

Abstract
Translocation of membrane impermeant molecules to the interior of living cells is a necessity for many biochemical investigations. Myristoylation was studied as a means to introduce peptides into living cells. Uptake of a myristoylated, fluorescent peptide was efficient in the B lymphocyte cell line BA/F3. In contrast, this cell line was resistant to peptide uptake using a cell penetrating peptide derived from the TAT protein. In BA/F3 cells, membrane association was shown to be rapid reaching a maximum within 30 minutes. Cellular uptake of the peptide lagged the membrane association, but occurred within a similar time frame. Experiments performed at 37°C vs. 4°C demonstrated profound temperature dependence in the cellular uptake of myristoylated cargo. Myristoylated peptides with either positive or negative charge were shown to load efficiently. In contrast to TAT-conjugated cargo, pyrenebutyrate did not enhance cellular uptake of the myristoylated peptide. The myristoylated peptide did not adversely affect cell viability at concentrations up to 100 μM. This assessment of myristoyl-based transport provides fundamental data needed in understanding the intracellular delivery of myristoylated peptide cargoes for cell-based biochemical studies.
The introduction of membrane-impermeant molecules into living cells plays an important role in the investigation of cellular behavior, thus there has long been a need to introduce such molecules without undue perturbation of the cell. Notable in this regard is the recent discovery of distinctive compounds that when conjugated to peptides, proteins and nucleotides lead to their uptake by living cells (1). Short peptide sequences known as cell penetrating peptides (CPPs, also known as protein transduction domains [PTDs]) have received the most attention in this category of molecules to internalize bioactive agents that would otherwise be excluded from cells (2). Although still a somewhat controversial topic, endocytosis appears to be the primary mechanism of cellular uptake mediated by CPPs (3). Association of the CPP with the cell membrane is a required first step in this cellular uptake process. Experimental evidence suggests that membrane association of TAT and other arginine-rich CPPs occurs through heparin sulfate proteoglycans (HSPGs) (4, 5). The transduction domain from the HIV TAT protein has perhaps been the most widely used and characterized CPP to date (2, 3). The ability of the TAT protein to cross the plasma membrane resides primarily in the highly basic region composed of the 9-amino acid residues 49-57 (RKKRRQRRR) (6, 7). Peptides incorporating this sequence have been conjugated to an extensive assortment of biomolecules for delivery into cells. These cargos range from small peptides and peptide nucleic acids to full-length proteins and nanoparticles (8-11). New CPPs continue to be discovered and include peptides derived from native proteins, synthetic peptides and small molecule mimics (12-15).
During the same time frame that extensive studies of CPPs have taken place, another approach to convey membrane permeability has been used, but poorly described-- the addition to the molecular cargo of the hydrophobic 14-carbon saturated fatty acid myristate. The myristoyl group is a naturally occurring post-translational modification that serves to target cytoplasmic proteins to intracellular membranes. In the cell, the enzyme N-myristoyl transferase catalyzes the covalent attachment of myristate to the N-terminal glycine residue of a number of proteins (16, 17). Myristoylation of an exogenous peptide or protein may also lead to membrane targeting (18). Surprisingly, unlike the CPPs, the cellular uptake of myristoylated cargos has received very little study. In early investigations of peptide inhibitors of protein kinases, O'Brian and co-workers found that myristoylation of a peptide substrate of protein kinase C resulted in an effective noncompetitive inhibitor of the kinase in vitro (19). Serendipitously, they found that the acylated peptide was taken up by live cells where it served to inhibit the target kinase (20). Since that time, myristoylated peptides have been used in a number of biochemical studies, yet the time course and mechanism of their cellular uptake remain poorly defined (20-23). Given the exciting potential for therapeutic delivery using this carrier (24-26), a better understanding of cellular uptake mediated by myristoylation of peptides is required.
A number of physicochemical studies have been performed on the behavior of myristoylated peptide insertion in lipid bilayers (27-33), while little has been published on the cellular uptake of such peptides (18). The in vitro studies in lipid bilayers and reconstituted cellular membranes suggest that the conjugated peptide inserts into the membrane by virtue of the myristoyl tag (28-31). Acyl phospholipids have been shown to translate across the lipid bilayer where the hydrophobic head group becomes exposed on the bilayer's opposite face (27, 34). A palmitoylated peptide has also been shown to flip-flop from the inner to the outer face of synthetic palmitoyl oleoyl phosphatidylcholine lipid vesicles (35). These findings suggest that peptide translation across the membrane may not require a carrier protein, since this phenomenon occurs in a purified lipid bilayer. Nevertheless, the molecular mechanism underlying myristoyl-based transport of peptide cargos into living cells has yet to be elucidated. The solitary mechanistic study of cellular uptake was performed by Ensenat-Waser et al (18). These investigators employed confocal microscopy and a fluorescently tagged 13-mer peptide containing a myristoylated N-terminus. After intact chromaffin cells were incubated for 12 hours in the presence of the myristoylated peptide, the fluorescence was distributed heterogeneously in the cytoplasmic compartment, but was excluded from the nucleus. Unfortunately, this single time point study did not provide data as to the kinetics of cellular uptake, temperature dependence of the uptake process, or role of the peptide cargo in the uptake process, parameters that would be of value in understanding cellular delivery of myristoylated cargos.
In the current work, the use of a myristate group has been evaluated as a means for cellular delivery of peptide cargos. This method is contrasted to the uptake of similar cargos conjugated to the TAT CPP. Furthermore, this comparison is carried out primarily in two cell types, a cervical carcinoma cell line which efficiently loads using TAT conjugates, and a B lymphocyte line which displays inefficient uptake of cargos conjugated to TAT. The kinetics of membrane association and cellular uptake of myristoylated peptide cargo are shown to be rapid. The delivery and intracellular release of a peptide cargo conjugated via a disulfide bond to the myristoyl domain is also studied as a means of delivering cargo that is spontaneously released in the cytoplasm upon reductive cleavage. The effects of temperature and the electrostatic charge of peptide cargos are also investigated, as is the impact of myristoylated peptide on cell viability. The current evaluation of myristoyl-based transport of peptides provides new data for an enhanced understanding of intracellular delivery of these increasingly important bioactive compounds.
Experimental Procedures
Materials
Trypsin and all tissue culture media and reagents were purchased from Invitrogen (Carlsbad, CA). Tris(2-carboxyethyl)-phosphate hydrochloride (TCEP) was purchased from Pierce (Rockford, IL), Oregon Green diacetate from Invitrogen, trypan blue, polylysine, pyrenebutyrate, and buffer reagents from Sigma. Purified ABL kinase was obtained Upstate (Temecula, CA). LavaCell was purchased from Active Motif (Carlsbad, CA).
Peptides
Peptides (Table I) were synthesized and purified by Anaspec (San Jose, CA). Peptides were fluorescently labeled with the 5′ isomer of fluorescein (5-FAM) on the N-terminus and the C-terminus was amidated unless otherwise noted. Myristoylated peptides were produced by incorporating a myristoylated lysine during solid phase synthesis. Peptides were dissolved in deionized water or DMSO (Sigma-Aldrich, St. Louis, MO), aliquoted, and stored at -70°C. The concentrations of the fluorescein-labeled peptides were determined by HPLC analysis at the Molecular Structure Facility at the University of California, Davis by adding a standard of known concentration to the acid hydrolyzed sample. Peptide charge was estimated at a pH of 7.4 using JaMBW 1.1 (http://www.bioinformatics.org/JaMBW/) taking into account the fluorescein charge.
Table I. Peptides Used in Current Studies.
| ABL | 5-FAM-Glu-Ala-Ile-Tyr-Ala-Ala-Pro-Phe-Ala-Lys-Lys-Lys-NH2 |
| ABL-ss-TAT |
|
| ABL-ss-Myr |
|
| ABL-Myr | 5-FAM-Glu-Ala-Ile-Tyr-Ala-Ala-Pro-Phe-Ala-Lys(Myr)-NH2 |
| Glu-Myr | 5-FAM-Glu-Glu-Glu-Glu-Glu-Glu-Glu-Lys(Myr)-OH |
| Arg-Myr | 5-FAM-Arg-Arg-Arg-Arg-Arg-Arg-Arg-Lys(Myr)-OH |
Cell Culture
HeLa, a human cervical carcinoma cell line, and a rat basophilic leukemia (RBL) cell line were grown in Dulbecco's modified eagle media (DMEM) supplemented with 10% fetal bovine serum (FBS), 4 mM L-glutamine, penicillin (100 units/mL), and streptomycin (100 μg/mL) at 37°C in 5% CO2. BA/F3, a mouse B-cell lymphoma cell line kindly provided by Junia Melo, and Jurkat cells, a human T-lymphocyte cell line, were cultured in RPMI supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL) at 37°C in 5% CO2. Disposable cell chambers were made by using Sylgard 184 (Dow Corning, Midland, MI) to attach a silicon “O” ring (15/16″ outer diameter) to a 25-mm, round, #1 glass coverslip. The cell chambers were pre-coated with polylysine to encourage cell adhesion to the glass surface and cells were plated and allowed to settle just prior to imaging.
