

Abstract
Endothelin-1 (ET-1) disrupts insulin-regulated glucose transporter GLUT4 trafficking. Since the negative consequence of chronic ET-1 exposure appears to be independent of signal disturbance along the insulin receptor substrate-1/phosphatidylinositol (PI) 3-kinase (PI3K)/Akt-2 pathway of insulin action, we tested if ET-1 altered GLUT4 regulation engaged by osmotic shock, a PI3K-independent stimulus that mimics insulin action. Regulation of GLUT4 by hyperosmotic stress was impaired by ET-1. Because of the mutual disruption of both insulin- and hyperosmolarity-stimulated GLUT4 translocation, we tested whether shared signaling and/or key phosphatidylinositol 4,5-bisphosphate (PIP2)-regulated cytoskeletal events of GLUT4 trafficking were targets of ET-1. Both insulin and hyperosmotic stress signaling to Cbl were impaired by ET-1. Also, plasma membrane PIP2 and cortical actin levels were reduced in cells exposed to ET-1. Exogenous PIP2, but not PI 3,4,5-bisphosphate, restored actin structure, Cbl activation, and GLUT4 translocation. These data show that ET-1-induced PIP2/actin disruption impairs GLUT4 trafficking elicited by insulin and hyperosmolarity. In addition to showing for the first time the important role of PIP2-regulated cytoskeletal events in GLUT4 regulation by stimuli other than insulin, these studies reveal a novel function of PIP2/actin structure in signal transduction.
Keywords: actin; Cbl; insulin resistance; phosphatidylinositol 4,5-bisphosphate
Insulin binding to the insulin receptor (IR) causes tyrosine autophosphorylation of the IR-β subunit, increasing the intrinsic tyrosine kinase activity of the IR [White and Kahn, 1994]. Two key targets of the activated IR are insulin receptor substrate-1 (IRS-1) and Cbl [Ahmed et al., 2000; Baumann et al., 2000], both of which provide docking sites for signaling proteins functioning to stimulate the translocation of the insulin-responsive glucose transporter GLUT4 from an intracellular compartment to the plasma membrane. Although details of Cbl signaling to TC10 and potentially actin are still being tested and explored, it is well established that IRS-1 signaling entails the downstream activation of a phosphatidylinositol (PI) 3-kinase (PI3K)-dependent signaling cascade involving phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3)[Watson and Pessin, 2001] and activation of a kinase cascade involving PIP3-dependent kinases (PDK1/2), Akt-2, and the atypical protein kinase isoforms λ and ζ (PKC-λ/ζ) [Kohn et al., 1996; Bandyopadhyay et al., 1997]. In addition to the pertinent signal transduction role of PIP2, recent studies have shown that this lipid, independent of its signaling activities, regulates cortical filamentous actin (F-actin) polymerization [Takenawa et al., 1999; Tsakiridis et al., 1999; Kanzaki et al., 2002, 2004; Chen et al., 2004; Hilpela et al., 2004]. In vitro [Kanzaki and Pessin, 2001; Kanzaki et al., 2001, 2002; Chen et al., 2004] and in vivo [Brozinick et al., 2004] observations clearly implicate the importance of the actin cytoskeletal network in GLUT4 regulation by insulin.
Interestingly, it is now clear that GLUT4 translocation can be activated by a variety of agents by a mechanism not involving PI3K. For example, exercise/contraction, hyperosmolarity, guanosine 5′-[γ-thio] triphosphate (GTPγS; a nonhydrolyzable GTP analog), endothelin-1 (ET-1), expression of constitutively active Gq (Gq/Q209L), and sphingomyelinase all have been reported to stimulate GLUT4 translocation and glucose transport by a novel PI3K-independent mechanism [Elmendorf, 2002]. Even though these alternative signaling pathways leading to GLUT4 translocation apparently bypass initial insulin signal transduction steps, they are likely to induce GLUT4 translocation through activation of a common convergent signal transduction step downstream and/or in parallel to PI3K.
It is recognized that chronic exposure of cells to ET-1, a key vasoactive peptide, may fuel insulin-resistance and cardiovascular disease states. For example, an association between elevated circulating levels of ET-1 has been described in subjects with insulin resistance associated with polycystic ovarian syndrome [Diamanti-Kandarakis et al., 2001], hypertension [Seljeflot et al., 1998], obesity [Ferri et al., 1995, 1997; Piatti et al., 1999, 2000; Irving et al., 2001], impaired glucose tolerance [Andronico et al., 1997], and type2diabetes [Mangiafico etal., 1998;Caballero et al., 1999; Ak et al., 2001; Sanchez et al., 2001]. Even more striking is that relatives of diabetic individuals display increased levels of circulating ET-1 [Caballero et al., 1999]. Mechanistically, ET-1 signals via phospholipase Cβ (PLCβ)-mediated hydrolysis of PIP2 and ET-1-induced disturbances in PI3K signaling have been reported to account for impaired insulin sensitivity [Jiang et al., 1999; Ishibashi et al., 2001]. However, we recently found that ET-1-induced PI3K abnormalities do not fully explain the insulin-resistant state [Strawbridge and Elmendorf, 2005]. Given that PIP2-regulated cortical F-actin events may be fundamentally important in GLUT4 regulation by osmotic shock, as it is insulin, we asked whether the insulin-mimetic/PI3K-independent activity of hyperosmolarity was inhibited by ET-1. Studies examined PIP2-regulated cytoskeletal mechanics and the PI3K-independent Cbl signaling pathway reported to be utilized in the regulation of GLUT4 translocation by both insulin and osmotic shock. The subsequent report provides a detailed account of these studies.
