

Abstract
Abnormal nuclear factor-κB (NF-κB) signaling has been attributed to the initiation and progression of cancer. Posttranslational modification of p65 facilitates optimal NF-κB signaling after activation. Here, we show that the phosphorylation of serine 536 was required for p65-mediated transcription and IκBα expression in fibroblasts. Furthermore, tumor necrosis factor (TNF) treatment slightly induced p65 phosphorylation, and both unphosphorylated and phosphorylated p65 translocated into the nucleus. The phosphorylation of serine 536 was not required for p65-mediated protection from TNF cytotoxicity and Traf1 induction in fibroblasts. Also, the corecruitment of p65 and RNA polymerase II to the Traf1 enhancer region did not require p65 phosphorylation. However, the corecruitment of p65 and RNA polymerase II to the Csf2 promoter required the phosphorylation of serine 536. These findings suggested that the requirement of serine phosphorylation at residue 536 and the distance between the NF-κB response element and the start of transcription may influence which genes will be transcribed.
Introduction
The role of nuclear factor-κB (NF-κB) signaling in the initiation and progression of cancer has been well documented (1). Studies have detected genetic alterations of the NF-κB genes by either amplification or chromosomal translocation, in various hematopoietic cancers (2). Further, normal or aberrant NF-κB signaling induces the expression of various genes that promote cellular proliferation, survival, inflammation, angiogenesis, and metastasis, which facilitate the development of cancer (3). Although targeting the NF-κB pathway in cancer treatment may be ideal, the normal physiologic role of NF-κB for the immune system and the protection from normal apoptosis is essential (4, 5). Thus, it is important to understand how the NF-κB signaling pathway coordinates the expression of the various genes.
Recent studies have shown that optimal NF-κB signaling involves the posttranslational modification of the NF-κB proteins after activation in addition to nuclear translocation (6, 7). The NF-κB signaling pathway can be activated through various means; however, the release of cytoplasmic NF-κB proteins from the inhibitor of κB (IκB) proteins and nuclear translocation are pivotal for signal transduction (8). Nuclear NF-κB proteins are posttranslationally modified, either in the translocation process or in the nuclear milieu, which has been shown to influence DNA binding, recruitment of the transcriptional machinery, or the export of the NF-κB proteins from the nucleus (9).
In particular, phosphorylation of p65 has been well documented and several target residues have been identified after NF-κB activation (10, 11). Within the rel homology domain, serine 276 has been shown to be phosphorylated by either protein kinase A (12) or mitogen- and stress-activated kinase-1/2 (13) after treatment with lipopolysaccharide or tumor necrosis factor (TNF), respectively. This modification promotes oligomerization, DNA binding, and interaction with the transcriptional coactivator p300/CBP (14). In addition, phosphorylation of serine 529 or 536 by either casein kinase II (15) or IκB kinases α and β (16), respectively, within the transactivation domain has been well documented. In addition to the aforementioned kinases, glycogen-synthase kinase-3β (17), Akt/phosphatidylinositol 3-kinase (18), and NF-κB activating kinase (19) are involved in the phosphorylation of p65 at serine 536, which promotes the recruitment transcriptional coactivators to facilitate transcription. The phosphorylation of serine 536 by the ribosomal subunit kinase-1 reduced the association of nuclear p65 from IκBα, which resulted in an increase in nuclear retention and thus enhanced transcription (20). However, several studies have shown that the substitution of alanine for either serine 529 or 536 did not affect p65 activity, which suggested that the phosphorylation of the serine residues is not required (21, 22).
We have analyzed several p65 mutants for the requirement of serine phosphorylation for p65 activity using RelA-/- mouse embryonic fibroblasts (MEF) as the target cell. The results showed that the substitution of serine 536 with alanine abrogated p65 transcriptional activity. However, protection from TNF-induced apoptosis and the induction of Traf1 expression by p65 did not require the phosphorylation of serine 536. Also, the corecruitment of p65 and RNA polymerase II to the Traf1 enhancer was independent of the phosphorylation status of serine 536. Thus, it seems that different phosphorylation patterns of p65 are associated with distinct functions for NF-κB.
Materials and Methods
Cells and reagents
RelA-/- MEF line (a generous gift from Dr. David Baltimore, California Institute of Technology, Pasadena, CA) was maintained in DMEM supplemented with fetal bovine serum and penicillin/streptomycin (Quality Biological). The wild-type (p65) and the various serine 536 mutants (S536A and S536D) were generated as described previously (23). Oligonucleotide primers were synthesized by Sigma. The DN-IκBα expression plasmid (Clontech) was subcloned into the pEF6/V5-His expression construct according to the protocol provided by the manufacturer (Invitrogen). The following antibodies were obtained from the indicated source: phospho-p65 serine 536 and Traf1 (Cell Signal Technology); p65, IκBα, RNA polymerase II, and p300 (Santa Cruz Biotechnology); β-tubulin (Sigma); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin (Abcam); V5 antibody (Invitrogen); and nucleoporin p65 (BD Bioscience; see Supplementary Table S1 for more information). The Traf1 and GAPDH short interfering RNA (siRNA) cocktail were obtained from Dharmacon. Recombinant mouse TNF was purchased from Peprotech.
Transfections
Transient transfections were performed with the FuGENE6 reagent according to the protocol provided by the manufacturer (Roche). In the siRNA experiments, reverse transfection protocol was performed with the siPORT NeoFX reagent (Ambion) with the following modifications. Cells were transiently transfected with the various p65 mutant expression plasmids at least 6h before the siRNA transfection. The transfectants were then transfected with the indicated siRNA and incubated for 48 h. The level of suppression by the indicated siRNA was assessed by real time semiquantitative reverse transcription-PCR.