Cell Loading
The lymphocytic cell line BA/F3 was loaded with fluorescent compounds as follows. Cells (5 × 105) were transferred to 1.7 mL centrifuge tubes and centrifuged for 2 minutes at 3000 × g. The supernatant was removed from the cell pellet, and the cells were resuspended and washed with an extracellular buffer (ECB; 135 mM NaCl, 5 mM KCl, 10 mM HEPES (pH 7.4), 1 mM MgCl2, and 1 mM CaCl2). The stock peptide solution in water or DMSO (1 – 10 mM) was diluted in ECB to achieve the desired final concentration of 20 μM (unless otherwise specified), and 20 μL of this solution was added to pelleted cells. The cells were incubated in the presence of the peptide for 30 minutes at 37°C, unless otherwise specified. The cells were then washed with ECB × 3. Cells were treated with trypsin for 1 minute. Trypsinization was halted by the addition of RPMI containing 10% FBS. Cells incubated with a peptide containing a disulfide linkage were treated additionally with the reducing agent TCEP (1 mM in ECB) to remove adsorbed extracellular fluorescent peptide (see Results). Cells were then resuspended in ECB, and plated in cell chambers. The cells were allowed to settle for 10 minutes, and then imaged (see below). For co-localization experiments using confocal imaging, BA/F3 cell membranes were stained with the vital membrane dye LavaCell (2 μM) by incubation at 37°C for 30 minutes per the manufacturer's protocol. This step was followed by loading of the myristoylated peptide as just described. In preliminary experiments, the cell lines RBL and HeLa were loaded while remaining adherent to the cell chambers in which they had been cultured overnight. Excessive adsorption of TAT-conjugated peptide to the glass substrate made quantitative fluorescence analysis of the cells difficult. For this reason, these adherent cell types were first washed with PBS, and then incubated with PBS containing 10 mM ethylenediamine tetraacetic acid (EDTA) until cells were released from the flask surface. This cell suspension was then treated using the protocol for the BA/F3 cells described above.
Cell Viability Assays
Cell viability was determined by measuring the retention of the viability dye Oregon green or exclusion of trypan blue. Cells were centrifuged and washed with ECB. Pelleted cells were resuspended in 25 μL of 10 μM Oregon Green diacetate and incubated at room temperature for 30 minutes. Cells were centrifuged and washed ×3 with ECB, treated with trypsin for 1 minute, and then plated in cell chambers and imaged. In experiments incorporating trypan blue exclusion, 20 μg/mL trypan blue was added to stain dead cells. Cells were imaged by transmitted light microscopy, and the ratio of dead-to-live cells was determined.
Fluorescence Microscopy and Image Analysis
Transmitted light and epifluorescence imaging were performed using a 40× air objective on an inverted microscope (Nikon TE2000, Melville, NY) fitted with a 100 W Hg arc lamp, 0.3 ND filter and standard FITC filter set (488 nm exc/535 nm em). Transmitted light and fluorescence images were obtained with a cooled CCD camera (Photometrix, Phoenix, AZ) using Metafluor software (Molecular Devices, Sunnyvale, CA). For all quantitative comparisons among cells under differing experimental conditions, camera gain and other relevant settings were kept constant. The freeware image analysis program Image J (http://rsb.info.nih.gov/ij/) was used for quantifying cellular fluorescence. Regions of interest (i.e. cells) were defined by hand using Image J. All images were corrected for background by subtracting the average background fluorescence (areas within the field of view not containing cells) from the region of interest. Confocal imaging was performed in the Michael Hooker Microscopy Facility at the University of North Carolina using a inverted laser scanning microscope (Zeiss 510 Meta, Thornwood, NY). Imaging was performed using a 63X, 1.4 NA, Plan-Apochromat, oil immersion objective. The two channel fluorescence excitation/emission ratios were 488 nm/518 nm for fluorescein-labeled peptides and 543 nm/585 nm for the LavaCell membrane dye. Co-localization analysis, including calculation of the Pearson's coefficient and Manders' overlap coefficient, was performed using the Image J plugin JACoP (http://rsb.info.nih.gov/ij/plugins/track/jacop.html). In some images, the contrast and brightness levels for the green channel were manipulated in Adobe PhotoShop to aid in the visualization of the green fluorescence signal for publication; however all co-localization values were generated using the raw, unadjusted data.
Capillary Electrophoresis
For single-cell analysis by capillary electrophoresis (CE), a customized CE system was used, as previously described (36). Briefly, chemical separations employed a 30 μM capillary (30 um inner diameter; effective length = 40 cm, total length = 53 cm) pretreated by washing with NaOH (0.3 M) for 12 h, H2O for 1 h, and then HCl (0.3 M) for 4 h, followed again by H2O for 24 h. Cell chambers were placed on an inverted microscope with an oil immersion 100× objective. Cells were maintained at 37°C and constantly perfused with warmed ECB. A cell was selected for analysis, lysed using a Nd:YAG-laser, and loaded into the capillary by electrophoresis (30 kV, negative polarity) as previously described (37). At 10 seconds after cell sampling, electrophoresis was halted, the capillary was moved to the run buffer (100 mM Tris, 100 mM tricine, pH = 8.0), and electrophoresis was continued. Peaks of fluorescent cellular analytes were detected by laser-induced fluorescence (LIF) and recorded as electrophoretic traces. The electrophoretic traces of cells were compared to standards composed of a peptide sample of known concentration and volume injected by gravity loading into the capillary and electrophoresed. CE standards were generated by treatment of the ABL-ss-Myr peptide (20 μM) with TCEP (1 mM) and then incubation with purified ABL kinase (100 ng, in 10mM MOPS, 2 mM ATP, 5 mM MgCl2, pH = 7.4; incubated at 37°C for 20 minutes) to produce in vitro phosphorylation of the ABL peptide.
Results
TAT does not load peptide cargo efficiently into lymphocyte-derived cells
Lymphocytes are regarded as difficult to transfect or to load with exogenous molecules (38). Electroporation is often the method chosen for transferring molecules into lymphocytes, but loss of cell viability and inefficiency of loading are significant drawbacks (38). CPPs have been used to load lymphocytes with cargo, but literature reports have been inconsistent in demonstrating successful transduction. The early report by Dowdy's group of TAT conjugated to the β-Gal protein for in vivo delivery in mice demonstrated very little β-Gal activity in the white pulp of the spleen suggesting poor uptake by lymphocytes (11). Other reports have also demonstrated a resistance of lymphocytes to take up the HIV TAT protein or molecular cargoes conjugated to this CPP (3, 39, 40). However, other reports have suggested that TAT or similar CPPs can be used for this purpose (38, 41). Some of these discrepancies in early studies may be explained by experimental artifacts arising from cell fixation when studying TAT uptake by immunofluorescence (3). Since TAT and other CPPs adhere avidly to the surface of many cell types, the adsorbed CPP:cargo must be removed from the extracellular surface before quantitative analysis for CPP uptake of cells (3, 10). To investigate the use of TAT for loading of a peptide cargo in lymphocytes, experiments were undertaken with the B lymphocyte cell line (BA/F3) and the T lymphocyte line (Jurkat). BA/F3 cells were incubated with up to 50 μM of a fluorescent peptide (ABL-ss-TAT) composed of an inhibitory peptide for the tyrosine kinase ABL (Glu-Ala-Ile-Tyr-Ala-Ala-Pro-Phe-Ala) (42) conjugated via a disulfide bond to the 9-amino-acid protein transduction domain derived from the TAT protein (Arg-Lys-Lys-Arg-Arg-Gln-Arg-Arg-Arg) (6, 43). After treatment with trypsin, the cells failed to display fluorescence intensity above cellular autofluorescence (Fig. 1A, B, E -- N.B. in this and all subsequent data, “control” refers to cells that have not been exposed to a fluorescent reagent and whose average fluorescence represents that cell type's native fluorescence intensity). Similar results were obtained in the Jurkat cell line (data not shown). On the other hand, BA/F3 cells readily took up the fluorescent dye Oregon green demonstrating their viability and metabolic activity (Fig. 1C) (44). In contrast, the adherent cell lines HeLa (Fig. 1D, E) and RBL (data not shown) were found to be readily loaded with the TAT-conjugated peptide under similar conditions.
Figure 1.
