

Abstract
Tetanus neurotoxin causes the spastic paralysis of tetanus by blocking neurotransmitter release at inhibitory synapses of the spinal cord. This is due to the penetration of the toxin inside the neuronal cytosol where it cleaves specifically VAMP/synaptobrevin, an essential component of the neuroexocytosis apparatus. Here we show that tetanus neurotoxin is internalized inside the lumen of small synaptic vesicles following the process of vesicle reuptake. Vesicle acidification is essential for the toxin translocation in the cytosol, which results in the proteolytic cleavage of VAMP/synaptobrevin and block of exocytosis.
Upon membrane depolarization and calcium entry into nerve terminals, synaptic vesicles (SVs) docked at active zones release their neurotransmitter content into the synaptic cleft (1–4). SVs are then rapidly recycled in an endocytic process that requires dynamin and clathrin (5, 6). The biochemical steps involved in vesicle docking, fusion, and reuptake are actively investigated. Recently, several protein components of the neuroexocytosis apparatus have been identified (4, 7–9), and a model for docking and fusion of vesicles with target membranes has been proposed (10). One of these proteins, a SV membrane protein, VAMP/synaptobrevin, is the specific target of the metalloproteinase activity of tetanus neurotoxin (TeNT) (11, 12). This neurotoxin consists of two disulfide-linked polypeptide chains: H (100 kDa) mediates the specific binding to the neuronal presynaptic membrane, while L (50 kDa) is responsible for the intracellular proteolytic activity. The spastic paralysis of tetanus is due to the specific intoxication of the spinal cord inhibitory interneurons by TeNT, which penetrates these cells and blocks the release of inhibitory neurotransmitters (13–16). In vitro TeNT is active also on other central nervous system (CNS) neurons: it inhibits γ-aminobutyrate (GABA)-mediated effects in rat hippocampal slices (17) and, when injected in rat ventral hippocampi, it causes an epileptiform syndrome (18, 19).
The steps involved in TeNT entry and trafficking inside peripheral and central neurons are not known. It is well documented that any form of nerve stimulation, and hence increase in synaptic activity, in the animal or in the phrenic nerve hemidiaphragm preparation, decreases the time of the onset of paralysis (20–24). Indirect evidence suggest that TeNT intoxication of spinal cord neurons involves TeNT transit through an intracellular acidic compartment (25, 26). To investigate the route of entry of TeNT inside CNS neurons, we have used primary cultures of hippocampal neurons, which represent a well-characterized experimental model (27). These neurons develop in vitro through a stereotyped sequence of events and eventually they form in vitro functional synaptic contacts, characterized by an active SV recycling (28, 29). SVs clustered at presynaptic sites of cultured hippocampal neurons internalize antibodies to the intravesicular domain of the SV protein synaptotagmin in a depolarization- and calcium-dependent manner (28, 30, 31). Hence, the pore that mediates neurotransmitter release is large enough to allow the passage of molecules of more than 150 kDa. We tested, therefore, the possibility that TeNT enters CNS neurons by means of SV endocytosis. Here we show that TeNT enters hippocampal neurons by means of the physiological process of SV recycling and that vesicle acidification is essential for the toxin translocation in the cytosol.
MATERIALS AND METHODS
Hippocampal Cell Culture.
Primary neuronal cultures were prepared from the hippocampi of 18-day-old fetal rats as described (27, 32).
Experimental Treatments.
Texas red (Pierce) was conjugated to TeNT by following the supplier’s recommendations, and the conjugate (TR-TeNT) was purified by chromatography on a Sephadex G-25 column. Neuronal cultures were incubated for 5–60 min in Krebs–Ringer solution, containing 20 nM TR-TeNT, in 5 or 55 mM KCl. In some cases, exposure was performed in 55 mM KCl, without extracellular calcium plus 1 mM EGTA. Cultures were then thoroughly washed, fixed, permeabilized, and double labeled for the SV protein SV2. In one set of experiments, cultures were pretreated with 150 nM bafilomycin A1 (Baf-A1) for 10 min before exposure to the toxin. For the VAMP-2 cleavage experiments, neurons were exposed to 10 nM TeNT for 5 min in Krebs–Ringer solution in the presence of 5 or 55 mM KCl. In one set of experiments, cultures were pretreated with 150 nM Baf-A1. Coverslips were then thoroughly washed and maintained in the incubator. After different periods of time (15, 30, 60, or 120 min), cultures were fixed, permeabilized, and double stained for VAMP-2 and synaptotagmin (or SV2). Some coverslips were photographed. In others, the number of VAMP-2-positive synapses was analyzed and expressed as a percentage of the total number of synapses, as evaluated by synaptotagmin or SV2 staining.