Immunoblot
Cell lysates were prepared with radioimmunoprecipitation assay buffer, and protein concentration was determined with the protein BCA assay (Pierce). Unless indicated otherwise in the figure legend, 5 μg of lysate were loaded into each lane, separated by SDS-PAGE, and transferred to a polyvinylidene difluoride membrane (Invitrogen). Membranes were probed with the various antibodies and visualized with enhanced chemifluorescence and the Typhoon scanner (GE Healthcare). Restore reagent (Pierce) was used to strip the membranes for subsequent analyses.
Reporter assay
The NF-κB reporter assay was performed according to the protocol provided by the manufacturer (Clontech). In general, cells were cotransfected with pNF-κB-SEAP reporter and pCMVβ plasmids. The cells were plated in triplicate in white 96-well tissue culture plates. After 48 h of incubation, the levels of secreted alkaline phosphatase and β-galactosidase activity were measured with the Quanti-BLUE (InvivoGen) and β-glo reagents (Promega). Differences in transfection efficiencies were normalized with the β-galactosidase activity. Results have been repeated at least thrice.
TNF cytotoxicity assay
After 24 h of incubation, transfectants were plated in triplicate and treated with various concentrations of TNF overnight. Cell viability was assessed with the WST cell viability assay (Roche). Percentage cell viability was calculated from the absorbance of TNF-treated transfectant versus untreated transfectant. Results have been repeated at least thrice.
Quantitation of mRNA transcripts
Total RNA was harvested with the RNeasy kit (Qiagen), and cDNA was obtained from the total RNA with the SuperScript First Stand Synthesis kit (Invitrogen). Transcript levels were measured with the iTaq SYBR green supermix (Bio-Rad) and gene-specific primers (see Supplementary Table S2; ref. 24). Real-time PCR products were subsequently evaluated with melting curve analyses and on agarose gels. The level of induction was calculated as described by Livak and Schmittgen (25). β-Actin and GAPDH were used as housekeeping genes for the calculations. Data points and error bars represent the average and SD, respectively, of induction. Results have been repeated at least thrice.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was conducted as previously described (23) with the following modifications. After cross-linking with formaldehyde, the transfectants were washed twice with cold PBS, scraped, and transferred to centrifuge tubes on ice. Soluble chromatin from the samples was obtained by sonication (Bronson sonicator 350, 10 cycles of 20 burst and 1 min rest on ice). The chromatin was incubated overnight with 2 μg of the indicated antibody. After washing and elution, the immunocomplexed DNA was isolated with Qiaquick columns (Qiagen) and analyzed by real-time PCR (see Supplementary Table S2). The level of increase was calculated as described previously (26). Depending on the gene, the corresponding negative primer set was used to calculate the increase.
Results
Phosphorylation of serine 536 is required for p65 transcription.
To investigate the role of p65 phosphorylation at serine 536 (phospho-p65), we transfected two serine 536 p65 mutants into RelA-/- MEF. The serine 536 to alanine (S536A) mutant represents a form of p65 that cannot be phosphorylated at position 536. In contrast, the serine 536 to aspartic acid (S536D) mutant is a phosphomimetic mutant that resembles phospho-p65 and is detected by antibody to phospho-p65. Both p65 mutants were transiently transfected at various amounts into the MEFs and the lysates were analyzed for phosphorylated p65 levels. As expected, the phosphoserine 536 p65 antibody detected the phosphomimetic S536D mutant (see Fig. 1A, lanes 10-12) but not the S536A mutant (lanes 7-9). It is important to note that a low level of phospho-p65 (top, lane 6) was detected in the lysate from the wild-type p65 transfectant. The lack of detection of the p65 and S536A mutant by the phospho-p65 antibody was not due to the absence of p65 or loading differences (lanes 4-9, Fig. 1A, middle and bottom, respectively). Furthermore, the same transfectants were analyzed for p65 transcriptional activity via the NF-κB reporter assay (Fig. 1B). The substitution of alanine for serine at residue 536 suppressed p65 transcriptional activity. In contrast, the phosphomimetic mutation (S536D) enhanced p65 transcriptional activity. As would be expected, NF-κB-regulated cellular genes were also up-regulated in cells transfected with either wild-type p65 or S536D; an increase in IκBα levels was observed with wild-type p65 and S536D (middle, Fig. 1A, lanes 4-6 and 7-9), but not with transfection of control (pEF) and S536A mutant constructs (lanes 1-3 and 7-9). These findings indicated that the phosphorylation of serine 536is required for p65 transcriptional activity.
Figure 1.
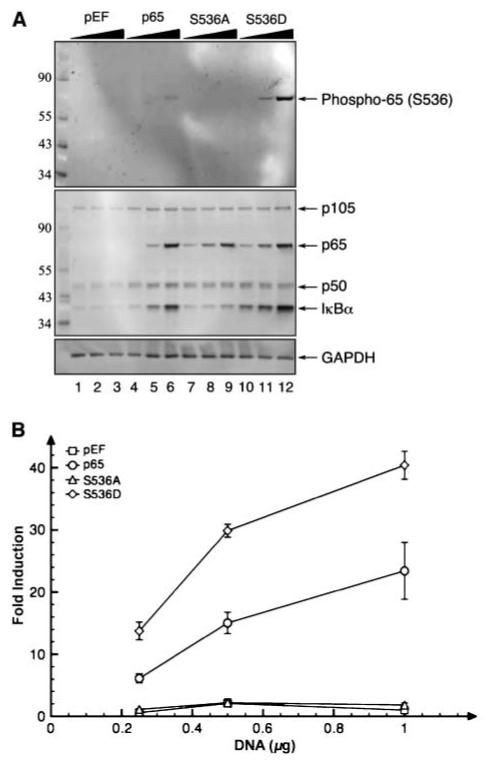
Phosphorylation of serine 536 was required for p65-mediated transcription. A, substitution of alanine for serine 536 inhibited induction of IκBα expression. Different concentrations of the p65 mutant expression plasmids were transfected into RelA-/- MEFs (black triangles, 0.25, 0.5, and 1 μg of plasmid DNA). Lysates from the transfects were probed with phospho-p65 serine 536 antibody (top; see Supplementary Fig. S7 for phospho-antibody neutralization study). The membrane was stripped and reprobed with an antibody cocktail of p105/50, p65, and IκBα antibodies (middle). The membrane was restripped and reprobed with GAPDH antibody to assess loading efficiency (bottom). Numbers on the left, molecular weight markers in kilodaltons. B, the substitution of alanine for serine 536 blocked p65 transcriptional activity. Cells transfected with 0.25, 0.5, and 1 μg of the various p65 mutant expression plasmids were analyzed for p65-mediated transcription by the NF-κB reporter assay. Points, average of the fold induction; bars, SD.
