is expressed in trophoblast giant cells of the placenta (24; 26; 29). That enhancers can be located both upstream and downstream of genes and can have effects on gene expression in an orientation and position independent manner makes this an intriguing possibility. KiSS1 encodes a 54 amino acid peptide termed metastin (or kisspeptin) which binds to a G-protein coupled receptor GPR54 thought to be involved in metastasis, trophoblast invasion and puberty (3; 6; 7). That neither KiSS1 nor hREN mRNA levels were altered by deletion of the CE suggests this hypothesis was not correct. This is further supported by our data showing that there was no alteration in the cellular localization of hREN in placenta.
Finally, we must consider the possibility that transcription factors from the mouse do not bind to the CE upstream of the human renin gene. Although this is extremely difficult to formally rule out, it should be noted that the level of expression of the hREN gene in the kidney of PAC160 mice in response to physiological cues which either stimulate or repress renin expression occurs identically in magnitude to changes in expression of the endogenous mouse renin gene. The distal, or kidney enhancer is highly homologous among mouse, rat and human renin genes, and its deletion in PAC160 has a similar effect as deletion of the homologous enhancer upstream of the mouse renin gene (10; 34). As there are now countless examples where human genes are appropriately expressed in transgenic mice, makes this argument untenable.
This leaves open to question the location and identity of regulatory elements which specify the expression of renin in a spatial and temporal manner. As stated above, it is possible that additional sequence surrounding the region we included in our deletion (−5777 to −5552) are required for renin expression. In considering other regions of the gene, it is important to note that transcription factors thought to be important developmentally have been identified to bind to the renin proximal promoter making this region of the gene a prime candidate (16; 20; 21). Similarly, Mrowka et al using a computational approach identified a conserved non-coding sequence 3 Kb further upstream of the distal enhancer (the KE) which now must also be considered a candidate regulatory region for renin expression (12). Recombineering methods similar to that described herein can be used to determine if these sequences are required to control human renin expression in vivo. The strength of the BAC-PAC-recombineering approach, although more laborious and time consuming, provides many advantages over standard promoter-reporter gene fusions. Depending on the size of the gene of interest, the BAC/PAC construct may contain neighboring genes which can be used very effectively as internal controls. Internal controls are generally lacking when promoter-reporter fusion transgenics are constructed. Promoter-reporter fusion constructs are also highly sensitive to position effects, that is, effects imparted by regulatory sequences at the site of insertion, whereas BAC/PAC constructs are generally considered to be immune from position effects. This immunity comes from the ability of large genomic constructs such as these to manipulate chromatin locally due to the presence of Locus Control Regions (LCRs). This is best exemplified by the elegant studies of Talbot et al (27) showing that the β-globin LCR consists of DNAse-I hypersensitive sites which confer high level, copy number-proportional and position-independent (i.e. immunity from position effects) expression in transgenic mice.
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL48058, HL61446, and HL55006). We acknowledge Deborah Davis for support with experiments involving Captopril treatment of mice and Ella Born for assistance with Southern blots. Transgenic mice were generated at the University of Iowa Transgenic Animal Facility directed by Curt D. Sigmund, Ph.D. and supported in part by grants from the NIH and from the Roy J. and Lucille A. Carver College of Medicine. We wish to thank Norma Sinclair, Patricia Yarolem, Amanda Decker and Joanne Schwarting for their technical expertise in generating transgenic mice. We also thank the University of Iowa Central Microscopy Facility. We gratefully acknowledge the generous research support of the Roy J. Carver Trust.