Immunocytochemistry.
After the experimental treatments, neurons were fixed in 4% formaldehyde in 0.1 M sodium phosphate buffer containing 0.12 M sucrose for 25 min at 37°C. Fixed neurons were permeabilized with 0.2% Triton X-100 and incubated with primary antibodies raised in rabbit against VAMP-2, followed by rhodamine-conjugated antibodies to rabbit immunoglobulin and/or with primary mouse antibodies directed against SV2 or synaptotagmin followed by fluorescein-conjugated antibodies to mouse immunoglobulin (28). Coverslips were mounted in 70% glycerol in phosphate buffer containing phenylenediamine at 1 mg/ml. Cells were photographed with Kodak TMAX 400 film on a Zeiss Axiophot microscope equipped with epifluorescence microscopy.
Preparation and Uptake of 125I-Transferrin.
Human transferrin was radiolabeled with 125I, following the procedure of Fraker and Speck (33), at a specific activity of 107 cpm/mg. Neuronal cultures were incubated for 5 min in Krebs–Ringer solution, containing 10 mCi (1 mCi = 37 MBq) of 125I-labeled transferrin (125I-transferrin), in 5 or 55 mM KCl followed by a 30-min incubation in the same medium with 5 mM KCl. After washings, cells were incubated for 5 min at 4°C in PBS, pH 2.0, and the medium was collected and its radioactivity was measured. Cells were treated with 0.1 M NaOH and the radioactivity of the lysate was measured.
Electron Microscopy.
Gold-conjugated TeNT was prepared by incubation of 400 mg of TeNT in 2 mM sodium phosphate/5 mM NaCl, pH 7, with 5 ml of 5-nm gold particles (British Biocell International, Cardiff) at room temperature for 10 min under mild agitation. Bovine serum albumin (BSA, 1% final concentration) was added and, after 10 min, the suspension was centrifuged at 21,000 rpm for 1 hr in a Beckman SW 40Ti rotor. The softly packed upper part of the pellet was resuspended in 0.5 ml of supernantant, 0.5 ml of 20 mM Tris·HCl/150 mM NaCl/1% BSA, pH 7, was added, and the mixture was stored at 4°C in the dark. Neurons were fixed for 1 hr at 24°C with 2% glutaraldehyde in culture medium, washed twice with 0.1 M sodium phosphate buffer, pH 7.3, and postfixed for 1 hr with 2% OsO4 in 0.1 M sodium phosphate buffer. After two quick changes of 0.1 M sodium Veronal buffer, pH 7.3, the cells were block-stained with 0.5% uranyl acetate in sodium Veronal buffer. The coverslips were then transferred to a clean glass Petri dish, dehydrated, and flat-embedded in Epon 812 as described (34). Prior to the resin polymerization step, the coverslips were positioned face up on glass slides with a smear of silicon grease. The glass coverslips were separated from the Epon blocks by incubation for 20–30 min with 48% hydrofluoric acid in a fume cupboard.
Antibodies.
Antibodies against VAMP-2 were generated in rabbit as previously described (35). Antibodies against TeNT were raised in horse, and the resulting antibody (100 international units/ml) was used at a dilution of 1:80. Antibodies against SV2 and synaptotagmin were a kind gift of K. Buckley (Harvard University) and R. Jahn (Yale University), respectively. Rhodamine-conjugated anti-rabbit antibodies were purchased from Boehringer Mannheim and fluorescein-conjugated anti-mouse antibodies were from Jackson ImmunoResearch.
RESULTS
TeNT Is Internalized at Nerve Terminals.