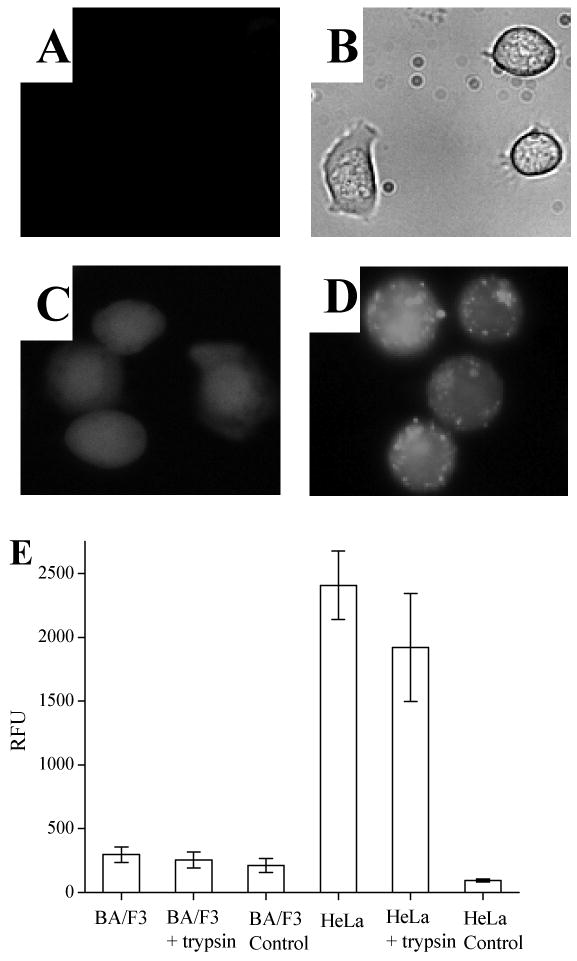
Cellular uptake of a fluorescent peptide conjugated to TAT in lymphocyte and non-lymphocyte cell lines. (A) Fluorescence micrograph of lymphocyte cells (BA/F3) loaded with ABL-ss-TAT followed by trypsinization to remove membrane-associated peptide. Minimal cellular fluorescence is present. (B) Transmitted-light image corresponding to “A”. (C) Fluorescence image of BA/F3 cells loaded with Oregon Green (10 μM). (D) Fluorescence image of nonlymphocyte cells (HeLa) loaded with ABL-ss-TAT followed by trypsinization shows that this cell type remains brightly fluorescent. (E) Histogram comparing the ABL-ss-TAT loading (relative fluorescence units [RFU]) between BA/F3 and HeLa cells with and without trypsin treatment. Histograms labeled “BA/F3 control” and “HeLa control” represent the cellular autofluorescence of the respective cell type.
A peptide cargo is efficiently loaded into lymphocyte-derived cells conjugated to the 14-carbon myristate group
To study the efficiency of loading lymphocyte cell lines using the myristate group, a myristoylated lysine was added to the C-terminus of the ABL substrate peptide (ABL-Myr). BA/F3 cells incubated with the ABL-Myr peptide followed by washing and trypsin exposure demonstrated a 12-fold greater fluorescence intensity (RFU 327 ± 88; n = 31) compared to cells not exposed to ABL-Myr (RFU 25 ± 3; n = 31) (Fig. 2A, C). Although cells loaded with the ABL-Myr peptide displayed fluorescence throughout the cell (see also confocal data, figure 6), in these experiments many cells displayed some evidence of cytoplasmic rimming of the fluorescence consistent with targeting of the peptide to the plasma membrane owing to the hydrophobic myristate. As a control, BA/F3 cells were also incubated in the presence of the ABL substrate domain lacking the myristate group. The fluorescence intensity of these cells was no different than the native BA/F3 autofluorescence (data not shown) demonstrating that cellular uptake was not driven by the peptide sequence used in these experiments. The myristoylated fluorescent peptide was also readily taken up by HeLa cells, where loaded cells showed a 5-fold increase (RFU 303 ± 81, n = 31) in fluorescence intensity compared to unloaded cells (RFU 58 ± 15, n = 31) (Fig. 2B, C).
Figure 2.
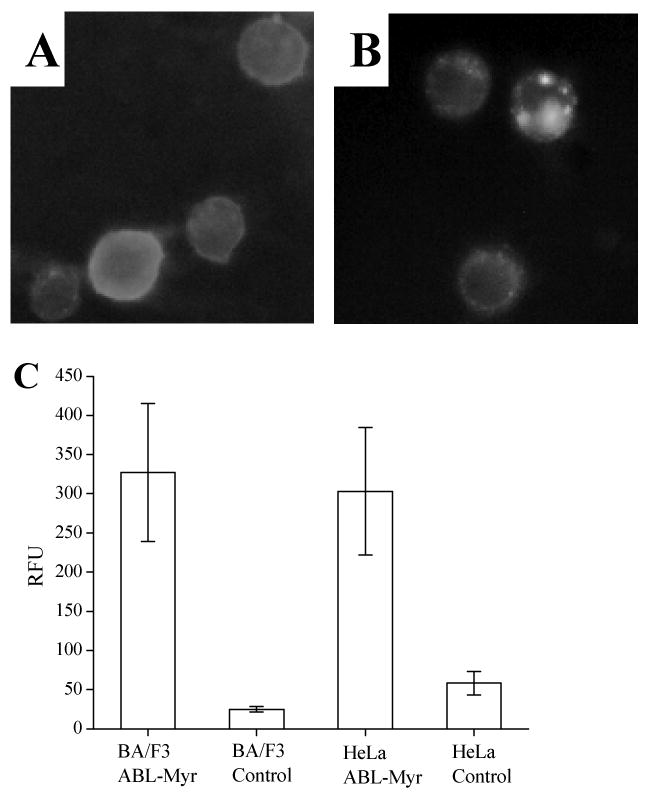
Comparison of peptide internalization mediated by myristate or TAT among lymphocyte and non-lymphocyte cell lines. (A) Fluorescence image of BA/F3 loaded with ABL-Myr followed by trypsinization. (B) Fluorescence micrograph of HeLa loaded with ABL-Myr followed by trypsin exposure. (C) Histogram comparing the cellular fluorescence of BA/F3 and HeLa cells loaded with the ABL-Myr conjugate, and the respective cell types without exposure to the fluorescent myristoylated peptide.
Figure 6.
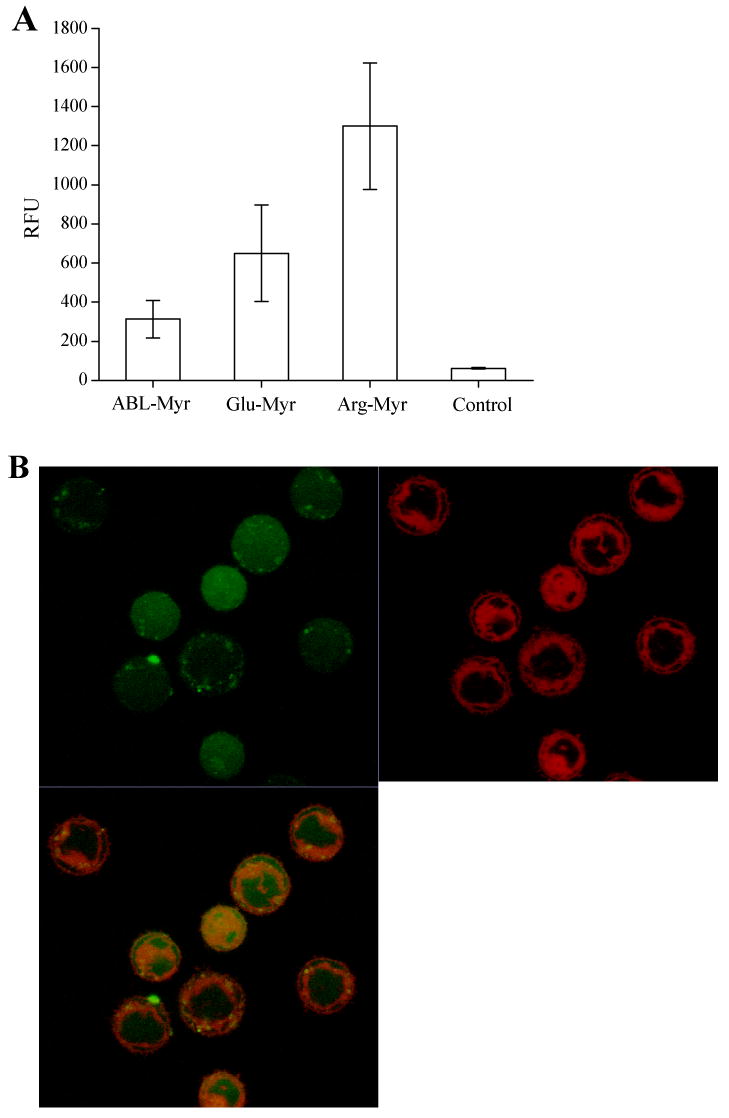
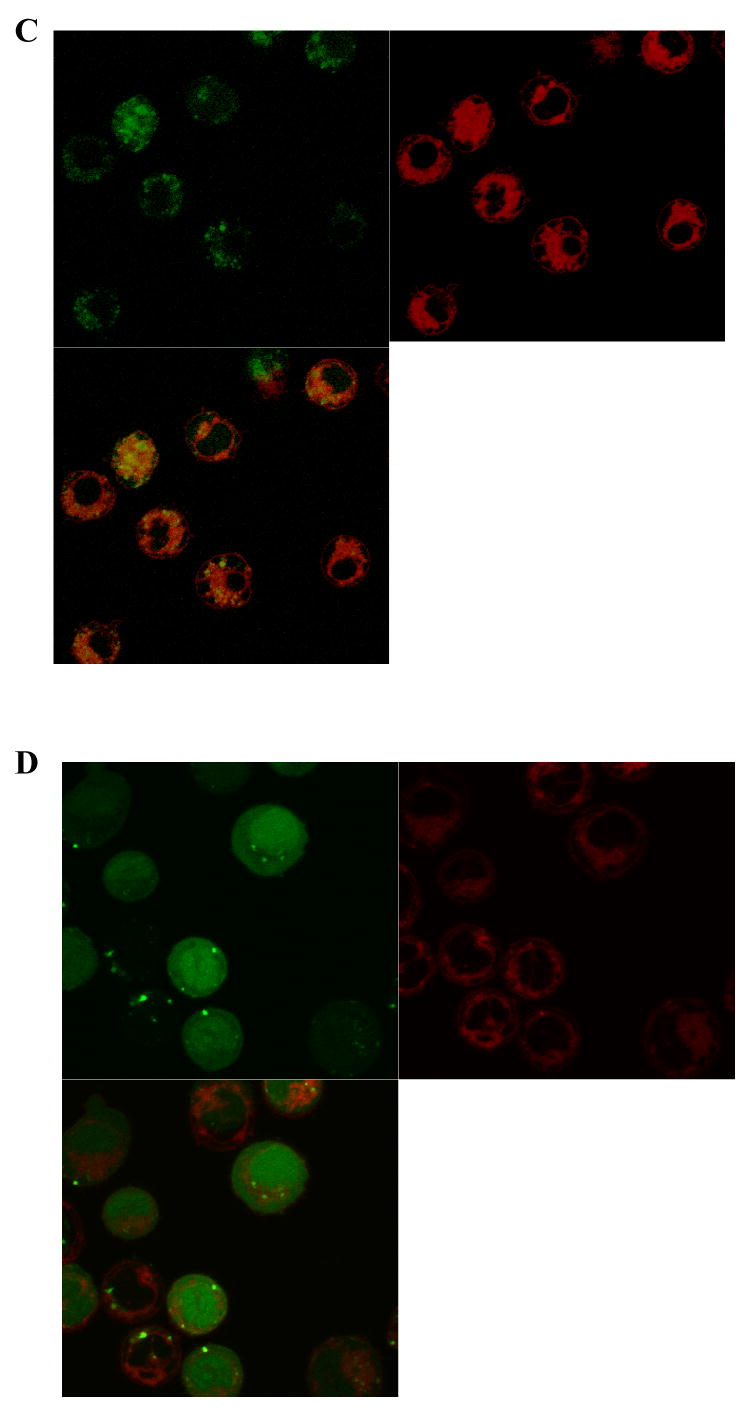
Effect of electrostatic charge on the intracellular delivery of a fluorescent peptide conjugated to the myristate group. A) Shown are histograms comparing the averages obtained from four independent experiments in which the cellular fluorescence of BA/F3 cells were determined after incubation with each of three myristoylated peptides of differing charges followed by trypsin treatment. B-D) Confocal two channel and overlay images of BA/F3 cells after staining with LavaCell (red fluorescence), and then loading with the myristoylated peptides as in “A”: (B) ABL-Myr, (C) Glu-Myr, or (D) Arg-Myr.