MATERIALS AND METHODS
Materials
Murine 3T3-L1 preadipocytes were from American Type Culture Collection (Manassas, VA). Dulbecco’s modified Eagle’s medium (DMEM) was from Invitrogen (Grand Island, NY). Fetal bovine serum (FBS) and bovine calf serum (BCS) were from Hyclone Laboratories, Inc. (Logan, UT). Latrunculin B was purchased from Calbiochem (San Diego, CA). Phosphatidylinositides (PtsIns(4,5)P2 diC16, PtsIns(3,4,5)P3 diC16) and histone carrier were purchased from Echelon Biosciences (Salt Lake City, UT). The Akt Kinase Assay Kit was from Cell Signaling Technology (Beverly, MA). Unless otherwise indicated, all other chemicals were from Sigma (St. Louis, MO).
Cell Culture and Treatments
Preadipocytes were cultured and differentiated to adipocytes as previously described [Kralik et al., 2002]. Studies were performed on adipocytes between 8 and 12 days post-differentiation. ET-1-induction of insulin resistance was performed by treating the cells in 10 nM ET-1/DMEM for 24 h as previously described [Strawbridge and Elmendorf, 2005]. Cells were either untreated or treated for 60 min with 20 μM latrunculin B and incubated for 30 min with 1.25 μM phosphatidylinositide: 0.625 μM histone complex. Unless otherwise indicated, cells were acutely stimulated for 30 min with 10 nM insulin or 600 mM sorbitol after pretreatments for GLUT4 translocation studies, or stimulated 5 min with either insulin or sorbitol for Cbl analyses.
Plasma Membrane Sheet Assay
Plasma membrane sheets were prepared as previously described [Kralik et al., 2002] with minor modifications. Briefly, following treatments, cells were placed on ice (GLUT4 trafficking analyses) and fixed as previously described [Kralik et al., 2002], or cells were kept at 37°C and fixed at room temperature (PI lipid analyses). Sheets were fixed for 20 min at 25°C in 2% paraformaldehyde/phosphate buffered saline (PBS), then blocked in 5% donkey serum for 60 min at 25°C, incubated for 1 h at 25°C with a 1:1,000 dilution of polyclonal rabbit GLUT4 antibody (provided by Dr. Jeffrey Pessin, SUNY, Stony Brook, NY), or for 60 min at 25°C with a 1:50 dilution of mouse PI 4,5-P2 antibody (Assay Designs, Inc., Ann Arbor, MI), followed by incubation at 25°C with 1:50 rhodamine red-X-conjugated anti-rabbit or anti-mouse antibody (Jackson Immunoresearch, Inc., West Grove, PA) for 60 or 45 min, respectively.
Whole Cell Immunofluorescence and Phalloidin Staining
Following treatment, adipocytes were fixed for 20 min at 25°C in 4% paraformaldehyde/0.2% Triton X-100/PBS. For labeling of actin after fixation, cells were incubated with 1:1,000 FITC-conjugated phalloidin for 2 h at 25°C. Samples were examined via a Zeiss LSM 510 NLO Confocal Microscope (Thornwood, NY). All microscopic and camera settings were identical within experiments, and representative images are shown.
GLUT4 Immunofluorescence Quantification
Whole cells or plasma membrane sheets were prepared and probed with primary antibodies to GLUT4 (Santa Cruz Biotechnology, Santa Cruz, CA). Caveolin-1 antibodies (Upstate, Waltham, MA and Santa Cruz Biotechnology) were used to normalize for protein. Caveolin-1 immunofluorescence was not statistically different between groups in any experiment (data not shown). Near infrared IRDye 800 and 700-conjugated anti-goat, anti-mouse, and anti-rabbit IgG secondary antibodies were purchased from Rockland (Gilbertsville, PA). Images were collected and quantitated with the Odyssey infrared imaging system (LI-COR, Lincoln, NE) as previously described [Razidlo et al., 2004; Wong, 2004].
Preparation of Total Cell Extracts and Immunoprecipitation
Total cell extracts were prepared and samples were immunoprecipitated with antibodies to Cbl-1 (Santa Cruz Biotechnology) as we have previously described [Kralik et al., 2002].
Electrophoresis and Immunoblotting
Immunoprecipitated fractions were separated by 7.5% SDS–PAGE. Then transferred to Immobilon P membrane (Millipore, Bedford, MA). Proteins were immunoblotted with either a monoclonal phosphotyrosine antibody (PY20: HRPO; Transduction Laboratories, San Diego, CA) or Cbl antibody (Santa Cruz Biotechnology). All immunoblots were subjected to enhanced chemiluminescence detection (Amersham, Piscataway, NJ) and densitometry (ImageJ v1.33u, NIH).
Statistical Analysis
All values are means ± SE. ANOVA was used to determine differences among groups. Where a significant difference was indicated, the Fisher’s Test was used to determine significant differences between groups. P <0.05 was considered statistically significant.
RESULTS
ET-1-Induces Resistance to Both Insulin- and Osmotic Shock-Stimulated GLUT4 ranslocation in 3T3-L1 Adipocytes
The ET-1-induced defect in GLUT4 regulation by insulin is apparent in 3T3-L1 adipocytes exposed to this vasoactive peptide for 24 h as we [Strawbridge and Elmendorf, 2005] and others [Ishibashi et al., 2001] have documented (Fig. 1A, panels 3 and 4). As expected, hyperosmotic incubation conditions (600 mM sorbitol) increased plasma membrane GLUT4 content (Fig. 1B, panels 1 and 3). Similar to the ET-1-induced loss of insulin-stimulated GLUT4 translocation, ET-1 treatment diminished the insulin-mimetic stimulatory action of hyperosmolarity (Fig. 1B, panels 3 and 4). Quantitation is presented as the ratio of GLUT4 to caveolin-1 immunofluorescence (Fig. 1D). As we previously reported [Strawbridge and Elmendorf, 2005], the ET-1-induced defect was clearly associated with a loss of plasma membrane PIP2, as assessed by PIP2 immuno-detection of plasma membrane sheets (Fig. 1C, main panels 1 and 2), and a concomitant drop in the cortical F-actin, as assessed by phalloidin staining of whole cells (Fig. 1C, inset panels 1 and 2).
Fig. 1.