References
- 1.Adams DJ, Head GA, Markus MA, Lovicu FJ, van der WL, Kontgen F, Arends MJ, Thiru S, Mayorov DN, Morris BJ. Renin enhancer is critical for control of renin gene expression and cardiovascular function. J Biol Chem. 2006;281:31753–31761. doi: 10.1074/jbc.M605720200. [DOI] [PubMed] [Google Scholar]
- 2.Baumann H, Wang Y, Richards CD, Jones CA, Black TA, Gross KW. Endotoxin-induced renal inflammatory response. Oncostatin M as a major mediator of suppressed renin expression. J Biol Chem. 2000;275:22014–22019. doi: 10.1074/jbc.M002830200. [DOI] [PubMed] [Google Scholar]
- 3.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs S, Philippe J, Germain S, Mathieu F, Jeunemaitre X, Corvol P, Pinet F. Functionality of two new polymorphisms in the human renin gene enhancer region. J Hypertens. 2002;20:2391–2398. doi: 10.1097/00004872-200212000-00018. [DOI] [PubMed] [Google Scholar]
- 5.Germain S, Bonnet F, Philippe J, Fuchs S, Corvol P, Pinet F. A novel distal enhancer confers chorionic expression on the human renin gene. J Biol Chem. 1998;273:25292–25300. doi: 10.1074/jbc.273.39.25292. [DOI] [PubMed] [Google Scholar]
- 6.Harms JF, Welch DR, Miele ME. KISS1 metastasis suppression and emergent pathways. Clin Exp Metastasis. 2003;20:11–18. doi: 10.1023/a:1022530100931. [DOI] [PubMed] [Google Scholar]
- 7.Hiden U, Bilban M, Knofler M, Desoye G. Kisspeptins and the placenta: regulation of trophoblast invasion. Rev Endocr Metab Disord. 2007;8:31–39. doi: 10.1007/s11154-007-9030-8. [DOI] [PubMed] [Google Scholar]
- 8.Jessen JR, Meng A, McFarlane RJ, Paw BH, Zon LI, Smith GR, Lin S. Modification of bacterial artificial chromosomes through chi-stimulated homologous recombination and its application in zebrafish transgenesis. Proc Natl Acad Sci U S A. 1998;95:5121–5126. doi: 10.1073/pnas.95.9.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X, Shi Q, Sigmund CD. Interleukin-1{beta} Attenuates Renin Gene Expression Via a Mitogen-Activated Protein Kinase Kinase-Extracellular Signal-Regulated Kinase and Signal Transducer and Activator of Transcription 3-Dependent Mechanism in As4.1 Cells. Endocrinology. 2006;147:6011–6018. doi: 10.1210/en.2006-0129. [DOI] [PubMed] [Google Scholar]
- 10.Markus MA, Goy C, Adams DJ, Lovicu FJ, Morris BJ. Renin Enhancer Is Crucial for Full Response in Renin Expression to an In Vivo Stimulus. Hypertension. 2007 doi: 10.1161/HYPERTENSIONAHA.107.096891. [DOI] [PubMed] [Google Scholar]
- 11.Moore N, Dicker P, O'Brien JK, Stojanovic M, Conroy RM, Treumann A, O'Brien ET, Fitzgerald D, Shields D, Stanton AV. Renin gene polymorphisms and haplotypes, blood pressure, and responses to renin-angiotensin system inhibition. Hypertension. 2007;50:340–347. doi: 10.1161/HYPERTENSIONAHA.106.085563. [DOI] [PubMed] [Google Scholar]
- 12.Mrowka R, Steege A, Kaps C, Herzel H, Thiele BJ, Persson PB, Bluthgen N. Dissecting the action of an evolutionary conserved non-coding region on renin promoter activity. Nucleic Acids Res. 2007;35:5120–5129. doi: 10.1093/nar/gkm535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nistala R, Sigmund CD. A reliable and efficient method for deleting operational sequences in PACs and BACs. Nucleic Acids Res. 2002;30:e41. doi: 10.1093/nar/30.10.e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan L, Black TA, Shi Q, Jones CA, Petrovic N, Loudon J, Kane C, Sigmund CD, Gross KW. Critical roles of a cyclic AMP responsive element and an E-box in regulation of mouse renin gene expression. J Biol Chem. 2001;276:45530–45538. doi: 10.1074/jbc.M103010200. [DOI] [PubMed] [Google Scholar]
- 15.Pan L, Glenn ST, Jones CA, Gronostajski RM, Gross KW. Regulation of renin enhancer activity by nuclear factor I and Sp1/Sp3. Biochim Biophys Acta. 2003;1625:280–290. doi: 10.1016/s0167-4781(03)00016-2. [DOI] [PubMed] [Google Scholar]
- 16.Pan L, Glenn ST, Jones CA, Gross KW. Activation of the rat renin promoter by HOXD10.PBX1b.PREP1, Ets-1, and the intracellular domain of notch. J Biol Chem. 2005;280:20860–20866. doi: 10.1074/jbc.M414618200. [DOI] [PubMed] [Google Scholar]
- 17.Pan L, Gross KW. Transcriptional regulation of renin: an update. Hypertension. 2005;45:3–8. doi: 10.1161/01.HYP.0000149717.55920.45. [DOI] [PubMed] [Google Scholar]