A peptide cargo conjugated to a myristoylated amino-acid sequence via a disulfide bond is readily loaded and released into lymphocyte-derived cells
As noted above, the retention of the myristoyl group on the peptide may enhance the localization of the peptide to cell membranes. In the event the target of the cargo is a cytosolic component, it may be desirable to release the cargo from the myristate, as separation of the cargo from the myristoyl tag will increase the cargo diffusion throughout the cytoplasm (43). In order to accomplish the intracellular release of the ABL peptide, a cysteine was added to the C-terminus of the ABL peptide. This cysteine-containing peptide was then conjugated to a myristoylated 5-amino-acid sequence (Cys-Lys(Myr)-Lys-Lys-Lys) via a disulfide bond formed between the cysteines of the two peptide domains. This multiple lysine sequence was used to generate a small myristoylated peptide with the necessary solubility characteristics for the disulfide conjugation. The resulting peptide's (ABL-ss-Myr) disulfide bond was reducible upon entering the cell, thus freeing the cargo from its membrane tether (43). BA/F3 cells exposed to the ABL-ss-Myr peptide displayed a 22-fold (RFU 1034 ± 204) increase in fluorescence intensity compared with unloaded cells (RFU 46 ± 8, n = 25) (Fig. 3A, C). BA/F3 cells were loaded using ABL-ss-Myr by the same protocol, but instead of the standard trypsin treatment, the cells were incubated with the reducing agent TCEP in order to cleave the disulfide bonds of any external fluorescent peptide to enhance its removal. After washing, these cells showed a similar fluorescence intensity to that of the trypsinized cells (RFU 1195 ± 206, n = 25) (Fig. 3B, C). BA/F3 cells treated with both trypsin and TCEP possessed a similar average fluorescence intensity to those treated with either agent alone (RFU 1147 ± 179, n = 25). Cytoplasmic rimming was not seen in cells loaded with the disulfide conjugate suggesting that partitioning of the cargo to the plasma membrane had been reduced.
Figure 3.
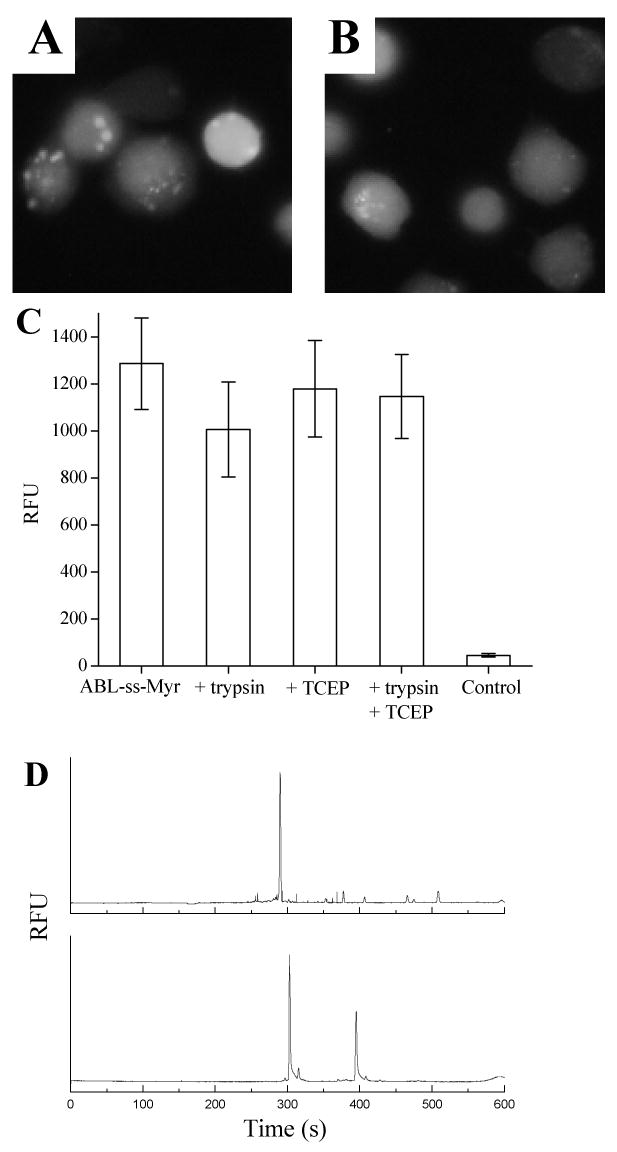
A fluorescent peptide conjugated to a myristoylated domain via a disulfide linkage is taken up by lymphocytes. (A) Fluorescence image of BA/F3 cells loaded with ABL-ss-Myr followed by trypsinization shows cellular fluorescence without cytoplasmic rimming. (B) Fluorescence image of BA/F3 cells loaded with the ABL-ss-Myr peptide and then washed with the reducing agent TCEP again shows homogenous cellular fluorescence. (C) Histograms comparing the cellular fluorescence of BA/F3 cells incubated with ABL-ss-Myr without any subsequent treatment with that of cells treated with either trypsin, TCEP, or both. (D) Electrophoretic trace showing the CE analysis of a single BA/F3 cell. BA/F3 cells were incubated with the ABL-ss-Myr peptide followed by combined trypsin and TCEP exposure. A single cell was then analyzed by CE with LIF detection of cellular contents (upper trace). The lower trace is of ABL-ss-Myr standards composed of the phosphorylated and non-phosphorylated forms of the ABL peptide.