Exposure of hippocampal neurons to a high-potassium medium induces extensive exocytosis and endocytosis of synaptic vesicles (28, 30, 31). Fig. 1 shows that, upon 5-min exposure of neurons to 55 mM KCl, TR-TeNT is internalized inside nerve terminals (Fig. 1B), as visualized by counterstaining the same culture after fixation and permeabilization with antibodies directed against the SV marker SV2 (Fig. 1A). TR-TeNT was used at 20 nM in most experiments for optimal visualization, but 1/10 the concentration of TeNT was sufficient to detect specific internalization, consistent with the Kd values for TeNT binding to rat brain membranes (36, 37). TR-TeNT was not taken up at a detectable level by nerve terminals when neurons were incubated in a medium free of calcium ions (Fig. 1 C and D) or in the absence of KCl (Fig. 1 E and F), two conditions in which SV recycling rate is strongly reduced. However, TR-TeNT internalization at synapses was detected, though at a lower extent, also in a medium free of depolarizing agents, provided that incubation was prolonged for more than 1 hr (not shown), a time period during which a sufficient number of synaptic vesicle exo–endocytosis cycles takes place (28). In contrast, depolarization-induced uptake of Texas red-labeled botulinum neurotoxins type A and B at synaptic terminals of hippocampal neurons was not detectable. Under the same conditions, internalization of fluid phase endocytosis markers such as Lucifer yellow and rhodamine-conjugated dextran was not observed. Moreover, a same amount of 125I-transferrin, a well-established endocytic marker, was internalized in 30 min by hippocampal neurons both in the presence and in the absence of KCl in the external medium (not shown). Altogether, these results suggest the possibility that TeNT is internalized in nerve terminals during SV recycling.
Figure 1.
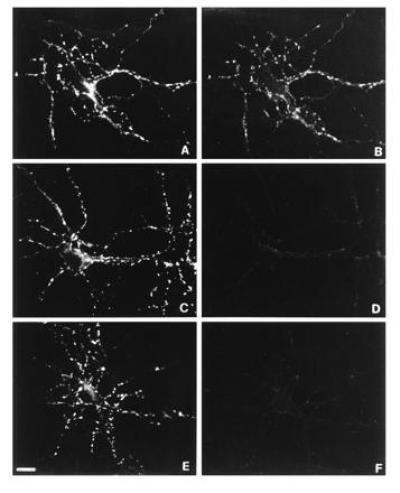
TR-TeNT labels presynaptic nerve terminals when added to living hippocampal neurons in primary cultures. Fifteen-day-old neurons were exposed to 20 nM TR-TeNT for 5 min in 55 mM KCl (A and B) or in 55 mM KCl in the absence of extracellular calcium (C and D) or in control medium (E and F). After fixation and permeabilization, neurons were counterstained for the SV protein SV2 followed by fluorescein-conjugated goat anti-mouse antibodies (A, C, and E). Puncta of SV2 immunoreactivity represent presynaptic nerve terminals, characterized by clusters of SVs, which outline perykaria and dendrites. TR-TeNT is internalized in nerve terminals only when the 5-min incubation is performed in the presence of 55 mM KCl (B). No internalization takes place in neurons exposed to the toxin in 55 mM KCl without calcium (D) or in control medium (F). (Bar = 13.8 μm.)
TeNT Uptake in Developing Hippocampal Neurons.
The property of forming clusters and undergoing regulated exocytosis is intrinsic to SVs and is not dependent upon the formation of a presynaptic specialization (31). SVs of hippocampal neurons undergo calcium-dependent exocytosis (31) and release glutamate by a calcium-dependent mechanism (38) even before synaptogenesis. However, the features of SV recycling appear to be a function of neuronal development. Indeed, a much higher rate of constitutive SV exocytosis takes place in isolated axons of developing neurons as compared with mature synaptic terminals (31, 39). Accordingly, in developing neurons, at stages preceding synapse formation, TR-TeNT was efficiently internalized already after 5 min in the absence of depolarization (Fig. 2 A and B). The internalization was more prominent in the axon, which represents the process where SVs are particularly concentrated and undergo active recycling (28). The pattern of labeling produced by the internalization of TR-TeNT was found to be coincident with that of the entire population of SVs, as shown by counterstaining the same neuron after fixation and permeabilization with antibodies directed against the SV marker SV2 (compare Fig. 2 A and C with B and D, respectively). These results represent a further support to the idea that TeNT is internalized inside the lumena of SVs during their exposure to the extracellular medium, as a consequence of the fusion with the plasma membrane.
Figure 2.