Induction of p65 phosphorylation by TNF
Various studies have shown that TNF treatment resulted in the phosphorylation of p65 at serine 536 (27, 28). As expected, the treatment of p65 transfectants with TNF resulted in an increase in phosphorylation of p65 (Fig. 2A, lanes 4-6). However, in comparison with the phosphomimetic S536D mutant (lane 2), the level of phosphorylation was very low. Moreover, when compared with total p65 levels (Fig. 2A, bottom), TNF treatment of wild-type p65 resulted in the phosphorylation of 10% of total p65. Although a small percentage of p65 was phosphorylated after TNF treatment, it is conceivable that the majority of the p65 population that translocates into the nucleus is phosphorylated. To address this, p65 transfectants were treated with TNF, and cytoplasmic and nuclear extracts were assessed for phospho-p65 and p65 levels (Supplementary Fig. S1). In accordance with Fig. 2A, <10% of the nuclear p65 was phosphorylated, which suggested that both phosphorylated and unphosphorylated p65 translocate into the nucleus after TNF treatment. Last, p65 transcriptional activity was analyzed in a NF-κB reporter assay after TNF treatment. As seen in Fig. 2B, the TNF treatment of the transfectants resulted in an increase in p65-mediated NF-κB transcriptional activity. This suggested that additional factors other than the phosphorylation of serine 536 were involved in p65 transcriptional activity after TNF treatment. The small increase in NF-κB transcriptional activity observed in the absence of p65 expression (pEF control) may be due to either c-Rel or other NF-κB protein transcriptional activity after TNF treatment. Thus, TNF treatment resulted in a small increase in the phosphorylation of p65 and the nuclear translocation of both unphosphorylated and phosphorylated p65.
Figure 2.
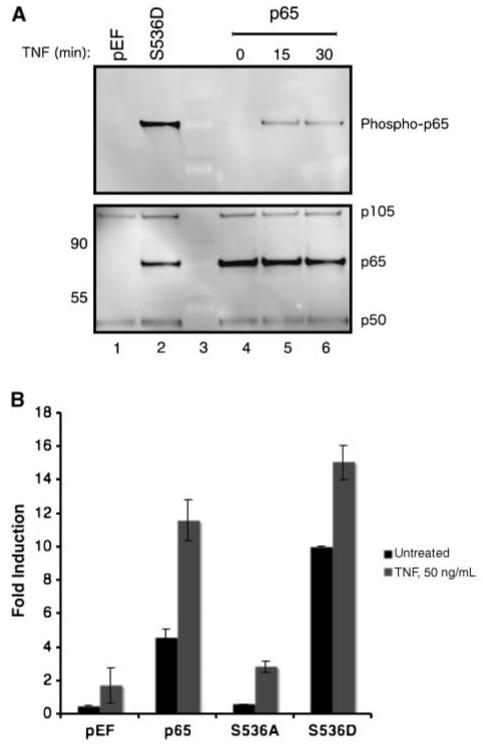
TNF induces p65 phosphorylation and increases p65-mediated transcription. A, induction of p65 phosphorylation after TNF treatment. Total lysates from cells transfected with 0.5 μg of p65 plasmid were treated with TNF (50 ng/mL) for 15 and 30 min and probed with phospho-p65 antibody (top). The samples were stripped/reprobed with an antibody cocktail of p105/50 and p65 antibodies. pEF and S536D samples were included as a phospho-p65 (compare lanes 2 and 4) and p65 (compare lanes 1 and 2) control. Equal levels of p105/50 served as loading controls for the blot. B, TNF treatment increases NF-κB transcriptional activity. Cells transfected with the various p65 mutants were treated with various amounts of TNF overnight and the induction of NF-κB activity was assessed by the reporter assay. Columns, average fold induction; bars, SD.
Protection from TNF does not require serine 536 phosphorylation
The role of p65 in protecting cells from TNF-mediated cytotoxicity has been well documented (29, 30). MEFs transfected with the various p65 mutants were treated with TNF, and the role of serine 536 phosphorylation in TNF resistance was analyzed. As seen in Fig. 3A, a decrease in cell viability was observed with TNF treatment in a dose-dependent manner in the absence of p65 expression. The expression of either transcriptionally active p65 or S536D resulted in an increase in cell viability with TNF treatment. In addition, the transcriptionally inactive p65 mutant S536A protected the cells from the cytotoxic effects of TNF. Because it is conceivable that S536A indirectly protects cells from TNF, the protective effects of the p65 mutants was measured in the presence of the dominant negative IκBα mutant (DN-IκBα). The DN-IκBα possesses two mutations (serine 32/36) that block IκBα phosphorylation and degradation, which prevent NF-κB nuclear translocation and transcriptional activity (Supplementary Fig. S2). As seen in Fig. 3B, the cotransfection of DN-IκBα resulted in the suppression of the p65-mediated TNF resistance in a dose-dependent manner. The level of protection conferred by the p65 mutants was reduced to the amount of protection seen in the absence of p65 with the cotransfection with the highest concentration of DN-IκBα. The findings indicate that the phosphorylation of serine 536 was not required for p65-mediated resistance to TNF effects.