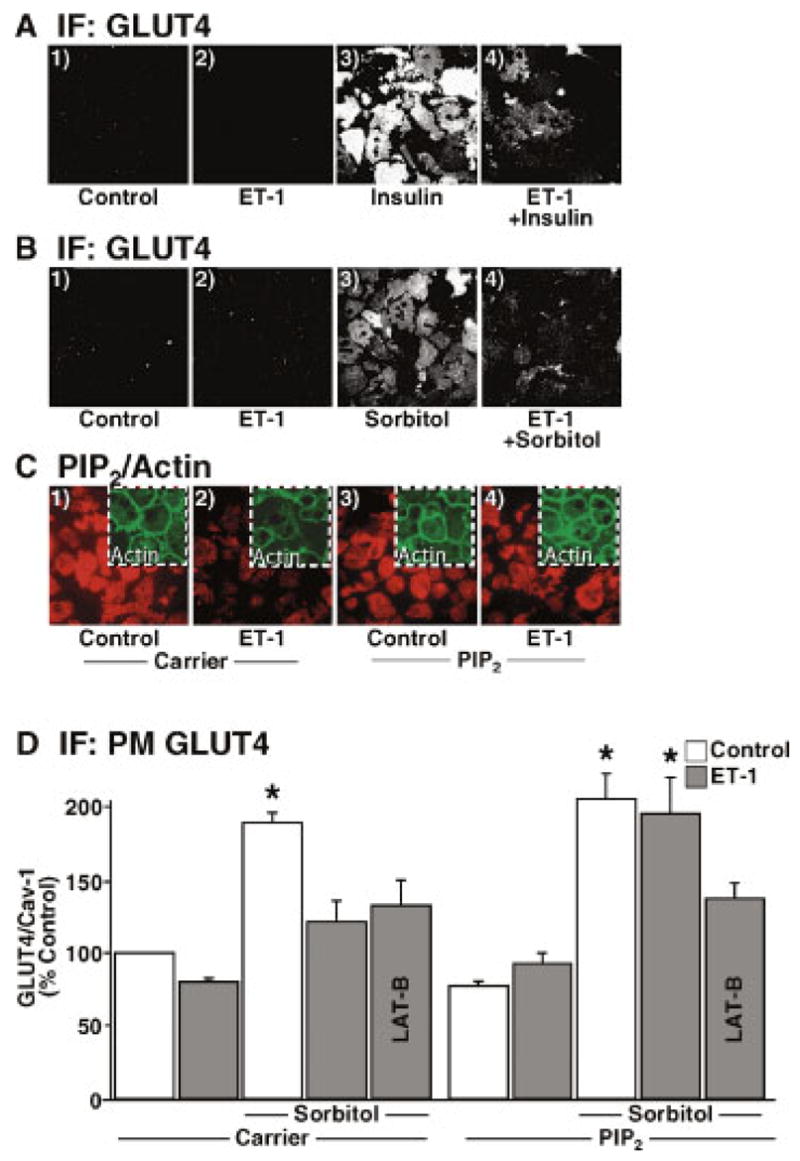
Endothelin-1 (ET-1) impairs insulin- and osmotic shock-stimulated GLUT4 translocation in a phosphatidylinositol 4,5-bisphosphate (PIP2)-dependent manner. 3T3-L1 adipocytes were incubated in the absence (panels 1 and 3) or presence (panels 2 and 4) of 10 nM ET-1 for 24 h. Following ET-1 exposure, cells were either left untreated (panels 1 and 2) or acutely (30 min) treated (panels 3 and 4) with (A) 10 nM insulin or (B) 600 mM sorbitol. Plasma membrane GLUT4 immunofluorescence was detected in membrane sheets as described in Materials and Methods. C: After ET-1 incubation, either 0.625 μM Histone H1 (Carrier) or 1.25 μM PIP2/0.625 μM Histone H1 (PIP2) was added to the medium for 1 h. Plasma membrane PIP2 immunofluorescence (red) was detected in membrane sheets (main panels) and cortical filamentous actin (F-actin) (green) was detected in whole cells (inset panels) as described in Materials and Methods. D: Prior to 600 mM sorbitol treatment, media was supplemented with carrier or PIP2 as indicated above. Some cells were also co-treated with Latrunculin B as described in Materials and Methods. GLUT4 immunofluorescence relative to caveolin-1 was determined using the LI-COR imaging system. Each bar is expressed as a percentage of control in the absence of PIP2 and represents the mean ± SEM of 5 determinations. (*P <0.003 versus control.) [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
PIP2 Restores Adipocyte Sensitivity to Osmotic Shock
Based on our previous studies teaching us that a component of ET-1-induced insulin resistance involves a loss of plasma membrane PIP2 [Strawbridge and Elmendorf, 2005], we next tested if exogenous PIP2 add-back could prevent the negative effect of ET-1 on GLUT4 translocation induced by hyperosmolarity. Using an established PIP2 replenishment procedure [Chen et al., 2004; Strawbridge and Elmendorf, 2005], we observed that carrier delivery of PIP2 into insulin-resistant adipocytes replenished plasma membrane PIP2 (Fig. 1C, compare main panels 2 and 4) and cortical F-actin (Fig. 1C, compare inset panels 2 and 4). This tactic sufficiently restored the ability of osmotic shock to stimulate GLUT4 translocation during chronic ET-1 exposure (Fig. 1D). As with insulin [Strawbridge and Elmendorf, 2005], this restoration was dependent upon F-actin integrity as the restorative effect did not occur if actin re-polymerization was blocked by latrunculin B co-treatment. Carrier alone was without effect on plasma membrane PIP2, cortical F-actin, and GLUT4 levels under all conditions tested.