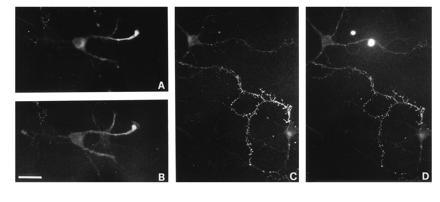
Internalization of TR-TeNT in isolated processes of developing hippocampal neurons. One-day- (A and B) and 7-day- (C and D) old neurons were exposed to 20 nM TR-TeNT for 5 min in control medium (B) or in 55 mM KCl (D). After fixation and detergent permeabilization, neurons were counterstained for SV2 (A and C). TR-TeNT produces a bright labeling of neuronal processes. The staining is more concentrated in the axon and has a finely punctate appearance. Note the identity of the fluorescent patterns produced by TR-TeNT and by the SV marker SV2. (Bar = 23.8 μm for A and B and 30 μm for C and D.)
TeNT Is Internalized Inside Small Synaptic Vesicles.
To obtain information on the type of intracellular compartment that mediates the uptake of TeNT inside synaptic terminals, the toxin was conjugated to gold particles. Neurons were exposed to the conjugated toxin under depolarizing conditions, fixed, and processed for electron microscopy. Virtually all intraneuronal gold particles were found in vesicular structures. Among these, 98% were vesicles with an appropriate size for synaptic vesicles (Fig. 3), the remaining being represented by vesicles with a larger diameter (more than 60 nm). A few gold particles were also observed at free cell surfaces (not shown). No gold particle was found in synaptic vesicles when neurons were incubated with gold-conjugated IgGs, which have the same size as gold-conjugated TeNT (not shown), indicating that the internalization of the gold-conjugated toxin is not mediated by a bulk endocytotic process.
Figure 3.
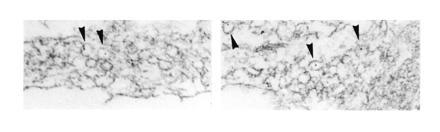
Internalization of TeNT by cultured hippocampal neurons revealed by electron microscopy immunocytochemistry. Neurons were exposed for 5 min under depolarizing conditions to 20 nM gold-conjugated TeNT. Gold particles are localized inside the lumena of small vesicles (arrowheads) which are organized in small clusters in neuronal processes. (Bar = 133 nm.)
TeNT Proteolysis of VAMP/Synaptobrevin in Hippocampal Neurons.
The L chain of TeNT is a zinc-endopeptidase specific for VAMP/synaptobrevin, which is cleaved at a single Gln-Phe peptide bond (11, 12). This leads to the release into the cytosol of the largest part of the cytosolic portion of VAMP, which includes its amino-terminal portion. In rats, only isoform 2 of VAMP and cellubrevin is cleaved efficiently by TeNT (12, 40). This cleavage can be visualized as a loss of immunostaining with a VAMP-2-specific antibody raised against the amino-terminal segment (Fig. 4). No VAMP-2 immunoreactivity is visible after 2 hr in neurons previously exposed to TeNT for 5 min in a depolarizing medium (Fig. 4D). On the other hand, VAMP-2 is present in synaptic terminals, stained with antibodies to synaptotagmin (Fig. 4 A, C, and E), both in control cells (Fig. 4B) and in neurons exposed to TeNT for 5 min in a medium with no KCl (Fig. 4F).
Figure 4.

VAMP-2 immunoreactivity after internalization of TeNT. Fifteen-day-old neurons were double labeled for VAMP-2 (B, D, and F) and synaptotagmin (A, C, and E) after different experimental treatments. (A and B) Control neurons. (C and D) Neurons examined 2 hr after a 5-min exposure to 10 nM TeNT under depolarization. (E and F) Neurons examined 2 hr after a 5-min exposure to 10 nM TeNT in Krebs–Ringer solution. In control neurons VAMP-2 immunoreactivity is present in virtually all synaptic terminals (B), as visualized by counterstaining for the SV protein synaptotagmin (A). VAMP-2 immunoreactivity is no longer visible in the large majority of nerve terminals (D) in cultures previously exposed to TeNT for 5 min under depolarizing conditions. VAMP-2 immunoreactivity is still present (F) in nerve terminals of neurons previously exposed to TeNT in control medium. (Bar = 13.8 μm.)
TeNT Cleavage of VAMP/Synaptobrevin Is Prevented by Baf-A1.