Figure 3.
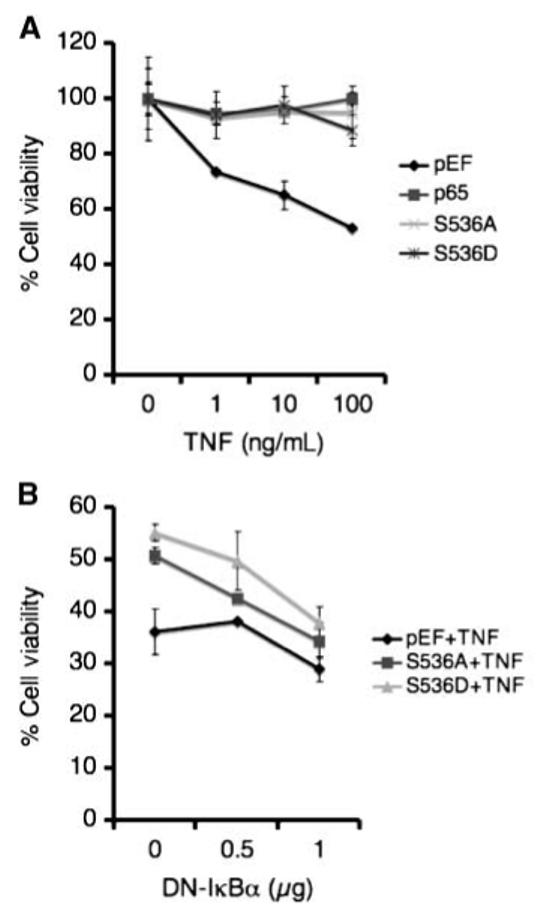
Phosphorylation of serine 536 was not required for p65-mediated TNF resistance. A, inhibition of TNF-mediated cytotoxicity with p65 mutant expression. Cells transfected with the various p65 mutants were treated with various doses of TNF overnight, and the level of cell viability was assessed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay. Points, average; bars, SD. B, inhibition of p65-mediated TNF resistance by DN-IκBα. Cells were cotransfected with the various p65 mutants with different amounts of DN-IκBα expression plasmid and treated with TNF (100 ng/mL) overnight. The percent cell viability was measured as described above.
Traf1 induction does not require serine 536 phosphorylation
Although the S536A mutant lacked p65 transcriptional activity, the data from the previous section suggested that S536A could be inducing the expression of a NF-κB-dependent antiapoptotic gene. To investigate this, various antiantiapoptotic gene candidates were analyzed after p65 mutant expression. Attention was given to genes that were induced by S536A because the expression profiles of wild-type p65 and S536D overlapped considerably (data not shown). Of the numerous NF-κB-dependent antiapoptotic genes (31, 32), Traf1 was consistently up-regulated in transfectants expressing the S536A mutant compared with the control pEF vector. As seen in Fig. 4A, Traf1 transcript levels were measured and compared with the control vector pEF. The expression of the various p65 mutants resulted in an increase in Traf1 transcripts. Furthermore, cotransfection of the DN-IκBα suppressor resulted in a decrease in the p65-mediated Traf1 induction. Similarly, an increase in Traf1 protein was observed with the expression of the various p65 mutants (Fig. 4B, top, lanes 4, 7, and 10). Also, the cotransfection of DN-IκBα with the p65 mutants resulted in the reduction of Traf1 expression in a dose-dependent manner (see lanes 4-12). In addition, TNF treatment further increased the p65-mediated Traf1 induction (Fig. 4C). It is notable that TNF treatment increased Traf1 transcript levels modestly in the absence of p65, which indicated that Traf1 transcription may involve other factor(s) in addition to p65. Thus, the induction of Traf1 expression by p65 does not require the phosphorylation of serine 536.
Figure 4.
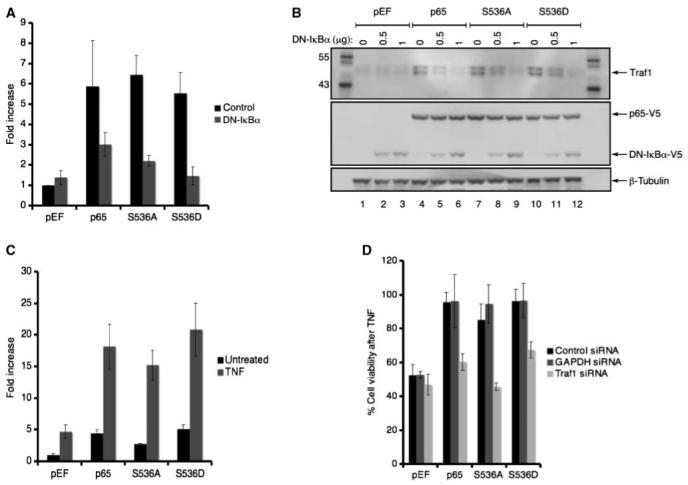
Phosphorylation of serine 536 was not required for Traf1 expression. A, the increase in Traf1 transcript levels with p65 mutant expression. cDNAfrom cells cotransfected with the various p65 mutants and either the control or DN-IκBα expression plasmid were analyzed for Traf1 transcript levels. The fold increase represents the amount of Traf1 induction over the pEF control. Columns, average; bars, SD. B, the induction of Traf1 protein with p65 mutant expression. Lysates from cells cotransfected with the various p65 mutants and different concentrations of DN-IκBα (numbers on top of the top panel) were probed with the Traf1 antibody (top). The membrane was stripped and reprobed with the V5 antibody (middle). The membrane was further reprobed with the β-tubulin antibody. C, TNF treatment increased Traf1 transcripts. Cells expressing the various p65 mutants were treated with TNF (50 ng/mL) for 4 h and Traf1 transcripts were analyzed as described above. D, Traf1 siRNA suppressed p65-mediated TNF resistance. Cells cotransfected with the p65 mutants and the indicated siRNA were treated with TNF (50 ng/mL) overnight and the percent cell viability was measured.