ET-1 Disrupts Insulin and Osmotic Shock Mediated Tyrosine Phosphorylation of Cbl
Parallel studies tested the effect of ET-1 on the rapid and transient insulin and osmotic shock-induced tyrosine phosphorylation of Cbl [Chen et al., 1997; Liu et al., 2003]. The extent of insulin-stimulated tyrosine phosphorylation of Cbl was markedly decreased at an early time point (5 min) and its transient level of phosphorylation remained reduced in cells treated with ET-1 (Fig. 2A). Remarkably, ET-1 induced a 45% decrease (P < 0.02) in Cbl tyrosine phosphorylation, which very closely parallels the ET-1-induced decrease in insulin-stimulated glucose transport previously reported [Strawbridge and Elmendorf, 2005]. In agreement with earlier studies [Chen et al., 1997; Kralik et al., 2002], osmotic shock markedly increased the tyrosine phosphorylation of Cbl under control conditions and the extent of phosphorylation at the 5-min time point was impaired by ET-1 as seen with insulin stimulation (Fig. 2B).
Fig. 2.
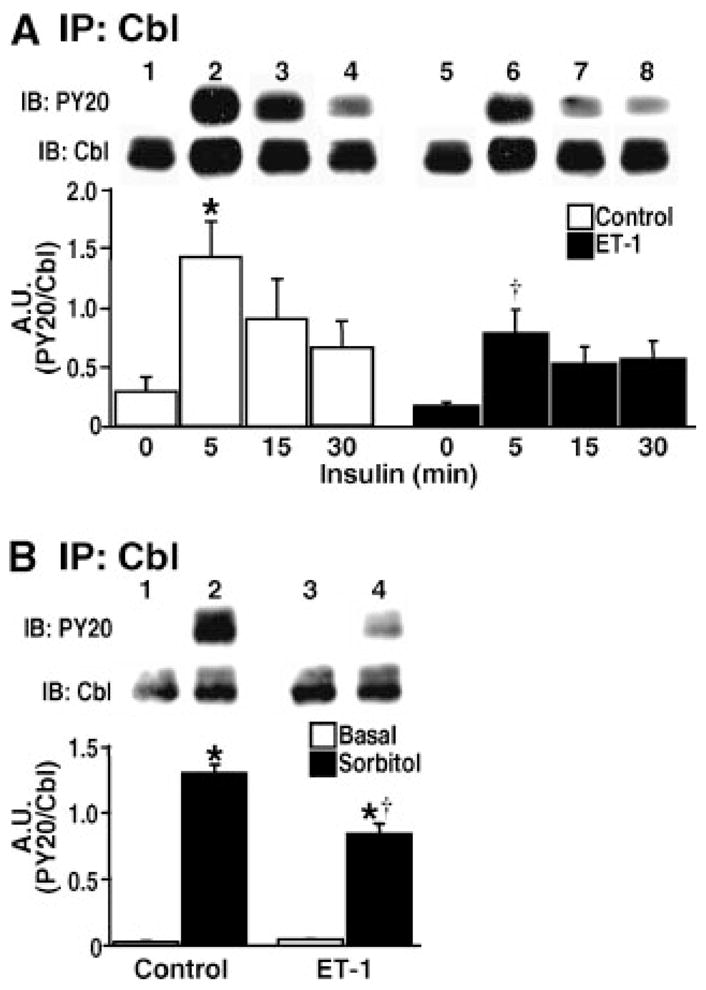
Insulin and phosphatidylinositol 3-kinase (PI3K)-independent Cbl signaling to GLUT4 is impaired by ET-1. Following control and ET-1 incubations, Cbl was immunoprecipitated and tyrosine phosphorylation assessed from cells that were left untreated or treated with (A) 10 nM insulin for the indicated times or (B) 600 mM sorbitol for 5 min. Densitometric values (A.U.) are from 3–5 independent experiments (*P <0.05 vs. unstimulated control, † P <0.02 vs. 5-min control time point).
PIP2, but not PIP3 Replenishment, Reverses ET-1-Induced Impairment of Insulin-Stimulated Cbl Tyrosine Phosphorylation
Since we have previously shown that the PIP2-dependent reversal of ET-1-induced insulin resistance is independent of the activities of Akt-1 and Akt-2 activity [Strawbridge and Elmendorf, 2005], we sought to determine if PIP2 replenishment would restore insulin-stimulated phosphorylation of Cbl. Consistent with a PIP2-induced defect in Cbl signal transduction, PIP2 add-back corrected the ET-1-induced defect in insulin-stimulated Cbl signaling (Fig. 3A), whereas PIP3 add-back did not reverse this defect (Fig. 3B).
Fig. 3.