Analysis of the fluorescent contents of individual BA/F3 cells loaded with the myristoylated, disulfide-conjugated peptide was undertaken using chemical cytometry. This CE-based method provides a quantitative measure of the fluorescent species obtained from single cells as previously described (37, 43, 45, 46). After trypsin and TCEP exposure, the electrophoretic traces obtained from these cells (n = 12) demonstrated predominately a single peak that co-migrated with the nonphosphorylated standard for the ABL peptide domain used in these experiments (Fig. 3D). These data indicate the intracellular presence of free peptide cargo since myristoylated peptide was not detected under the electrophoretic conditions employed (data not shown).
Kinetics of cellular uptake of the myristoyl-conjugated cargo
The rate at which the peptide cargo was loaded into the cell was determined in BA/F3 cells. The aim of the initial set of experiments was to measure the myristoyl-conjugated peptide's on-rate to the cell membrane combined with its cellular uptake. BA/F3 cells were incubated with ABL-ss-Myr for varying times, washed without trypsin or TCEP exposure, and fluorescently imaged. The average fluorescence intensity of the cells increased rapidly with a 27-fold increase (RFU 330 ± 58, n = 27) by 10 minutes of incubation, and a maximal intensity reached by 30 minutes of 43-fold compared to unloaded cells (RFU 515 ± 87, n = 27 vs. RFU 12 ± 2; n = 27) (Fig. 4A).
Figure 4.
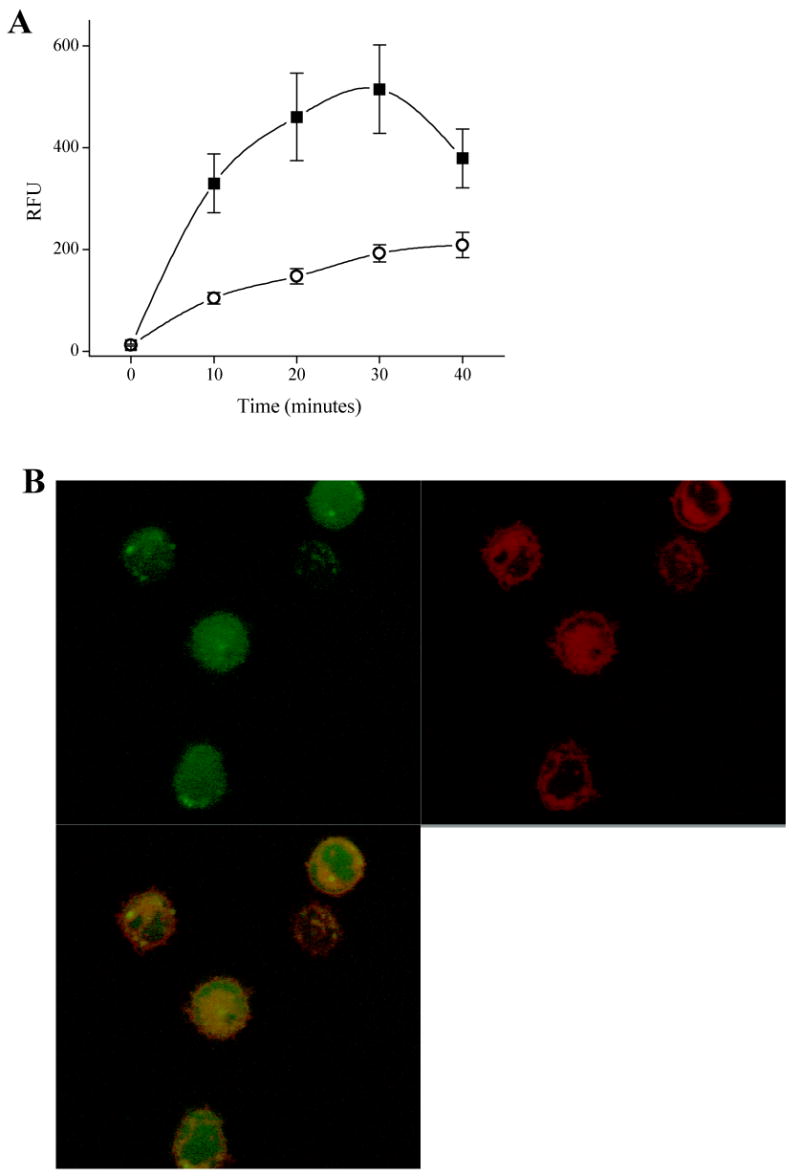
Time-courses of ABL-ss-Myr cell membrane on-rate and internalization in BA/F3 cells. A) BA/F3 cells were incubated for varying times with ABL-ss-Myr, washed in ECB and then imaged (“■”). In a second experiment, BA/F3 cells were incubated for varying times with ABL-ss-Myr in the same manner after which the cells were immediately washed with ECB containing the reducing agent TCEP (1 mM) (“○”) to remove extracellular fluorescent peptide (see text). B) Confocal two channel and overlay images of BA/F3 cells stained with LavaCell (red fluorescence), then loaded for 10 minutes with ABL-ss-Myr (green fluorescence) and washed with TCEP as above.
A second set of experiments was conducted to determine the rate at which the peptide was internalized. BA/F3 cells were loaded in an identical manner to those just described, but underwent treatment with TCEP immediately after incubation with ABL-ss-Myr. The addition of TCEP reduced the disulfide bond of any extracellular ABL-ss-Myr releasing its fluorescein-labeled ABL-substrate domain. This step effectively stopped further uptake of the fluorescent cargo by removal of any peptide bound to the external plasma membrane. After exposure to trypsin and TCEP, the cells were washed and imaged. The fluorescence intensity of the cells was 8-fold (RFU 104 ± 23, n = 23) that of unloaded control cells (RFU 12 ± 2, n = 23) after 10 minute incubation with the myristoylated disulfide conjugate and reached 16-fold (RFU 192 ± 34, n = 23) after a 30 minute incubation (Fig. 4A). These data are consistent with a rapid on-rate to the extracellular side of the cell membrane, and a more prolonged time course for intracellular uptake of the myristoylated peptide. To better assess localization of the internalized peptide, confocal imaging was undertaken in BA/F3 cells loaded and washed identically to those above, except for that the cells were pre-stained with the vital membrane dye LavaCell (54). As seen in Figure 4B, the green channel fluorescence representing the peptide was for the most part homogeneously distributed throughout the cell by 10 minutes. Intensity correlation coefficient-based statistical analysis of co-localization between the peptide and the LavaCell in five independent experiments provided a Pearson's coefficient of 0.69 ± 0.03 (mean ± std. dev.) and a Manders' overlap coefficient of 0.41 ± 0.07. These data were similar at 30 minutes where the Pearson's coefficient was 0.62 ± 0.13 and the Manders' coefficient was 0.28 ± 0.18. These findings are consistent with distribution of the peptide in the cytosol and nucleus as well as in cell membranes (55). Whether the partial overlap between peptide and membrane signals was due to persistence of the myristoyl group targeting a portion of the peptide to the membrane or was due to some degree of endosomal uptake cannot be determined from the current data. The similarity in the correlation coefficients at the ten and thirty minute timepoints suggests that the peptide distribution throughout the cell occurs in less than 10 minutes in support of a non-endosomal process.
Temperature dependence of myristoylated cargo entry into the cell
To assess the effect of temperature on the transport of peptide cargo with the myristate group, cell loading experiments were conducted with BA/F3 cells at 4°C and 37°C. BA/F3 cells were incubated with ABL-ss-Myr (20 μM) for 30 minutes. For experiments conducted at 4°C, incubation and treatment with trypsin and TCEP was performed on ice with all reagents and buffers pre-chilled to 4°C. At the end of the incubation period, the cells were immediately placed on the microscope and imaged at room temperature. Similarly, for experiments conducted at 37°C, cells and buffers were maintained at 37°C at all times up to the moment of imaging. Under these conditions, cells loaded at 4°C demonstrated reduced uptake of the myristoylated peptide with fluorescence intensity 2.5-fold (RFU 121 ± 19, n = 23) over that of unloaded controls (RFU 46 ± 8, n = 23) (Fig. 5). In contrast, BA/F3 cells loaded at 37°C had a significantly increased uptake with a 25-fold increase in fluorescence intensity (RFU 1138 ± 186, n = 23). Analogous experiments were performed in HeLa cells with both ABL-ss-Myr and ABL-ss-TAT, and a similar result was observed (Fig. 5B). HeLa cells loaded with ABL-ss-Myr at 4°C (RFU 859 ± 244, n = 31) demonstrated a 3-fold increase over unloaded cells (RFU 248 ± 51, n = 31), while HeLa cells loaded with ABL-ss-Myr at 37°C (RFU 2726 ± 47, n = 31) showed an 11-fold increase. Consistent with recent reports in the literature regarding temperature dependence of TAT-mediated cell uptake, the HeLa cells loaded with ABL-ss-TAT at 4°C showed minimal loading (RFU 545 ± 195, n = 31 vs. RFU 248 ± 51, n = 31 for control cells). HeLa cells incubated with the ABL-ss-TAT at 37°C showed an 8-fold increase in fluorescence intensity (RFU 1923 ± 297, n = 31) compared with controls. These data indicate that the cellular uptake of the myristoyl conjugate was a temperature dependent process.