It is conceivable that the increase of Traf1 transcript and protection from TNF by S536A may not reflect a causal relationship. To address this, the amount of protection from TNF was analyzed in transfectants expressing the various p65 mutants with siRNA against Traf1. TNF treatment of the cells without p65 resulted in a loss of cell viability, and the expression of any of the p65 mutants protected the cells from TNF as seen as an increased the cell viability (Fig. 4D). The cotransfection of the Traf1 siRNA resulted in a decrease in Traf1 levels (Supplementary Fig. S3) and the loss of p65-mediated protection from TNF as seen by a decrease in cell viability. As a control, siRNA against GAPDH did not affect the p65-mediated protection from TNF. The data suggested that Traf1 expression plays a major role in protecting the cells against TNF. Collectively, the data suggest that the induction of Traf1 expression may be responsible at least in part for the S536A-mediated protection from TNF cytotoxicity.
Serine 536 phosphorylation is not required for the corecruitment of RNA polymerase II to Traf1 gene
Previous studies have identified two NF-κB response elements and a Sp1 element as being required for optimal CD40-induced Traf1 expression in murine B cells (33). It was determined that the individual NF-κB response elements acted as enhancer elements, which could not independently induce Traf1 expression. The NF-κB enhancer elements are located either distal or intronic to the Traf1 gene. It is possible that the S536A mutant could be acting as an enhancer to facilitate Traf1 transcription. To investigate this, ChIP analyses were conducted to detect the recruitment of the S536A mutant to either NF-κB enhancer element of the Traf1 gene. As seen in Fig. 5A, three sets of primers were used. Two sets of primers were designed to span either the distal (-1,597/-1,392) or the intronic (6,984/7,216) NF-κB enhancer element of the Traf1 gene. The third primer set (-2,309/-2,081) was designed around a region 0.5 kb upstream from the distal element, which does not contain a NF-κB response element and served as a negative control. In addition to measuring the recruitment of p65 to the Traf1 gene after TNF treatment, the corecruitment of RNA polymerase II was included to assess the transcriptional status. The β-actin promoter primer set was added as a positive control for the recruitment of RNA polymerase II (34). With the exception of the recruitment of RNA polymerase II to the promoter of β-actin, none of the Traf1 enhancer elements were occupied before or after TNF treatment in the pEF transfectant without p65 (Fig. 5B). In contrast, a 4-fold increase in S536A was recruited to the distal Traf1 enhancer element after TNF treatment (Fig. 5C). Also, RNA polymerase II was corecruited to the same site after TNF treatment. The increase in recruitment of S536A and RNA polymerase II was specific to TNF treatment because the levels of RNA polymerase II recruitment to β-actin did not significantly increase after treatment. Similarly, the corecruitment of S536D and RNA polymerase II to the distal Traf1 enhancer element was increased after TNF treatment (Fig. 5D). Wild-type p65 and RNA polymerase II were also corecruited to the distal enhancer element of the Traf1 gene after TNF treatment (see Supplementary Fig. S4). The findings suggested that p65, regardless of its phosphorylation status at serine 536, is recruited to the distal NF-κB enhancer element of the Traf1 gene after TNF treatment. Furthermore, the occupancy of the distal site by p65 promotes the corecruitment of RNA polymerase II.
Figure 5.
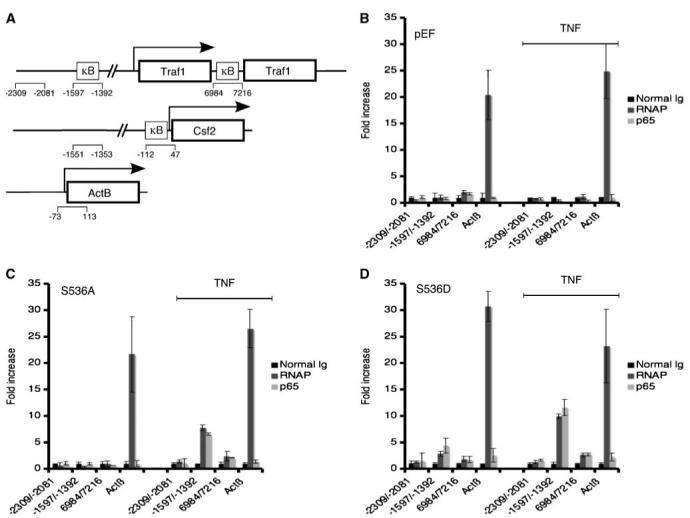
Recruitment of p65 and RNApolymerase II to Traf1 gene did not require serine 536 phosphorylation. A, primers of the Traf1, Csf2,and ActB genes used in the ChIP experiments. B to D, corecruitment of p65 and RNApolymerase II (RNAP) to the Traf1 distal enhancer after TNF treatment. As described in Materials and Methods, immunoprecipitated soluble chromatin complexes were isolated from cells transfected with the p65 mutants (B, pEF; C, S536A; D, S536D) that were either untreated or treated with 50 ng/mL of TNF for 1 h (line on the right side of the histogram), and the level of DNA enrichment was assessed by semiquantitative real-time PCR. The fold increase in recruitment to a certain site represents an increase compared with the negative region of the same gene. Columns, average; bars, SD.