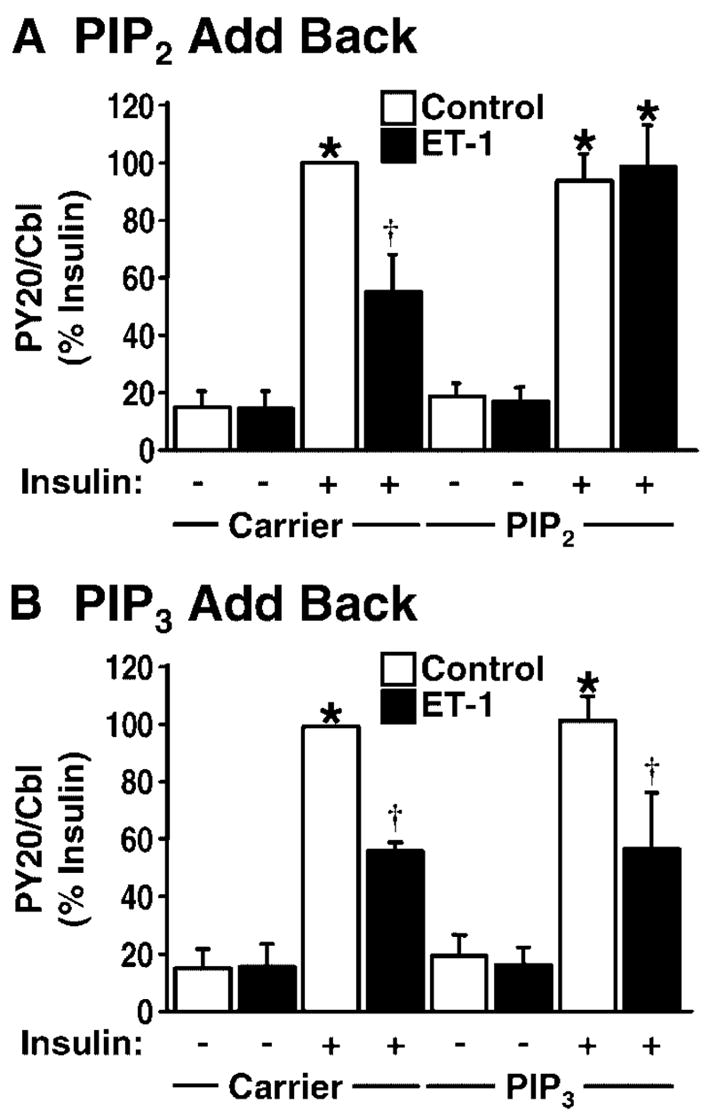
PIP2, but not phosphatidylinositol 3,4,5-trisphosphate (PIP3), replenishment reverses ET-1-induced impairment of insulin-stimulated Cbl tyrosine phosphorylation. PIP2 and PIP3 levels were replenished and insulin-stimulated Cbl tyrosine phosphorylation was assessed in the absence (open bars) and presence (shaded bars) of ET-1. Carrier or phosphoinositide-treated cells were left unstimulated (−) or stimulated with insulin (+) for 5 min. Cbl protein was immunoprecipitated, resolved, and analyzed for tyrosine phosphorylation. A: Cbl phosphorylation following replenishment of PIP2 or (B) PIP3 into cells is reported as mean 3SE from 4–5 independent experiments. A: *P <0.0001 versus unstimulated control, † P <0.005 versus all groups, (B) *P <0.0001 versus unstimulated control, † P <0.01 versus all groups except insulin-treated ET-1 group.
DISCUSSION
In recent years, there has been increasing awareness of the potential role of ET-1 in the development of insulin resistance. Studies from several laboratories have reported that (i) circulating levels of ET-1 are elevated in insulin resistance associated with aging [Sayama et al., 1999], metabolic syndrome X [Ferri et al., 1997], obesity [Ferri et al., 1995, 1997], polycystic ovary syndrome [Diamanti-Kandarakis et al., 2001], and type 2 diabetes [Takahashi et al., 1990; Ferri et al., 1997]; (ii) ET-1 administration in vivo leads to insulin resistance in rats [Juan et al., 1996; Wilkes et al., 2003], and humans [Teuscher et al., 1998]; (iii) blockade of the endothelin type-A (ET-A) receptor prevents ET-1-induced reduction in insulin sensitivity in humans [Ottosson-Seeberger et al., 1997], as well as in vitro [Chou et al., 1994; Jiang et al., 1999; Idris et al., 2001; Ishibashi et al., 2001] and in vivo [Teuscher et al., 1998; Wilkes et al., 2003] models of ET-1-induced insulin resistance; and (iv) key signal transduction mechanisms of insulin action in skeletal muscle [Idris et al., 2001; Wilkes et al., 2003], smoothmuscle [Jiang et al., 1999], and fat [Chou et al., 1994; Idris et al., 2001; Ishibashi et al., 2001] cells are impaired following chronic ET-1 treatment. The results of our studies are in full agreement with these observations and further demonstrate ET-1-induced defects in lipid membrane, actin cytoskeletal, and signaling events regulating GLUT4 trafficking.
Using hyperosmolarity as a molecular tool to better understand targets of ET-1 action, this report found that ET-1 disturbs PIP2-regulated cortical F-actin polymerization and tyrosine phosphorylation of Cbl. With regards to PIP2/actin, these data further demonstrate the essential role of membrane lipids and cellular cytoskeleton in GLUT4 regulation and dysregulation, as we have recently published. Unlike the recognized importance of PIP2/actin, a role for Cbl signaling in GLUT4 regulation remains controversial [Minami et al., 2003; Ahn et al., 2004; Mitra et al., 2004]. Nonetheless, Gual et al. [2002] recently suggested that like insulin, osmotic shock-induced glucose transport involves the Cbl pathway. We found that the abilities of insulin and osmotic shock to elicit the tyrosine phosphorylation of Cbl in 3T3-L1 adipocytes were impaired following ET-1 exposure. Moreover, the corrective effect of exogenous PIP2 on actin structure and GLUT4 translocation concomitantly occurred with a restoration in Cbl phosphorylation. Although regulation of the actin filament network by PIP2 is supported by several studies [Shibasaki et al., 1997; Sechi and Wehland, 2000; Kanzaki et al., 2004], a role for PIP2 and/or cortical F-actin in the spatial regulation of Cbl has not been investigated. The presence of PIP2, actin, and Cbl in cholesterol-dependent microdomains of the plasma membrane [Baumann et al., 2000; Laux et al., 2000; Kanzaki and Pessin, 2002; Kwik et al., 2003] highlight the possible importance of these structural and signaling components in ET-1-induced insulin resistance. Interestingly, the anti-diabetic actions of metformin have been demonstrated to influence cellular cholesterol levels [Hsueh and Law, 1998] and the metabolic profile of women with polycystic ovary syndrome and high levels of ET-1 [Diamanti-Kandarakis et al., 2001]. A model whereby plasma membrane cholesterol and PIP2 jointly influence insulin action and where ET-1 disturbs this putatively critical lipid environment for insulin action is intriguing and we are in the process of testing this.
Based upon the findings herein and recent findings demonstrating the importance of PIP2, cortical F-actin, and Cbl in insulin action [Kanzaki and Pessin, 2001, 2002; Jiang et al., 2002], these studies provide key insight into the cellular mechanisms involved in ET-1-induced insulin resistance. We speculate that ET-1 action not only negatively impacts phosphoinositide regulation of cortical F-actin polymerization, but also membrane-based signal transduction events. Tests examining whether disruption of PIP2 by ET-1 disturbs Cbl recruitment to the plasma membrane (where it apparently undergoes tyrosine phosphorylation [Baumann et al., 2000]) are underway. In conclusion, this study advances our understanding of the pertinent role membrane lipids play in cytoskeletal structure and insulin action, and reveals a novel PIP2-dependent Cbl-signaling disturbance in insulin action. Further studies designed to deepen our understanding of membrane abnormalities associated with insulin-resistant states will have a fundamental impact on diabetes research.