SVs are endowed with a vacuolar-type ATPase proton pump, which acidifies the vesicle lumen and creates an electrochemical gradient that drives neurotransmitter uptake (41, 42). Baf-A1, a specific inhibitor of the vacuolar ATPase (43), inhibits TeNT intoxication in phrenic nerve hemidiaphragm and in spinal cord neurons in culture (25, 26, 44). These results have been taken as an indication that TeNT has to enter an acidic intracellular compartment to intoxicate cells. However, Baf-A1 could have affected the toxin uptake by the cells or have inhibited its metalloproteinase activity. Fig. 5 A and B shows that the internalization of TR-TeNT in SVs is not affected by Baf-A1. However, the internalized TeNT is unable to cleave VAMP-2 in Baf-A1-treated neurons (Fig. 5 C, D, and E). Control experiments of TeNT proteolysis of VAMP on SVs isolated from rat brain cortex showed that Baf-A1 has no effect on the proteolytic activity of TeNT (not shown). Thus, acidification of neuronal vesicles is indeed necessary for the membrane translocation of the L chain into the cytosol, where VAMP-2 cleavage takes place.
Figure 5.

Vesicle acidification is required for TeNT proteolysis of VAMP-2 but not for TeNT penetration. (A and B) TR-TeNT is internalized in synaptic vesicles of Baf-A1-treated neurons. A bright labeling of nerve terminals, as visualized by counterstaining for synaptotagmin (A), is produced by internalization of TR-TeNT (B). (C and D) Neurons pretreated with Baf-A1, exposed to 10 nM TeNT for 5 min under depolarization, and examined 2 hr later for VAMP-2 immunoreactivity. VAMP-2 immunoreactivity is still present in Baf-A1-treated nerve terminals, exposed to TeNT in 55 mM KCl and counterstained for synaptotagmin. (Bar = 13.8 μm.) (E) Quantitative analysis of VAMP-2 cleavage at different times after exposure to 10 nM TeNT for 5 min in 55 mM KCl. VAMP-2 proteolysis is strongly reduced when the neurons are exposed to the toxin in 55 mM KCl without extracellular calcium. VAMP-2 cleavage is almost completely prevented by neuronal pretreatment with Baf-A1.
DISCUSSION
Hippocampal neurons are a very-well-characterized model of CNS neurons in culture in terms of synapse formation and of synaptic vesicle recycling (28, 39, 45). TeNT is active on hippocampal neurons in vivo, where it produces an inhibition of the γ-aminobutyrate-mediated effects (17) and causes an epileptiform syndrome (18, 19). Here, primary cultures of hippocampal neurons were used to investigate the possibility that TeNT enters CNS neurons by means of SV recycling. Such a mode of internalization would explain previous findings that animals injected with TeNT and exercised develop paralytic symptoms faster than animals at rest (20–22). Similarly, it would account for the more rapid inhibition of neuromuscular transmission by TeNT upon nerve stimulation (23, 24). Gold-labeled TeNT was occasionally seen inside vesiscles indistinguishable from small SVs (46, 47), though in most cases the toxin was detected in coated vesicles, endosome, and tubules (47), and in one study the toxin was reported to enter cells via noncoated invaginations (48). Though indicative of TeNT internalization inside intracellular compartments, these studies have not established any relation between TeNT entry into neurons and the process of SV recycling.
We have shown here that TeNT enters synaptic terminals by mean of an activity-dependent mechanism. The depolarization-dependent increase of TR-TeNT uptake reflects stimulation of exo-endocytosis of SVs. Accordingly, the increase in TR-TeNT uptake is crucially dependent on the presence of calcium in the extracellular medium. Antibodies directed against the intravesicular domain of the SV protein synaptotagmin, which have a molecular mass comparable with that of clostridial neurotoxins, are internalized inside the lumen of SVs under the same experimental conditions (28, 30, 31).
The finding that fluid phase markers are not internalized (or, at least, are internalized at levels which are below fluorescence detection) under the same experimental conditions rules out the possibility that TR-TeNT stains nerve terminals by means of the fluid phase uptake associated to SV endocytosis. The presence of gold-conjugated TeNT inside the lumen of SVs, as shown by electron microscopy, provides a clear evidence that TeNT becomes internalized in neuronal cells during the short time of exposure of the vesicle lumen to the external medium. The present findings do not preclude the possibility that TeNT proceeds thereafter into endosomal compartments, following the membrane traffic pathway believed to take place in nerve terminals (reviewed in refs. 3, 45, and 49). However, they clearly indicate that the first step of entry of TeNT into hippocampal neuronal cells is mediated by SV uptake.