In contrast to Traf1, the expression of Csf2 (granulocyte macrophage colony-stimulating factor) by p65 required the phosphorylation of serine 536. As seen in Fig. 6A, Csf2 transcript levels increased with the expression of p65 and S536D, but not with S536A, and the levels further increased with TNF treatment. It is notable that the magnitude of Csf2 induction by p65 was dependent on the level of serine 536 phosphorylation, an observation that was not seen with the IκBα expression. ChIP experiments were conducted on cells transfected with either S536A or S536D before and after TNF treatment to determine if S536D was selectively recruited to the Csf2 promoter. Two primer sets were designed (see Fig. 5A) where one set encompasses a region that does not contain an NF-κB response element (-1,551/-1,353) and the other one is proximal to the transcription start which does contain a NF-κB response element (-112/47; ref. 35). As seen in Fig. 6B, S536D and RNA polymerase II were recruited to the Csf2 promoter only after TNF treatment. S536A was not. Holloway et al. (35) have shown that the induction of Csf2 gene requires the chromatin rearrangement of the Csf2 promoter. Based on our findings, it was possible that the lack of recruitment of the S536A mutant to the Csf2 promoter was due to chromatin inaccessibility. Ito et al. (36) have shown that the acetylation of histones by p300 is required for chromatin rearrangement of the Csf2 promoter for p65-medieated Csf2 induction. Furthermore, Chen et al. (37) have shown that the phosphorylation of p65 at serine 536 facilitates the association between p300 and p65. It was possible that the Csf2 promoter was inaccessible to the S536A mutant. To address this, we have performed ChIP analysis with anti-p300 antibody on the chromatin samples used in Fig. 6B. As seen with S536D and RNA polymerase, but not S536A, p300 was recruited to the proximal region of Csf2 gene after TNF treatment (Fig. 6C). The findings were in agreement with the aforementioned studies where phosphorylation of p65 promotes the association with p300, and p300 is required for chromatin accessibility of p65 to the Csf2 gene. Thus, the lack of recruitment of S536A mutant to the Csf2 gene was due to the inability of S536A to associate with p300, which thereby prevented chromatin accessibility. Taken together, the results indicated that the phosphorylation of p65 was required for the Csf2 gene where the NF-κB response element is proximal to the start of transcription. By contrast, as seen in Traf1 expression, the capacity of p65 to function as an enhancer at distal sites may not require phosphorylation of serine 536.
Figure 6.
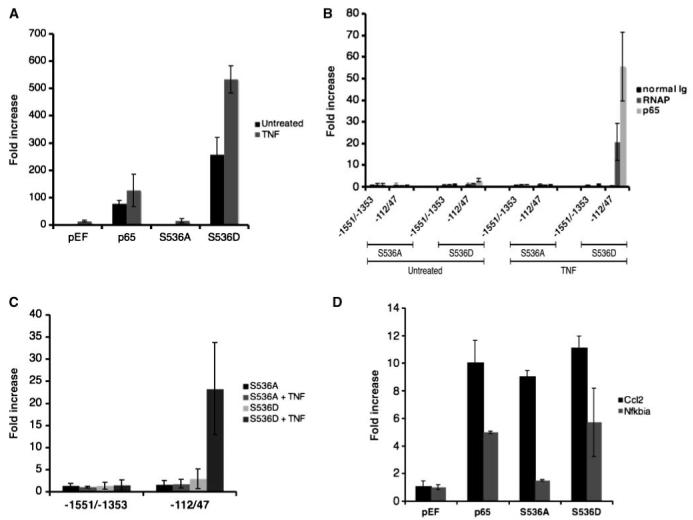
Phosphorylation of serine 536 was required for Csf2 transcription. A, an increase in p65-mediated Csf2 transcript levels required serine 536 phosphorylation. Csf2 transcript levels were measured either before or after TNF treatment (50 ng/mL) for 4 h in cDNA isolated from cells transfected with the p65 mutants.B, corecruitment of p65 and RNA polymerase II after TNF treatment required serine 536 phosphorylation. ChIP analyses were conducted on cells transfected with the p65 mutants either before or after TNF using the Csf2 promoter primers as described in Fig. 5A. (See Supplementary Fig. S6 for gel images.) C, corecrutiment of p300 to the proximal region of the Csf2 gene after TNF treatment. ChIP analyses were conducted with anti-p300 antibody. D, an increase in p65-mediated Nfkbia, but not Ccl2, transcript levels required serine 536 phosphorylation. Ccl2 and Nfkbia transcripts were measured from total RNAi solated from cells transfected with the p65 mutants.
The relationship between the requirement for serine 536 phosphorylation for p65-mediated transcription and the location of the NF-κB response element was not unique to the induction of Traf1 and Csf2. Similar to Traf1, the induction of Ccl2 transcription, which has a distal NF-κB-dependent enhancer element (38), did not require the phosphorylation of serine 536 (Fig. 6D). Alternatively, the phosphorylation of p65 was essential for the induction for Nfkbia transcription, which has a proximal NF-κB response element (39). Taken together, these findings were in accordance with those of theTraf1 and Csf2 results where the distance of the NF-κB cis-acting element to the transcription start site determines the requirement for p65 phosphorylation.