Acknowledgments
We are grateful to both Dr. Ping Liu and Dr. Guoli Chen for excellent technical assistance.
Grant sponsor: American Diabetes Association Research Award; Grant number: 7-05-RA-37; Grant sponsor: American Heart Association Predoctoral Fellowship; Grant number: 0410042Z; Grant sponsor: National Institutes of Health/National Center for Complementary and Alternative Medicine; Grant number: R01-AT001846.
Abbreviations used
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- ET-1
endothelin-1
- ET-A
endothelin type-A receptor
- F-actin
filamentous actin
- G-actin
globular actin
- IR
insulin receptor
- IRS
insulin receptor substrate
- PDK
PIP3-dependent kinase
- PLC
phospholipase C
- PKC
protein kinase C
- PI3K
phosphatidylinositol 3-kinase
- PBS
phosphate buffered saline
- FBS
fetal bovine serum
- BCS
bovine calf serum
- DMEM
Dulbecco’s modified Eagle’s medium
- 2-DG
2-deoxyglucose
- GSK-3
glycogen synthase kinase-3
References
- Ahmed Z, Smith BJ, Pillay TS. The APS adapter protein couples the insulin receptor to the phosphorylation of c-Cbl and facilitates ligand-stimulated ubiquitination of the insulin receptor. FEBS Lett. 2000;475:31–34. doi: 10.1016/s0014-5793(00)01621-5. [DOI] [PubMed] [Google Scholar]
- Ahn MY, Katsanakis KD, Bheda F, Pillay TS. Primary and essential role of the adaptor protein APS for recruitment of both c-Cbl and its associated protein CAP in insulin signaling. J Biol Chem. 2004;279:21526–21532. doi: 10.1074/jbc.M307740200. [DOI] [PubMed] [Google Scholar]
- Ak G, Buyukberber S, Sevinc A, Turk HM, Ates M, Sari R, Savli H, Cigli A. The relation between plasma endothelin-1 levels and metabolic control, risk factors, treatment modalities, and diabetic microangiopathy in patients with Type 2 diabetes mellitus. J Diabetes Complications. 2001;15:150–157. doi: 10.1016/s1056-8727(01)00137-4. [DOI] [PubMed] [Google Scholar]
- Andronico G, Mangano M, Ferrara L, Lamanna D, Mule G, Cerasola G. In vivo relationship between insulin and endothelin role of insulin-resistance. J Hum Hypertens. 1997;11:63–66. doi: 10.1038/sj.jhh.1000386. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay G, Standaert ML, Zhao L, Yu B, Avignon A, Galloway L, Karnam P, Moscat J, Farese RV. Activation of protein kinase C (alpha, beta, and zeta) by insulin in 3T3/L1 cells. Transfection studies suggest a role for PKC-zeta in glucose transport. J Biol Chem. 1997;272:2551–2558. doi: 10.1074/jbc.272.4.2551. [DOI] [PubMed] [Google Scholar]
- Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- Brozinick JT, Jr, Hawkins ED, Strawbridge AB, Elmendorf JS. Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. J Biol Chem. 2004;279:40699–40706. doi: 10.1074/jbc.M402697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero AE, Arora S, Saouaf R, Lim SC, Smakowski P, Park JY, King GL, LoGerfo FW, Horton ES, Veves A. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes. 1999;48:1856–1862. doi: 10.2337/diabetes.48.9.1856. [DOI] [PubMed] [Google Scholar]
- Chen D, Elmendorf JS, Olson AL, Li X, Earp HS, Pessin JE. Osmotic shock stimulates GLUT4 translocation in 3T3L1 adipocytes by a novel tyrosine kinase pathway. J Biol Chem. 1997;272:27401–27410. doi: 10.1074/jbc.272.43.27401. [DOI] [PubMed] [Google Scholar]
- Chen G, Raman P, Bhonagiri P, Strawbridge AB, Pattar GR, Elmendorf JS. Protective effect of phosphatidylinositol 4,5-bisphosphate against cortical filamentous actin loss and insulin resistance induced by sustained exposure of 3T3-L1 adipocytes to insulin. J Biol Chem. 2004;279:39705–39709. doi: 10.1074/jbc.C400171200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YC, Perng JC, Juan CC, Jang SY, Kwok CF, Chen WL, Fong JC, Ho LT. Endothelin-1 inhibits insulin-stimulated glucose uptake in isolated rat adipocytes. Biochem Biophys Res Commun. 1994;202:688–693. doi: 10.1006/bbrc.1994.1985. [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E, Spina G, Kouli C, Migdalis I. Increased endothelin-1 levels in women with polycystic ovary syndrome and the beneficial effect of metformin therapy. J Clin Endocrinol Metab. 2001;86:4666–4673. doi: 10.1210/jcem.86.10.7904. [DOI] [PubMed] [Google Scholar]
- Elmendorf JS. Signals that regulate GLUT4 translocation. J Membr Biol. 2002;190:167–174. doi: 10.1007/s00232-002-1035-3. [DOI] [PubMed] [Google Scholar]
- Ferri C, Bellini C, Desideri G, Di Francesco L, Baldoncini R, Santucci A, De Mattia G. Plasma endothelin-1 levels in obese hypertensive and normotensive men. Diabetes. 1995;44:431–436. doi: 10.2337/diab.44.4.431. [DOI] [PubMed] [Google Scholar]
- Ferri C, Bellini C, Desideri G, Baldoncini R, Properzi G, Santucci A, De Mattia G. Circulating endothelin-1 levels in obese patients with the metabolic syndrome. Exp Clin Endocrinol Diabetes. 1997;105:38–40. doi: 10.1055/s-0029-1211794. [DOI] [PubMed] [Google Scholar]