One consequence of the present findings is that the nerve terminal receptor of TeNT has to be a lumenal membrane component of SVs. Its identification has escaped a variety of experimental approaches, including chemical cross-linking, immunoprecipitation, and immunoaffinity chromatography performed on rat CNS neurons and synaptosomes (G.S., O.R., and C.M., unpublished work). Synaptotagmin in complex with gangliosides has been recently indicated as the neuronal receptor for botulinum neurotoxin type B (50). Internalization of TeNT in hippocampal neurons is not prevented or reduced by the contemporary exposure of the neurons to antibodies directed against the intravesicular domain of synaptotagmin I (unpublished observations). These results strongly argue against the possibility that synaptotagmin I may be involved in TeNT internalization as well. It should be mentioned, however, that, even if synaptotagmin I is present at high concentrations in hippocampal neuron SVs (28), internalization of type B botulinum neurotoxin by an SV-mediated process was never observed (unpublished observation).
After its penetration inside the lumen of SVs, TeNT is translocated into the neuronal cytosol, where it produces the proteolytic cleavage of its substrate, the SV protein VAMP-2 (12, 16). Baf-A1, a specific and potent inhibitor of the vacuolar-type H+-ATPase, prevents neuron intoxication by TeNT (25, 26). Here, we excluded the possibilities that TeNT internalization inside the vesicles or the metalloproteinase activity is affected by Baf-A1 treatment. What appears to be prevented by Baf-A1 is the translocation of the catalytic L chain across the vesicle membrane into the cytosol. The Baf-A1 effect does not clarify whether TeNT L chain enters the cytosol from SVs or from endosomal compartments, since both of them are endowed with a vacuolar-type ATPase. However, this is an additional evidence that TeNT, as well as the other clostridial neurotoxins, belongs to a group of toxins including diphtheria toxin, exotoxin A, and the anthrax toxic complex, which penetrate into cells via a low-pH intracellular compartment. These toxins are characterized by a three-domain structural organization (reviewed in refs. 16 and 51). The catalytic activity is associated with a domain (termed A) that is disulfide linked to the other two domains, involved in cell binding (termed R) and in membrane translocation (termed T). A common property of the three-domain toxins is their ability to form ion channels in planar lipid bilayers at low pH. Acid pH triggers a conformational change of the toxin molecule that enables the T domain to penetrate the lipid bilayer and translocate the A domain across the membrane. The A domain is released into the cytosol after reduction of the interdomain disulfide bond (51, 52). This latter part of TeNT trafficking inside CNS neurons appears to be the rate-limiting step of the entire process. Indeed, a virtually complete cleavage of VAMP-2 is obtained only after 2 hr from the exposure to the toxin, whereas the binding and the internalization of the toxin take place in only 5 min.
The present results clearly trace the pathway followed by TeNT to entry CNS neurons and deliver the catalytic L chain in their cytosol. TeNT parasitizes the physiological process of SV recycling at the nerve terminal. The toxin binds to the inner surface of SVs during the short time of exposure of the lumen to the external medium and is internalized by vesicle endocytosis. The acidification of the lumen of SVs (and/or endosomes) is required for the translocation of the L chain in the cytosol and the low-pH-induced conformational change of TeNT that leads to its insertion in the lipid bilayer is well documented (16, 53, 54). This is, to our knowledge, the first example of a pathogen that uses SV recycling to enter into neurons. However, this pathway may be followed by other physiological or pathogenic ligands, including neurotropic viruses.
Acknowledgments
We thank Drs. K. Buckley (Harvard University) and R. Jahn (Yale University) for the gift of antibodies against SV2 and synaptotagmin, respectively. This study was supported by grants from Telethon-Italia, no. 763 (C.M.) and no. 672 (M.M.), and from the Consiglio Nazionale delle Ricerche.
Footnotes
Abbreviations: SV, synaptic vesicle; TeNT, tetanus neurotoxin; TR-TeNT, conjugate of Texas red and TeNT; CNS, central nervous system; Baf-A1, bafilomycin A1.