- 18.Pan L, Jones CA, Glenn ST, Gross KW. Identification of a novel region in the proximal promoter of the mouse renin gene critical for expression. Am J Physiol Renal Physiol. 2004;286 doi: 10.1152/ajprenal.00319.2003. [DOI] [PubMed] [Google Scholar]
- 19.Pan L, Wang Y, Jones CA, Glenn ST, Baumann H, Gross KW. Enhancer-Dependent Inhibition of Mouse Renin Transcription by Inflammatory Cytokines. Am J Physiol Renal Physiol. 2005;288:F117–F124. doi: 10.1152/ajprenal.00333.2003. [DOI] [PubMed] [Google Scholar]
- 20.Pan L, Xie Y, Black TA, Jones CA, Pruitt SC, Gross KW. An Abd-B class HOX.PBX recognition sequence is required for expression from the mouse Ren-1c gene. J Biol Chem. 2001;276:32489–32494. doi: 10.1074/jbc.M011541200. [DOI] [PubMed] [Google Scholar]
- 21.Sequeira Lopez ML, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Dev Cell. 2004;6:719–728. doi: 10.1016/s1534-5807(04)00134-0. [DOI] [PubMed] [Google Scholar]
- 22.Shi Q, Gross KW, Sigmund CD. NF-Y antagonizes renin enhancer function by blocking stimulatory transcription factors. Hypertension. 2001;38:332–336. doi: 10.1161/01.hyp.38.3.332. [DOI] [PubMed] [Google Scholar]
- 23.Shi Q, Gross KW, Sigmund CD. Retinoic acid-mediated activation of the mouse renin enhancer. J Biol Chem. 2001;276:3597–3603. doi: 10.1074/jbc.M008361200. [DOI] [PubMed] [Google Scholar]
- 24.Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev Biol. 2007;304:567–578. doi: 10.1016/j.ydbio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 25.Sinn PL, Davis DR, Sigmund CD. Highly Regulated Cell-Type Restricted Expression of Human Renin in Mice Containing 140 Kb or 160 Kb P1 Phage Artificial Chromosome Transgenes. J Biol Chem. 1999;274:35785–35793. doi: 10.1074/jbc.274.50.35785. [DOI] [PubMed] [Google Scholar]
- 26.Takimoto-Ohnishi E, Saito T, Ishida J, Ohnishi J, Sugiyama F, Yagami K, Fukamizu A. Differential roles of renin and angiotensinogen in the feto-maternal interface in the development of complications of pregnancy. Mol Endocrinol. 2005;19:1361–1372. doi: 10.1210/me.2004-0158. [DOI] [PubMed] [Google Scholar]
- 27.Talbot D, Collis P, Antoniou M, Vidal M, Grosveld F, Greaves DR. A dominant control region from the human beta-globin locus conferring integration site-independent gene expression. Nature. 1989;338:352–355. doi: 10.1038/338352a0. [DOI] [PubMed] [Google Scholar]
- 28.Tamura K, Chen YE, Tanaka Y, Sakai M, Tsurumi Y, Koide Y, Kihara M, Pratt RE, Horiuchi M, Umemura S, Dzau VJ. Nuclear receptor LXRalpha is involved in cAMP-mediated human renin gene expression. Mol Cell Endocrinol. 2004;224:11–20. doi: 10.1016/j.mce.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Terao Y, Kumano S, Takatsu Y, Hattori M, Nishimura A, Ohtaki T, Shintani Y. Expression of KiSS-1, a metastasis suppressor gene, in trophoblast giant cells of the rat placenta. Biochim Biophys Acta. 2004;1678:102–110. doi: 10.1016/j.bbaexp.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Todorov VT, Desch M, Schmitt-Nilson N, Todorova A, Kurtz A. Peroxisome Proliferator-Activated Receptor-{gamma} Is Involved in the Control of Renin Gene Expression. Hypertension. 2007 doi: 10.1161/HYPERTENSIONAHA.107.092817. [DOI] [PubMed] [Google Scholar]
- 31.Todorov VT, Volkl S, Friedrich J, Kunz-Schughart LA, Hehlgans T, Vermeulen L, Haegeman G, Schmitz ML, Kurtz A. Role of CREB1 and NF{kappa}B-p65 in the down-regulation of renin gene expression by tumor necrosis factor {alpha} J Biol Chem. 2005;280:24356–24362. doi: 10.1074/jbc.M502968200. [DOI] [PubMed] [Google Scholar]
- 32.Ying L, Morris BJ, Sigmund CD. Transactivation of the human renin promoter by the cyclic AMP/ Protein Kinase A pathway is mediated by both CREB-dependent and CREB-independent mechanisms in Calu-6 cells. J Biol Chem. 1997;272:2412–2420. doi: 10.1074/jbc.272.4.2412. [DOI] [PubMed] [Google Scholar]
- 33.Yuan W, Pan W, Kong J, Zheng W, Szeto FL, Wong KE, Cohen R, Klopot A, Zhang Z, Li YC. 1,25-dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem. 2007 doi: 10.1074/jbc.M705495200. [DOI] [PubMed] [Google Scholar]
- 34.Zhou X, Davis DR, Sigmund CD. The human renin kidney enhancer is required to maintain baseline renin expression but is dispensable for tissue-specific, cell-specific and regulated expression. J Biol Chem. 2006;281:35296–35304. doi: 10.1074/jbc.M608055200. [DOI] [PubMed] [Google Scholar]