Figure 5.

The intracellular delivery of a fluorescent peptide mediated by myristoylation is temperature dependent. (A) Histogram of the cellular fluorescence of BA/F3 cells exposed to the ABL-ss-Myr peptide at 4°C and 37°C. (B) Histogram of the cellular fluorescence of HeLa cells incubated with the ABL-ss-Myr peptide or the ABL-ss-TAT peptide at 4°C and 37°C.
Effect of peptide charge on cell loading
To study the impact of the peptide cargo on cell uptake employing the myristoylated domain, uptake of three peptides differing in overall charge at pH 7 (ABL-Myr [-2], Glu-Myr [-8], Arg-Myr [+4], see Table I) were studied. BA/F3 cells were incubated with 20 μM of peptide for 30 minutes at 37°C, and then treated with trypsin and washed. While all peptides loaded into the cells, the more hydrophilic peptides were taken up to a greater degree (Fig. 6A). Cells exposed to the ABL-Myr peptide displayed a fluorescence intensity on average 5-fold over that of controls (RFU 313 ± 96 vs. RFU 61 ± 4, n = averaged RFUs from 4 independent experiments). Cells incubated with the negatively charged myristoylated polyglutamate peptide possessed a fluorescence intensity 10-fold over background (RFU 650 ± 246). It should be noted that in some of the individual experiments (n ≥ 25 cells analyzed for each peptide), the polyglutamate peptide loaded as well or better than the polyarginine peptide; nevertheless, overall the myristoylated polyarginine peptide displayed the highest fluorescence intensity of the three peptides tested (RFU 1300 ± 320). To analyze co-localization of these peptides with intracellular membranes, confocal imaging was again performed. BA/F3 cells pre-stained with the vital membrane dye LavaCell were loaded and trypsin treated as above (Fig. 6B-D). Similar to the earlier experiment using the disulfide-linked ABL-ss-Myr, the green channel fluorescence was for the most part homogeneously distributed throughout the cell, although cells loaded with the Glu-Myr peptide appeared more punctate (Fig. 6D) and Arg-Myr peptide showed some nuclear enhancement (Fig. 6D). In three independent experiments, the ABL-Myr peptide had a Pearson's coefficient of 0.57 ± 0.06 (mean ± std. dev.) and a Manders' coefficient of 0.37 ± 0.02. For the Myr-Glu peptide, the Pearson's coefficient was 0.49 ± 0.07 and the Manders' coefficient was 0.09 ± 0.04. The Myr-Arg peptide had a Pearson's coefficient of 0.35 ± 0.03 and the Manders' coefficient of 0.49 ± 0.09. As noted, the confocal images of the Myr-Arg peptide demonstrated greater nuclear enhancement than was present with the other peptides. This finding is likely due to nuclear targeting of peptides containing polyarginine domains (3). As was seen above, the confocal data are consistent with only partial co-localization of the myristoylated peptides with intracellular membranes.
Influence of pyrenebutyrate on peptide loading
Large multi-ring molecules such as pyrenebutyrate (PB) aid in TAT-mediated internalization (47). Therefore, experiments were performed to compare the use of PB treatment in myristoyl-mediated and TAT-mediated internalization. PB treatment followed the protocol previously described by Takeuchi et al (47). BA/F3 cells were pre-incubated with PB (50 μM) in ECB with glucose for two minutes at 37°C. Cells were then incubated in the presence of either the ABL-ss-TAT or ABL-ss-Myr peptide with 25 μM PB for 30 minutes at 37°C. Control cells were treated in the same manner, but in the absence of PB. As seen in Figure 7A, BA/F3 cells exposed to the myristoylated peptide ABL-ss-Myr loaded equally well in the presence (RFU = 1727 ± 253, n = 37) or absence (RFU = 1464 ± 197, n = 37) of PB. BA/F3 cells exposed to the TAT-conjugate ABL-ss-TAT failed to show cellular uptake of the peptide even after treatment with PB. In these experiments, the fluorescence intensity of BA/F3 cells treated with PB (RFU = 61 ± 15, n = 37) or untreated (RFU 66 ± 12, n = 37) was identical to control cells that were not exposed to the fluorescent peptide conjugate (RFU 60 ± 11, n = 37). In contrast when these same experiments were undertaken with HeLa cells, cells treated with PB and incubated in the presence of ABL-ss-TAT displayed a 9-fold increase in the uptake of the peptide (RFU 2,523 ± 412, n = 33) over cells loaded in the same manner, but in the absence of PB (RFU 278 ± 34, n = 33) (Fig. 7B). As with the BA/F3 experiments, treating the cells with PB did not appear to influence the cellular uptake of the ABL-ss-Myr peptide. Indeed, HeLa cells treated with PB and exposed to the myristoylated peptide displayed an average fluorescence intensity (RFU 713 ± 95, n = 33) which was slightly less than PB untreated cells (RFU 1033 ± 270, n = 33).
Figure 7.

The effect of pyrenebutyrate (PB) on the intracellular delivery of a fluorescent peptide conjugated to either the myristate group or to TAT. (A) Histograms of the average cellular fluorescence of BA/F3 cells after incubation with either ABL-ss-Myr (“Myr”) or ABL-ss-TAT (“TAT”) with and without pyrenebutyrate. (B) Histograms of the average cellular fluorescence of HeLa cells incubated under the same conditions.
Evaluation of cell viability after loading
BA/F3 cells were incubated with ABL-ss-Myr to determine the viability of cells after loading at varying concentrations (0 – 100 μM) of the myristoyl-conjugated peptide domain. After 30 minute incubation at 37°C and washing, the cells were stained with trypan blue and evaluated for exclusion of the dye as a measure of cell viability. In the presence of the highest concentrations of the ABL-ss-Myr, BA/F3 cells showed no evidence of cell membrane permeability to trypan blue (99 % viability, n = 200) compared with a viability rate in controls of 99.5 % (n = 200).
Discussion
Cell penetrating peptides are extremely valuable tools for conveying molecular cargos to the interior of cells. Nevertheless, no single CPP appears to be a ubiquitous carrier for all cell types, perhaps related to the presence or absence of specific cell surface molecules directing cell membrane association (3). On the other hand, the myristoyl group is known to insert into purified lipid bilayers. Insertion occurs via direct hydrophobic interaction without the need of supplementary moieties for adherence to the cell surface (48). This property may allow a greater repertoire of cell types to be loaded using myristoylation as the transducing domain. In the present study cells exposed to the non-myristoylated peptide cargo alone did not possess fluorescence intensity greater than controls. Membrane association and translocation were primarily due to the myristate group rather than to the peptide cargo. Furthermore, uptake of myristoylated peptide in both the HeLa and the TAT-resistant BA/F3 cells supports the potential of myristoyl-based loading as a ubiquitous means to confer intracellular uptake of membrane impermeant peptides.
The punctate intracellular fluorescence noted in HeLa cells after exposure to the TAT-conjugated peptide (Fig. 1) is consistent with uptake by an endocytic process (49). While some punctate fluorescence was seen in confocal and epifluorescence imaging of HeLa and BA/F3 cells exposed to the myristoyl-conjugates, in general intracellular fluorescence was more homogeneous than that seen with the TAT-linked cargos. Although the current studies do not rule out endocytosis as one means of entry, the rapid establishment of diffuse cellular fluorescence by confocal microscopy suggests that alternative pathways for peptide translocation may be present. Flip-flop diffusion of the myristoyl-conjugated peptide is one potential mechanism for this translocation to the interior of the cell (35). The experiments in HeLa cells show that the uptake process is temperature dependent for both myristoyl- and TAT-conjugated peptides. While this result could suggest the presence of an active process such as endocytosis, alternative explanations are possible. Translocation of the cargo through the lipid bilayer by the process of flip-flop diffusion would face an energy barrier, and thus could be expected to be slowed by lowering the temperature. Additionally, peptide flip-flop may also be drastically reduced due to the fluid-gel transition of the mammalian plasma membrane at temperatures below 20°C (30, 50). Future studies to clarify the molecular mechanism of cargo translocation across the plasma membrane will help elucidate this issue.