References
- 1.De Camilli P, Jahn R. Annu Rev Physiol. 1990;52:625–645. doi: 10.1146/annurev.ph.52.030190.003205. [DOI] [PubMed] [Google Scholar]
- 2.Burgoyne R D, Morgan A. Biochem J. 1993;293:305–316. doi: 10.1042/bj2930305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kelly, R. B. (1993) Cell 72, Suppl., 43–53. [DOI] [PubMed]
- 4.Sudhof T C. Nature (London) 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 5.Robinson P J, Liu J P, Powell K A, Fykse E M, Sudhof T C. Trends Neurosci. 1994;17:348–353. doi: 10.1016/0166-2236(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 6.Takei K, McPherson P S, Schmid S L, De Camilli P. Nature (London) 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- 7.Sollner T, Whiteheart S W, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman J E. Nature (London) 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 8.Ferro-Novick S, Jahn R. Nature (London) 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- 9.Bennett M K, Scheller R H. Annu Rev Biochem. 1994;63:63–100. doi: 10.1146/annurev.bi.63.070194.000431. [DOI] [PubMed] [Google Scholar]
- 10.Rothman J E, Warren G. Curr Biol. 1994;4:220–233. doi: 10.1016/s0960-9822(00)00051-8. [DOI] [PubMed] [Google Scholar]
- 11.Schiavo G, Poulain B, Rossetto O, Benfenati F, Tauc L, Montecucco C. EMBO J. 1992;11:3577–3583. doi: 10.1002/j.1460-2075.1992.tb05441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, Das Gupta B R, Montecucco C. Nature (London) 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 13.Mellanby J, Green J. J Neurosci. 1981;6:281–300. doi: 10.1016/0306-4522(81)90123-8. [DOI] [PubMed] [Google Scholar]
- 14.Wellhoner H H. Rev Physiol Biochem Pharmacol. 1982;93:1–68. doi: 10.1007/BFb0032668. [DOI] [PubMed] [Google Scholar]
- 15.Halpern J L, Neale E A. Curr Top Microbiol Immunol. 1995;195:221–241. doi: 10.1007/978-3-642-85173-5_10. [DOI] [PubMed] [Google Scholar]
- 16.Montecucco C, Schiavo G. Q Rev Biophys. 1995;28:423–472. doi: 10.1017/s0033583500003292. [DOI] [PubMed] [Google Scholar]
- 17.Calabresi P, Benedetti M, Mercuri N B, Bernardi G. Neuroscience. 1989;30:663–670. doi: 10.1016/0306-4522(89)90159-0. [DOI] [PubMed] [Google Scholar]
- 18.George G, Mellanby J. J Exp Neurol. 1982;75:678–689. doi: 10.1016/0014-4886(82)90034-6. [DOI] [PubMed] [Google Scholar]
- 19.Brace H M, Jefferys J G R, Mellanby J. J Physiol (London) 1985;368:343–357. doi: 10.1113/jphysiol.1985.sp015861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponomarev A W. Z Ges Exp Med. 1928;61:93–106. [Google Scholar]
- 21.Kryzhanovsky G N. Bull Exp Biol Med. 1958;44:1456–1464. English transl. [Google Scholar]
- 22.Wellhoner H H, Seib U C, Hensel B. Naunyn-Schmiedebergs Arch Pharmacol. 1973;276:387–394. doi: 10.1007/BF00499891. [DOI] [PubMed] [Google Scholar]
- 23.Habermann E, Dreyer F, Bigalke H. Naunyn-Schmiedebergs Arch Pharmacol. 1980;311:33–40. doi: 10.1007/BF00500299. [DOI] [PubMed] [Google Scholar]
- 24.Simpson L L. J Pharmacol Exp Ther. 1985;234:100–105. [PubMed] [Google Scholar]
- 25.Simpson L L, Coffield J A, Bakry N. J Pharmacol Exp Ther. 1994;269:256–262. [PubMed] [Google Scholar]
- 26.Williamson L C, Neale E A. J Neurochem. 1994;63:2342–2345. doi: 10.1046/j.1471-4159.1994.63062342.x. [DOI] [PubMed] [Google Scholar]
- 27.Goslin G, Banker G. In: Culturing Nerve Cells. Banker G, Goslin K, editors. Cambridge, MA: MIT Press; 1991. pp. 251–282. [Google Scholar]