Discussion
The data collectively showed that the phosphorylation of serine 536 was not required for the induction of Traf1 expression and protection from TNF-mediated cytotoxicity. In this situation, p65 functions as an enhancer element in gene transcription. In contrast, the ability of p65 to induce transcription directly as seen in the NF-κB reporter assay or the expression of IκBα relied on the phosphorylation of serine 536. In addition, although a small percentage of p65 was phosphorylated at serine 536 after TNF treatment, both populations of unphosphorylated and phosphorylated p65 translocated into the nucleus. Depending on the phosphorylation status of p65 and the gene promoter, a selective recruitment and induction of transcription were observed after TNF treatment. Although it has been generally understood that the phosphorylation of p65 results in an increase of NF-κB-dependent gene transcription, the data described here suggested that the location of the NF-κB binding region in the context of the transcription start site may influence which genes will be induced by nonphosphorylated or phosphorylated p65.
In addition to Traf1 induction by the various p65 mutants, other antiapoptotic genes were induced after p65 expression with or without TNF treatment. XIAP, c-IAP 1 and 2, and Traf2 are some of the antiapoptotic genes that were increased with p65, and S536D expression as detected in cDNA microarray analyses (see Supplementary Table S3). The induction of these antiapoptotic genes by p65 has been well documented by others (19). The induction of Traf1 by the nontranscriptionally active, nonphosphorylated p65 (S536A mutant) has not been reported. Although an increase in sensitivity to TNF-mediated cytotoxicity was observed with all of the p65 mutants after Traf1 suppression (as seen in Fig. 4D), S536A was the most sensitive. It is possible that the other antiapoptotic genes protected the cells expressing either the wild-type or phosphorylated p65 from TNF-induced apoptosis. It is notable that other genes, which are not involved in regulating apoptosis, were also induced by p65 and S536D as detected with the cDNA microarray analyses. Furthermore, the level of Traf1 and Csf2 induced by TNF treatment in the exogenously expressed p65 in the RelA-/- MEF line were similar to that observed with endogenous p65 in 3T3 fibroblasts (see Supplementary Fig. S5).
The contradictory results where S536A lacked transcriptional activity but induced Traf1 transcription could be explained by the dual role of p65 in inducing gene expression. It is clear from the evidence that the S536A mutant did not induce either IκBα or Csf2 expression, and the S536A mutant did not have any transcriptional activity as seen in the NF-κB reporter assay (Fig. 1B). In this case, the NF-κB response elements are proximal to the start of transcription for these genes. It is likely that phospho-p65 plays a pivotal role in establishing the preinitiation complex by recruiting transcriptional cofactors, which may regulate chromatin accessibility or assembly of the transcription complex. In this situation, phosphorylation of p65 at serine 536 is critical. As shown by Chen et al. (37), the association of p65 and p300 requires the phosphorylation of serine 536. Also, Ito et al. (36) have shown that the accessibility of the Csf2 promoter to p65 involves p300 activity. In accordance, we have observed the corecruitment of p300 with S536D and RNA polymerase II to the Csf2 promoter after TNF treatment (Fig. 6C). In contrast, as with Traf1 gene, the NF-κB response element is distal to the start of transcription and other transcription factors, such as Sp1, have been described to play a role in transcription (40). Although the chromatin architecture at the Traf1 gene after TNF treatment is not entirely known, it is conceivable that p65 plays a supportive role by stabilizing the formation of the enhanceosome complex from a distal site to facilitate transcription (41, 42). This function of p65 does not require the phosphorylation of serine 536. In accordance with p65 playing a supportive role in Traf1 transcription, the magnitude of Traf1 induction by p65 was far less than that seen with Csf2. Similarly, the level of p65 recruitment and corecruitment of RNA polymerase II to the Traf1 enhancer element was far less than that to the Csf2 promoter.
As seen in with the Traf1 gene, phosphorylation of p65 at serine 536was not necessary for Ccl2 transcription. The Traf1 and Ccl2 genes share a common feature in that a distal NF-κB-dependent enhancer and a proximal Sp1-dependent promoter have been shown to regulate gene transcription. The induction of both genes by p65 was independent of serine 536 phosphorylation. Teferedegne et al. (38) have shown that the truncation of the transactivation domain of p65, which contains serine 536, did not inhibit p65-mediated Ccl2 induction after TNF treatment. It is plausible that the location of the NF-κB response element from the transcription initiation site could predict whether phosphorylation of p65 at serine 536 is required for the induction of transcription.
The phosphorylation of p65 has been shown to be required for gene transcription. The evidence presented here also confirms the need to phosphorylate serine 536 for optimal p65 transcriptional activity, and IκBα and Csf2 gene expression. However, p65 mediated resistance to TNF cytotoxicity involved the induction of Traf1 expression, and this was independent of the phosphorylation of serine 536. Furthermore, phosphorylation of p65 at serine 536 was not essential for the corecruitment of p65 and RNA polymerase II to the Traf1 enhancer. These differences correlate with the proximity of the NF-κB binding site to the transcriptional start site. Thus, the formation of an enhanceosome complex at the distal region and the induction of Traf1 transcription by p65 did not require the phosphorylation of serine 536. We propose that the location of the NF-κB binding site could predict the requirement for the phosphorylation of serine 536in regulating p65-mediated gene transcription. It is conceivable that both unphosphorylated and phosphorylated p65 induce different genes that contribute to the initiation and progression of cancer development.
Supplementary Material
Acknowledgments
Grant support: Intramural Research Program of the NIH, National Institute on Aging.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Dr. Ranjan Sen for the critical discussions.