The study evaluating the effect of the peptide cargo's electrostatic charge yielded a clear ranking in the order of efficiency of loading the three peptides. Although ABL, the most hydrophobic cargo tested, was efficiently loaded, charged cargos loaded to a greater degree. Overall, cells exposed to the myristoylated polyarginine peptide displayed the highest fluorescence intensity. This result is consistent with experimental evidence that multiple arginine residues alone can bring about cellular uptake (3, 10, 51). The increased loading of the polyarginine conjugate over the other peptide cargos tested suggests that its uptake may be driven by both the myristate tag and the arginine-rich domain. In contradistinction to studies using TAT and other arginine-rich peptides, the myristoyl-conjugated polyglutamate cargo was also efficiently loaded into cells. Thus, the C14 domain can mediate cellular uptake in the presence of a highly negatively charged cargo without the requirement for positive charge. Since the hydrophobic character of the C14 tag was the major characteristic driving uptake, the myristoylated ABL cargo should have been most efficiently loaded; however, this was not the case. A likely explanation for this finding was that the ABL-Myr peptide was less soluble in physiologic buffers than the hydrophilic peptides. Although every effort was made to maintain consistent concentrations among the comparative experiments, the lower solubility of the ABL peptide conjugate may underlie its apparent reduced uptake.
PB is a negatively-charged counterion with high hydrophobicity. It is believed to increase the intracellular uptake of arginine-rich CPPs by ion pairing with the guanidinium cations of arginine residues, thus increasing the CPP's hydophobicity and facilitating the direct translocation across the lipid bilayer (47). While PB significantly enhanced loading of HeLa cells exposed to the ABL-TAT similar to previously reported data (47), it did not have a salutary effect on uptake of this same TAT-conjugated peptide in the BA/F3 cell line. PB also failed to enhance myristate-mediated cellular internalization of the ABL peptide in either HeLa or BA/F3 cells. These data support that uptake of myristoylated peptides occurs through a process unlike TAT-mediated uptake.
The myristate group may provide advantages to CPPs, particularly TAT, when the desire is to deliver an inhibitory-peptide or an enzyme-substrate cargo to a membrane-bound protein. Anchoring of the cargo to the membrane through the myristoyl tag can be expected to enhance the local concentration of the cargo in the vicinity of the cellular target which should augment the cargo's biological effect (48). Reduced compartmentalization of the cargo compared with TAT, as was seen in the cell types studied here, enables more effective distribution of the cargo within the cell. Furthermore, unlike some CPPs which possess a nuclear localization sequence (52), no propensity was found for the myristate group to convey the cargo to the nuclear compartment, except in the case of conjugation to a cargo with nuclear tropism. The incorporation of a cleavable bond between the myristoyl domain and the cargo provides a further advantage by enabling release of the cargo to enhance partitioning to the cytoplasm. In addition to the disulfide linker described here, a photo-cleavable linker can be employed if temporal control of cargo release is desired (43). Although fatty acids such as myristate have been reported to be toxic under conditions of continual exposure (53), no evidence of reduced viability was seen under the conditions required for cell loading in these studies.
Myristoylation is a useful tool for the biologist seeking to transport molecular cargos to the interior of living cells. To properly utilize this method for cell-based studies, knowledge of the mechanics of cell uptake, the kinetics of cellular internalization, and the subcellular localization of myristoyl-conjugated molecules is important. Despite more than a decade of use, the understanding of these properties for myristate-mediated cellular import has been limited. The current studies have begun to elucidate the characteristics of this transport for peptide cargos; however, the precise manner as to how membrane binding and cellular uptake are influenced by the myristoyl domain in combination with the hydrophobicity and electrostatic charge of the peptide cargo merits further biophysical studies. In addition, further study in various cell types and with non-peptide cargoes will be needed to fully clarify the range of cell types and cargoes for which this method is optimally suited. These additional studies are called for as myristoylation conveys a number of advantages including the potential for broad application in cell types not amenable to TAT-based translocation.
Abbreviations
- ABL
a 9 amino acid inhibitory peptide of the tyrosine kinase ABL
- CE
capillary electrophoresis
- CPP
cell penetrating peptide
- ECB
extracellular buffer
- Myr
the 14-carbon fatty acid myristate
- PB
pyrenebutyrate
- PTD
protein transduction domain
- -ss-
a disulfide bond
- TAT
9 amino acid CPP derived from the HIV TAT protein
- TCEP
tris(2-carboxyethyl)-phosphate hydrochloride
Footnotes
This work was supported by grants from the National Institutes of Health to C.E.S. and N.L.A.
References
- 1.Stephens DJ, Pepperkok R. The many ways to cross the plasma membrane. PNAS. 2001;98:4295–4298. doi: 10.1073/pnas.081065198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murriel CL, Dowdy SF. Influence of protein transduction domains on intracellular delivery of macromolecules. Expert Opinion in Drug Delivery. 2006;3:739–746. doi: 10.1517/17425247.3.6.739. [DOI] [PubMed] [Google Scholar]
- 3.Chauhan A, Tikoo A, Kapur AK, Singh M. The taming of the cell penetrating domain of the HIV Tat: Myths and realities. Journal of Controlled Release. 2007;117:148–162. doi: 10.1016/j.jconrel.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 Tat requires cell surface heparan sulfate proteoglycans. Journal of Biological Chemistry. 2001;276:3254–3261. doi: 10.1074/jbc.M006701200. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. Journal of Biological Chemistry. 2002;277:2437–2443. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 6.Vogel BE, Lee SJ, Hildebrand A, Craig W, Pierschbacher MD, Wong-Staal F, Ruoslahti E. A novel integrin specificity exemplified by binding of the α vs β 5 integrin to the basic domain of the HIV TAT protein and vitronectin. Journal of Cell Biology. 1993;121:461–468. doi: 10.1083/jcb.121.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. PNAS. 2000;97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green I, Christison R, Voyce CJ, Bundell KR, Lindsay MA. Protein transduction domains: Are they delivering? Trends in Pharmacological Science. 2003;24:213–215. doi: 10.1016/S0165-6147(03)00076-2. [DOI] [PubMed] [Google Scholar]
- 9.Lewin M, Carlesso N, Tung CH, Tang XW, Cory D, Scadden DT, Weissleder R. TAT peptide derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nature Biotechnology. 2000;18:410–414. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 10.Richard JP, Melikov K, Vives E, Ramos C, Verbeure B, Gait MJ, Chernomordik LV, Lebleu B. Cell penetrating peptides. A re-evaluation of the mechanism of cellular uptake. Journal of Biological Chemistry. 2003;278:585–590. doi: 10.1074/jbc.M209548200. [DOI] [PubMed] [Google Scholar]
- 11.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 12.Okuyama M, Laman H, Kingsbury SR, Visintin C, Leo E, Eward KL, Stoeber K, Boshoff C, Williams GH, Selwood DL. Small-molecule mimics of an α-helix for efficient transport of proteins into cells. Nature Methods. 2007 doi: 10.1038/NMETH997. [DOI] [PubMed] [Google Scholar]
- 13.Reshetnyak YK, Andreev OA, Lehnert U, Engelman DM. Translocation of molecules into cells by pH-dependent insertion of a transmembrane helix. PNAS. 2006;103:6460–6465. doi: 10.1073/pnas.0601463103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhee M, Davis P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. Journal of Biological Chemistry. 2006;281:1233–1240. doi: 10.1074/jbc.M509813200. [DOI] [PubMed] [Google Scholar]
- 15.Yandek LE, Pokorny A, Floren A, Knoelke K, Langel U, Almeida PFF. Mechanism of the cell-penetrating peptide transportan 10 permeation of lipid bilayers. Biophysical Journal. 2007;92:2434–2444. doi: 10.1529/biophysj.106.100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schultz AM, Henderson LE, Oroszlan S. Fatty acylation of proteins. Annual Reviews of Cell Biology. 1988;4:611–647. doi: 10.1146/annurev.cb.04.110188.003143. [DOI] [PubMed] [Google Scholar]
- 17.Towler DA, Gordon JI, Adams SP, Glaser L. The biology and enzymology of eukaryotic protein acylation. Annual Reviews of Biochemistry. 1988;57:69–99. doi: 10.1146/annurev.bi.57.070188.000441. [DOI] [PubMed] [Google Scholar]