- Gual P, Shigematsu S, Kanzaki M, Gremeaux T, Gonzalez T, Pessin JE, Le Marchand-Brustel Y, Tanti JF. A Crk-II/TC10 signaling pathway is required for osmotic shock-stimulated glucose transport. J Biol Chem. 2002;277:43980–43986. doi: 10.1074/jbc.M203042200. [DOI] [PubMed] [Google Scholar]
- Hilpela P, Vartiainen MK, Lappalainen P. Regulation of the actin cytoskeleton by PI(4,5)P2 and PI(3,4,5)P3. Curr Top Microbiol Immunol. 2004;282:117–163. doi: 10.1007/978-3-642-18805-3_5. [DOI] [PubMed] [Google Scholar]
- Hsueh WA, Law RE. Cardiovascular risk continuum: Implications of insulin resistance and diabetes. Am J Med. 1998;105:4S–14S. doi: 10.1016/s0002-9343(98)00205-8. [DOI] [PubMed] [Google Scholar]
- Idris I, Patiag D, Gray S, Donnelly R. Tissue- and time-dependent effects of endothelin-1 on insulin-stimulated glucose uptake. Biochem Pharmacol. 2001;62:1705–1708. doi: 10.1016/s0006-2952(01)00815-2. [DOI] [PubMed] [Google Scholar]
- Irving RJ, Noon JP, Watt GC, Webb DJ, Walker BR. Activation of the endothelin system in insulin resistance. Qjm. 2001;94:321–326. doi: 10.1093/qjmed/94.6.321. [DOI] [PubMed] [Google Scholar]
- Ishibashi KI, Imamura T, Sharma PM, Huang J, Ugi S, Olefsky JM. Chronic endothelin-1 treatment leads to heterologous desensitization of insulin signaling in 3T3-L1 adipocytes. J Clin Invest. 2001;107:1193–1202. doi: 10.1172/JCI11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang ZY, Zhou QL, Chatterjee A, Feener EP, Myers MG, White MF, King GL. Endothelin-1 modulates insulin signaling through phosphatidylinositol 3-kinase pathway in vascular smooth muscle cells. Diabetes. 1999;48:1120–1130. doi: 10.2337/diabetes.48.5.1120. [DOI] [PubMed] [Google Scholar]
- Jiang ZY, Chawla A, Bose A, Way M, Czech MP. A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J Biol Chem. 2002;277:509–515. doi: 10.1074/jbc.M108280200. [DOI] [PubMed] [Google Scholar]
- Juan CC, Fang VS, Huang YJ, Kwok CF, Hsu YP, Ho LT. Endothelin-1 induces insulin resistance in conscious rats. Biochem Biophys Res Commun. 1996;227:694–699. doi: 10.1006/bbrc.1996.1571. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Pessin JE. Insulin-stimulated GLUT4 translocation in adipocytes is dependent upon cortical actin remodeling. J Biol Chem. 2001;276:42436–42444. doi: 10.1074/jbc.M108297200. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Pessin JE. Caveolin-associated filamentous actin (cav-actin) defines a novel F-actin structure in adipocytes. J Biol Chem. 2002;277:25867–25869. doi: 10.1074/jbc.C200292200. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Watson RT, Khan AH, Pessin JE. Insulin stimulates actin comet tails on intracellular GLUT4-containing compartments in differentiated 3T3L1 adipocytes. J Biol Chem. 2001;276:49331–49336. doi: 10.1074/jbc.M109657200. [DOI] [PubMed] [Google Scholar]
- Kanzaki M, Watson RT, Hou JC, Stamnes M, Saltiel AR, Pessin JE. Small GTP-binding protein TC10 differentially regulates two distinct populations of filamentous actin in 3T3L1 adipocytes. Mol Biol Cell. 2002;13:2334–2346. doi: 10.1091/mbc.01-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzaki M, Furukawa M, Raab W, Pessin JE. Phosphatidylinositol-4, 5-bisphosphate (PI4,5P2) regulates adipocyte actin dynamics and GLUT4 vesicle recycling. J Biol Chem. 2004;279:30622–30633. doi: 10.1074/jbc.M401443200. [DOI] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, Roth RA. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J Biol Chem. 1996;271:31372–31378. doi: 10.1074/jbc.271.49.31372. [DOI] [PubMed] [Google Scholar]
- Kralik SF, Liu P, Leffler BJ, Elmendorf JS. Ceramide and glucosamine antagonism of alternate signaling pathways regulating insulin- and osmotic shock-induced glucose transporter 4 translocation. Endocrinology. 2002;143:37–46. doi: 10.1210/endo.143.1.8606. [DOI] [PubMed] [Google Scholar]
- Kwik J, Boyle S, Fooksman D, Margolis L, Sheetz MP, Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci USA. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laux T, Fukami K, Thelen M, Golub T, Frey D, Caroni P. GAP43, MARCKS, and CAP23 modulate PI(4,5)P(2) at plasmalemmal rafts, and regulate cell cortex actin dynamics through a common mechanism. J Cell Biol. 2000;149:1455–1472. doi: 10.1083/jcb.149.7.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, DeYoung SM, Hwang JB, O’Leary EE, Saltiel AR. The roles of Cbl-b and c-Cbl in insulin-stimulated glucose transport. J Biol Chem. 2003;278:36754–36762. doi: 10.1074/jbc.M300664200. [DOI] [PubMed] [Google Scholar]
- Mangiafico RA, Malatino LS, Santonocito M, Spada RS. Plasma endothelin-1 concentrations in non-insulin-dependent diabetes mellitus and nondiabetic patients with chronic arterial obstructive disease of the lower limbs. Int Angiol. 1998;17:97–102. [PubMed] [Google Scholar]