References
- 1.Basseres DS, Baldwin AS. Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–30. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 2.Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–43. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 3.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF-κB. Oncogene. 2006;25:6800–16. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 4.Luo JL, Kamata H, Karin M. IKK/NF-κB signaling: balancing life and death—a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-κB signaling module. Oncogene. 2006;25:6706–16. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 6.Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 7.Perkins ND. Post-translational modifications regulat- ing the activity and function of the nuclear factor κB pathway. Oncogene. 2006;25:6717–30. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 8.Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 9.Chen LF, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 10.Viatour P, Merville MP, Bours V, Chariot A. Phos- phorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 11.Li CC, Korner M, Ferris DK, Chen E, Dai RM. Longo DL. NF-κB/Rel family members are physically associated phosphoproteins. Biochem J. 1994;303:499–506. doi: 10.1042/bj3030499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–24. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 13.Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–24. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong H, May MJ, Jimi E, Ghosh S. The phosphor- ylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–36. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 15.Bird TA, Schooley K, Dower SK, Hagen H, Virca GD. Activation of nuclear transcription factor NF-κB by interleukin-1 is accompanied by casein kinase II- mediated phosphorylation of the p65 subunit. J Biol Chem. 1997;272:32606–12. doi: 10.1074/jbc.272.51.32606. [DOI] [PubMed] [Google Scholar]
- 16.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–6. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 17.Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2002;283:G204–11. doi: 10.1152/ajpgi.00016.2002. [DOI] [PubMed] [Google Scholar]
- 18.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-κB through utilization of the IκB kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–40. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 19.Fujita F, Taniguchi Y, Kato T, et al. Identification of NAP1, a regulatory subunit of IκB kinase-related kinases that potentiates NF-κB signaling. Mol Cell Biol. 2003;23:7780–93. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 Induces NF-κB activation by an IκB kinase-independent mechanism involving phosphorylation of p65 by ribo- somal S6 kinase 1. J Biol Chem. 2004;279:26115–25. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- 21.Okazaki T, Sakon S, Sasazuki T, et al. Phosphorylation of serine 276 is essential for p65 NF-κB subunit- dependent cellular responses. Biochem Biophys Res Commun. 2003;300:807–12. doi: 10.1016/s0006-291x(02)02932-7. [DOI] [PubMed] [Google Scholar]
- 22.Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and IL-1-inducible phosphoryla- tion of p65 NF-kB at serine 536 is mediated by multiple protein kinases including IKKα, IKKbβ, IKKε, TBK1 and an unknown kinase and couples p65 to TAFII31-mediated IL-8 transcription. J Biol Chem. 2004;22:55633–43. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- 23.Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphor- ylation of RelA/p65 on serine 536 defines an I{κ}B{α}- independent NF-{κ}B pathway. J Biol Chem. 2005;280:34538–47. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- 24.Overbergh L, Giulietti A, Valckx D, Decallonne R, Bouillon R, Mathieu C. The use of real-time reverse transcriptase PCR for the quantification of cytokine gene expression. J Biomol Tech. 2003;14:33–43. [PMC free article] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔC(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Geisberg JV, Struhl K. Quantitative sequential chro- matin immunoprecipitation, a method for analyzing co-occupancy of proteins at genomic regions in vivo. Nucleic Acids Res. 2004;32:e151. doi: 10.1093/nar/gnh148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D, Westerheide SD, Hanson JL, Baldwin AS., Jr. Tumor necrosis factor α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275:32592–7. doi: 10.1074/jbc.M001358200. [DOI] [PubMed] [Google Scholar]
- 28.Sakurai H, Suzuki S, Kawasaki N, et al. Tumor necrosis factor-α-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–23. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- 29.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 30.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004;23:2934–49. doi: 10.1038/sj.onc.1207515. [DOI] [PubMed] [Google Scholar]
- 31.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr. NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 32.Schwenzer R, Siemienski K, Liptay S, et al. The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-κB and c-Jun N-terminal kinase. J Biol Chem. 1999;274:19368–74. doi: 10.1074/jbc.274.27.19368. [DOI] [PubMed] [Google Scholar]
- 33.Dunn IF, Sannikova TY, Geha RS, Tsitsikov EN. Identification and characterization of two CD40-induc- ible enhancers in the mouse TRAF1 gene locus. Mol Immunol. 2000;37:961–73. doi: 10.1016/s0161-5890(01)00015-3. [DOI] [PubMed] [Google Scholar]
- 34.Fujii-Yamamoto H, Kim JM, Arai K, Masai H. Cell cycle and developmental regulations of replication factors in mouse embryonic stem cells. J Biol Chem. 2005;280:12976–87. doi: 10.1074/jbc.M412224200. [DOI] [PubMed] [Google Scholar]
- 35.Holloway AF, Rao S, Chen X, Shannon MF. Changes in chromatin accessibility across the GM-CSF promoter upon T cell activation are dependent on nuclear factor κB proteins. J Exp Med. 2003;197:413–23. doi: 10.1084/jem.20021039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1κ-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen LF, Williams SA, Mu Y, et al. NF-κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–75. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teferedegne B, Green MR, Guo Z, Boss JM. Mecha- nism of action of a distal NF-κB-dependent enhancer. Mol Cell Biol. 2006;26:5759–70. doi: 10.1128/MCB.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-α is critical for cytokine-induced gene expression. Nature. 2003;423:655–9. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 40.Dunn IF, Geha RS, Tsitsikov EN. Structure of the murine TRAF1 gene. Mol Immunol. 1999;36:611–7. doi: 10.1016/s0161-5890(99)00075-9. [DOI] [PubMed] [Google Scholar]
- 41.George AA, Sharma M, Singh BN, Sahoo NC, Rao KV. Transcription regulation from a TATA and INR-less promoter: spatial segregation of promoter function. EMBO J. 2006;25:811–21. doi: 10.1038/sj.emboj.7600966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edelstein LC, Lagos L, Simmons M, Tirumalai H, Gelinas C. NF-κ B-dependent assembly of an enhanceosome-like complex on the promoter region of apoptosis inhibitor Bfl-1/A1. Mol Cell Biol. 2003;23:2749–61. doi: 10.1128/MCB.23.8.2749-2761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.