- 18.Ensenat-Waser R, Martin F, Barahona F, Vazquez J, Soria B, Reig JA. Direct visualization by confocal fluorescent microscopy of the permeation of myristoylated peptides through the cell membrane. IUBMB Life. 2002;54:33–36. doi: 10.1080/15216540213823. [DOI] [PubMed] [Google Scholar]
- 19.Ioannides CG, Freedman RS, Liskamp RM, Ward NE, O'Brian CA. Inhibition of IL-2 receptor induction and IL-2 production in the human leukemic cell line Jurkat by a novel peptide inhibitor of protein kinase C. Cell Immunology. 1990;131:242–252. doi: 10.1016/0008-8749(90)90250-u. [DOI] [PubMed] [Google Scholar]
- 20.O'Brian CA, Ward NE, Liskamp RM, de Bont DB, Earnest LE, van Boom JH, Fan D. A novel N-myristylated synthetic octapeptide inhibits protein kinase C activity and partially reverses murine fibrosarcoma cell resistance to Adriamycin. Investigational New Drugs. 1991;9:169–179. doi: 10.1007/BF00175084. [DOI] [PubMed] [Google Scholar]
- 21.Harris TE, Persaud SJ, Jones PM. Pseudosubstrate inhibition of cyclic AMP-dependent protein kinase in intact pancreatic islets: effects on cyclic AMP-dependent and glucose-dependent insulin secretion. Biochemical and Biophysical Research Communications. 1997;232:648–651. doi: 10.1006/bbrc.1997.6344. [DOI] [PubMed] [Google Scholar]
- 22.Ishida A, Fujisawa H. Stabilization of calmodulin-dependent protein kinase II through the autoinhibitory domain. Journal of Biological Chemistry. 1995;270:2163–2170. doi: 10.1074/jbc.270.5.2163. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto A, Kawamata T, Ninomiya T, Omote K, Namiki A. Endothelin-1 enhances capsaicin-evoked intracellular Ca2+ response via activation of endothelin A receptor in a protein kinase Cε-dependent manner in dorsal root ganglion neurons. Neuroscience. 2006;137:949–960. doi: 10.1016/j.neuroscience.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 24.Chuang VTG, Kragh-Hansen U, Otagiri M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharmaceutical Research. 2002;19:569–577. doi: 10.1023/a:1015396825274. [DOI] [PubMed] [Google Scholar]
- 25.Dasgupta P, Mukherjee R. Lipophilization of somatostatin analog RC-160 with long chain fatty acid improves its antiproliferative and antiangiogenic activity in vitro. British Journal of Pharmacology. 2000;129:101–109. doi: 10.1038/sj.bjp.0702990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veuillez F, Rieg FF, Guy RH, Deshusses J, Buri P. Permeation of a myristoylated dipeptide across the buccal mucosa: topological distribution and evaluation of tissue integrity. International Journal of Pharmaceutics. 2002;231:1–9. doi: 10.1016/s0378-5173(01)00850-x. [DOI] [PubMed] [Google Scholar]
- 27.Kubelt J, Menon AK, Muller P, Herrmann A. Transbilayer movement of fluorescent phospholipid analogues in the cytoplasmic membrane of Escherichia coli. Biochemistry. 2002;41:5605–5612. doi: 10.1021/bi0118714. [DOI] [PubMed] [Google Scholar]
- 28.Losonczi JA, Tian F, Prestegard JH. Nuclear magnetic resonance studies of the N-terminal fragment of adenosine diphosphate ribosylation factor 1 in micelles and bicelles: influence of N-myristoylation. Biochemistry. 2000;39:3804–3816. doi: 10.1021/bi9923050. [DOI] [PubMed] [Google Scholar]
- 29.Reig F, Haro I, Polo D, Egea MA, Alsina MA. Interfacial interactions of hydrophobic peptides with lipid bilayers. Journal of Colloid and Interface Science. 2002;246:60–69. doi: 10.1006/jcis.2001.8056. [DOI] [PubMed] [Google Scholar]
- 30.Sankaram MB. Membrane interaction of small N-myristoylated peptides: implications for membrane anchoring and protein-protein association. Biophysical Journal. 1994;67:105–112. doi: 10.1016/S0006-3495(94)80459-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Victor K, Cafiso DS. Structure and position of the N-terminal membrane-binding domain of pp60src at the membrane interface. Biochemistry. 1998;37:3402–3410. doi: 10.1021/bi9721501. [DOI] [PubMed] [Google Scholar]
- 32.Ben-Tal N, Honig B, Peitzsch RM, Denisov G, McLaughlin S. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophysical Journal. 1996;71:561–575. doi: 10.1016/S0006-3495(96)79280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peitzsch RM, McLaughlin S. Binding of acylated peptides and fatty acids to phospholipid vesicles: pertinence to myristoylated proteins. Biochemistry. 1993;32:10436–10443. doi: 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- 34.Siegmund A, Grant A, Angeletti C, Malone L, Nichols JW, Rudolph HK. Loss of Drs2p does not abolish transfer of fluorescence-labeled phospholipids across the plasma membrane of Saccharomyces cerevisiae. Journal of Biological Chemistry. 1998;273:34399–34405. doi: 10.1074/jbc.273.51.34399. [DOI] [PubMed] [Google Scholar]
- 35.Eisele F, Kuhlmann J, Waldmann H. Synthesis and membrane binding properties of a lipopeptide fragment from influenza virus A hemagglutinin. Chem Eur J. 2002;8:3362–3376. doi: 10.1002/1521-3765(20020802)8:15<3362::AID-CHEM3362>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 36.Nelson AR, Allbritton NL, Sims CE. Rapid sampling for single-cell analysis by capillary electrophoresis. Methods in Cell Biology. 2007;82:709–722. doi: 10.1016/S0091-679X(06)82026-1. [DOI] [PubMed] [Google Scholar]
- 37.Sims CE, Meredith GD, Krasieva TB, Berns MW, Tromberg BJ, Allbritton NL. Laser-micropipet combination for single-cell analysis. Analytical Chemistry. 1998;70:4570–4577. doi: 10.1021/ac9802269. [DOI] [PubMed] [Google Scholar]
- 38.Fenton M, Bone N, Sinclair AJ. The efficient and rapid import of a peptide into primary B and T lymphocytes and a lymphoblastoid cell line. Journal of Immunologic Methods. 1998;212:41–48. doi: 10.1016/s0022-1759(97)00208-1. [DOI] [PubMed] [Google Scholar]
- 39.Leifert JA, Harkins S, Whitton JL. Full-length proteins attached to the HIV tat protein transduction domain are neither transduced between cells, nor exhibit enhanced immunogenicity. Gene Therapy. 2002;9:1422–1428. doi: 10.1038/sj.gt.3301819. [DOI] [PubMed] [Google Scholar]
- 40.Leifert JA, Whitton JL. “Translocatory proteins” and “protein transduction domains”: A critical analysis of their biological effects and the underlying mechanisms. Molecular Therapy. 2003;8:13–20. doi: 10.1016/s1525-0016(03)00151-5. [DOI] [PubMed] [Google Scholar]
- 41.Ho A, Schwarze SR, Mermelstein SJ, Waksman G, Dowdy SF. Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo. Cancer Research. 2001;61:474–477. [PubMed] [Google Scholar]
- 42.Songyang Z, Carraway KL, Eck MJ, Harrison SC, Feldman RA, Mohammed M, Schlessinger J, Hubbard SR, Smith DP, Eng C, Lorenzo MJ, Ponder BAJ, Mayer BJ, Cantley LC. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 43.Soughayer JS, Wang Y, Li H, Cheung SH, Rossi FM, Stanbridge EJ, Sims CE, Allbritton NL. Characterization of TAT-mediated transport of detachable kinase substrates. Biochemistry. 2004;43:8528–8540. doi: 10.1021/bi036296d. [DOI] [PubMed] [Google Scholar]
- 44.Haugland R. Handbook of Fluorescent Probes and Research Chemicals. 6th. Molecular Probes, Inc; Eugene, OR: 1996. [Google Scholar]
- 45.Li H, Sims CE, Kaluzova M, Stanbridge EJ, Allbritton NL. A quantitative single-cell assay for protein kinase B reveals important insights into the biochemical behavior of an intracellular substrate peptide. Biochemistry. 2004;43:1599–1608. doi: 10.1021/bi035597k. [DOI] [PubMed] [Google Scholar]
- 46.Meredith GM, Sims CE, Soughayer JS, Allbritton NL. Measurement of kinase activation in single mammalian cells. Nature Biotechnology. 2000;18:309–312. doi: 10.1038/73760. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, Sakai N, Matile S, Futaki S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chemical Biology. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 48.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. TIBS. 1995;20:272–276. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 49.Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. Journal of Biological Chemistry. 2003;278:34141–34149. doi: 10.1074/jbc.M303045200. [DOI] [PubMed] [Google Scholar]
- 50.Moore AI, Squires EL, Graham JK. Adding cholesterol to the stallion sperm plasma membrane improves cryosurvival. Cryobiology. 2005;51:241–249. doi: 10.1016/j.cryobiol.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. Journal of Biological Chemistry. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 52.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. Journal of Biological Chemistry. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 53.Gutknecht J. Proton conductance caused by long-chain fatty acids in phospholipid bilayer membranes. Journal of Membrane Biology. 1988;106:83–93. doi: 10.1007/BF01871769. [DOI] [PubMed] [Google Scholar]
- 54.Choi HY, Veal DA, Karuso P. Epicocconone, a new cell-permeable long Stokes' shift fluorescent stain for live cell imaging and multiplexing. Journal of Fluorescence. 2006;16:475–482. doi: 10.1007/s10895-005-0010-7. [DOI] [PubMed] [Google Scholar]
- 55.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. Journal of Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]