- Minami A, Iseki M, Kishi K, Wang M, Ogura M, Furukawa N, Hayashi S, Yamada M, Obata T, Takeshita Y, Nakaya Y, Bando Y, Izumi K, Moodie SA, Kajiura F, Matsumoto M, Takatsu K, Takaki S, Ebina Y. Increased insulin sensitivity and hypoinsulinemia in APS knockout mice. Diabetes. 2003;52:2657–2665. doi: 10.2337/diabetes.52.11.2657. [DOI] [PubMed] [Google Scholar]
- Mitra P, Zheng X, Czech MP. RNAi-based analysis of CAP, Cbl, and CrkII function in the regulation of GLUT4 by insulin. J Biol Chem. 2004;279:37431–37435. doi: 10.1074/jbc.C400180200. [DOI] [PubMed] [Google Scholar]
- Ottosson-Seeberger A, Lundberg JM, Alvestrand A, Ahlborg G. Exogenous endothelin-1 causes peripheral insulin resistance in healthy humans. Acta Physiol Scand. 1997;161:211–220. doi: 10.1046/j.1365-201X.1997.00212.x. [DOI] [PubMed] [Google Scholar]
- Piatti P, Fragasso G, Monti LD, Caumo A, Van Phan C, Valsecchi G, Costa S, Fochesato E, Pozza G, Pontiroli AE, Chierchia S. Endothelial and metabolic characteristics of patients with angina and angiographically normal coronary arteries: Comparison with subjects with insulin resistance syndrome and normal controls. J Am Coll Cardiol. 1999;34:1452–1460. doi: 10.1016/s0735-1097(99)00379-4. [DOI] [PubMed] [Google Scholar]
- Piatti PM, Monti LD, Galli L, Fragasso G, Valsecchi G, Conti M, Gernone F, Pontiroli AE. Relationship between endothelin-1 concentration and metabolic alterations typical of the insulin resistance syndrome. Metabolism. 2000;49:748–752. doi: 10.1053/meta.2000.6257. [DOI] [PubMed] [Google Scholar]
- Razidlo GL, Kortum RL, Haferbier JL, Lewis RE. Phosphorylation regulates KSR1 stability, ERK activation, and cell proliferation. J Biol Chem. 2004;279:47808–47814. doi: 10.1074/jbc.M406395200. [DOI] [PubMed] [Google Scholar]
- Sanchez SS, Aybar MJ, Velarde MS, Prado MM, Carrizo T. Relationship between plasma Endothelin-1 and glycemic control in type 2 diabetes mellitus. Horm Metab Res. 2001;33:748–751. doi: 10.1055/s-2001-19137. [DOI] [PubMed] [Google Scholar]
- Sayama H, Nakamura Y, Saito N, Konoshita M. Does the plasma endothelin-1 concentration reflect atherosclerosis in the elderly? Gerontology. 1999;45:312–316. doi: 10.1159/000022111. [DOI] [PubMed] [Google Scholar]
- Sechi AS, Wehland J. The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci. 2000;113(Pt 21):3685–3695. doi: 10.1242/jcs.113.21.3685. [DOI] [PubMed] [Google Scholar]
- Seljeflot I, Moan A, Aspelin T, Tonnessen T, Kjeldsen SE, Arnesen H. Circulating levels of endothelin-1 during acute hyperinsulinemia in patients with essential hypertension treated with type 1 angiotensin receptor antagonist or placebo. Metabolism. 1998;47:292–296. doi: 10.1016/s0026-0495(98)90259-1. [DOI] [PubMed] [Google Scholar]
- Shibasaki Y, Ishihara H, Kizuki N, Asano T, Oka Y, Yazaki Y. Massive actin polymerization induced by phosphatidylinositol-4-phosphate 5-kinase in vivo. J Biol Chem. 1997;272:7578–7581. doi: 10.1074/jbc.272.12.7578. [DOI] [PubMed] [Google Scholar]
- Strawbridge AB, Elmendorf JS. Phosphatidylinositol 4,5-bisphosphate reverses endothelin-1-induced insulin resistance via an actin-dependent mechanism. Diabetes. 2005;54:1698–1705. doi: 10.2337/diabetes.54.6.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Ghatei MA, Lam HC, O’Halloran DJ, Bloom SR. Elevated plasma endothelin in patients with diabetes mellitus. Diabetologia. 1990;33:306–310. doi: 10.1007/BF00403325. [DOI] [PubMed] [Google Scholar]
- Takenawa T, Itoh T, Fukami K. Regulation of phosphatidylinositol 4,5-bisphosphate levels and its roles in cytoskeletal re-organization and malignant transformation. Chem Phys Lipids. 1999;98:13–22. doi: 10.1016/s0009-3084(99)00014-6. [DOI] [PubMed] [Google Scholar]
- Teuscher AU, Lerch M, Shaw S, Pacini G, Ferrari P, Weidmann P. Endothelin-1 infusion inhibits plasma insulin responsiveness in normal men. J Hypertens. 1998;16:1279–1284. doi: 10.1097/00004872-199816090-00009. [DOI] [PubMed] [Google Scholar]
- Tsakiridis T, Tong P, Matthews B, Tsiani E, Bilan PJ, Klip A, Downey GP. Role of the actin cytoskeleton in insulin action. Microsc Res Tech. 1999;47:79–92. doi: 10.1002/(SICI)1097-0029(19991015)47:2<79::AID-JEMT1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Watson RT, Pessin JE. Subcellular compartmentalization and trafficking of the insulin-responsive glucose transporter, GLUT4. Exp Cell Res. 2001;271:75–83. doi: 10.1006/excr.2001.5375. [DOI] [PubMed] [Google Scholar]
- White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- Wilkes JJ, Hevener A, Olefsky J. Chronic endothelin-1 treatment leads to insulin resistance in vivo. Diabetes. 2003;52:1904–1909. doi: 10.2337/diabetes.52.8.1904. [DOI] [PubMed] [Google Scholar]
- Wong SK. A 384-well cell-based phospho-ERK assay for dopamine D2 and D3 receptors. Anal Biochem. 2004;333:265–272. doi: 10.1016/j.ab.2004.05.011. [DOI] [PubMed] [Google Scholar]