- 28.Matteoli M, Takei K, Perin M S, Sudhof T C, De Camilli P. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan T A, Smith S J. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 30.Mundigl O, Verderio C, Kraszewski K, De Camilli P, Matteoli M. Eur J Cell Biol. 1995;66:246–256. [PubMed] [Google Scholar]
- 31.Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Banker G A, Cowan W M. Brain Res. 1977;126:379–425. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 33.Fraker P J, Speck J C. Biochem Biophys Res Commun. 1978;80:849–857. doi: 10.1016/0006-291x(78)91322-0. [DOI] [PubMed] [Google Scholar]
- 34.Ceccarelli B, Hurlbut W P, Mauro A. J Cell Biol. 1973;57:499–524. doi: 10.1083/jcb.57.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossetto O, Gorza L, Schiavo G, Schiavo N, Scheller R H, Montecucco C. J Cell Biol. 1996;132:167–179. doi: 10.1083/jcb.132.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogers T R, Snyder S H. J Biol Chem. 1981;256:2402–2407. [PubMed] [Google Scholar]
- 37.Bakry N, Kamata Y, Sorensen R, Simpson L L. J Pharmacol Exp Ther. 1991;258:613–619. [PubMed] [Google Scholar]
- 38.Verderio C, Coco S, Fumagalli G, Matteoli M. Proc Natl Acad Sci USA. 1995;92:6449–6453. doi: 10.1073/pnas.92.14.6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matteoli M, Verderio C, Krawzeski K, Mundigl O, Coco S, Fumagalli G, De Camilli P. J Physiol (Paris) 1995;89:51–55. doi: 10.1016/0928-4257(96)80551-1. [DOI] [PubMed] [Google Scholar]
- 40.McMahon H T, Ushkaryov Y A, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Sudhof T C. Nature (London) 1993;364:287–289. doi: 10.1038/364346a0. [DOI] [PubMed] [Google Scholar]
- 41.Moriyama Y, Maeda M, Futai M. J Exp Biol. 1992;172:171–178. doi: 10.1242/jeb.172.1.171. [DOI] [PubMed] [Google Scholar]
- 42.Schuldiner S, Shirvan A, Llinial M. Physiol Rev. 1995;75:369–392. doi: 10.1152/physrev.1995.75.2.369. [DOI] [PubMed] [Google Scholar]
- 43.Bowman E J, Siebers A, Altendorf K. Proc Natl Acad Sci USA. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson L L. J Pharmacol Exp Ther. 1983;225:546–552. [PubMed] [Google Scholar]
- 45.Parton R G, Simons K, Dotti C G. J Cell Biol. 1992;119:123–137. doi: 10.1083/jcb.119.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwab M E, Suda K, Thoenen H. J Cell Biol. 1979;82:798–810. doi: 10.1083/jcb.82.3.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parton R G, Ockleford C D, Critchley D R. J Neurochem. 1987;49:1057–1066. doi: 10.1111/j.1471-4159.1987.tb09994.x. [DOI] [PubMed] [Google Scholar]
- 48.Montesano R, Roth J, Robert A, Orci L. Nature (London) 1982;296:651–653. doi: 10.1038/296651a0. [DOI] [PubMed] [Google Scholar]
- 49.Sudhof T C, Jahn R. Neuron. 1991;6:665–677. doi: 10.1016/0896-6273(91)90165-v. [DOI] [PubMed] [Google Scholar]
- 50.Nishiki T, Kamata Y, Nemoto Y, Omori A, Ito T, Takahashi M, Kozaki S. J Biol Chem. 1994;269:10498–10503. [PubMed] [Google Scholar]
- 51.Montecucco C, Papini E, Schiavo G. FEBS Lett. 1994;346:92–98. doi: 10.1016/0014-5793(94)00449-8. [DOI] [PubMed] [Google Scholar]
- 52.Parker M W, Pattus F. Trends Biochem Sci. 1993;18:391–395. doi: 10.1016/0968-0004(93)90096-6. [DOI] [PubMed] [Google Scholar]
- 53.Boquet P, Duflot E. Proc Natl Acad Sci USA. 1982;79:7614–7618. doi: 10.1073/pnas.79.24.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montecucco C, Schiavo G, Brunner J, Duflot E, Boquet P, Roa M. Biochemistry. 1986;25:919–924. doi: 10.1021/bi00352a027. [DOI] [PubMed] [Google Scholar]