

Abstract
Intermediate Q, the methane-oxidizing species of soluble methane monooxygenase, is proposed to have an [FeIV2(μ-O)2] diamond core. In an effort to obtain a synthetic precedent for such a core, bulk electrolysis at 900 mV (versus Fc+/0) has been performed in MeCN at −40°C on a valence-delocalized [FeIIIFeIV(μ-O)2(Lb)2]3+ complex (1b) (E1/2 = 760 mV versus Fc+/0). Oxidation of 1b results in the near-quantitative formation of a deep red complex, designated 2b, that exhibits a visible spectrum with λmax at 485 nm (9,800 M−1·cm−1) and 875 nm (2,200 M−1·cm−1). The 4.2 K Mössbauer spectrum of 2b exhibits a quadrupole doublet with δ = −0.04(1) mm·s−1 and ΔEQ = 2.09(2) mm·s−1, parameters typical of an iron(IV) center. The Mössbauer patterns observed in strong applied fields show that 2b is an antiferromagnetically coupled diiron(IV) center. Resonance Raman studies reveal the diagnostic vibration mode of the [Fe2(μ-O)2] core at 674 cm−1, downshifting 30 cm−1 upon 18O labeling. Extended x-ray absorption fine structure (EXAFS) analysis shows two O/N scatterers at 1.78 Å and an Fe scatterer at 2.73 Å. Based on the accumulated spectroscopic evidence, 2b thus can be formulated as [FeIV2(μ-O)2(Lb)2]4+, the first synthetic complex with an [FeIV2(μ-O)2] core. A comparison of 2b and its mononuclear analog [FeIV(O)(Lb)(NCMe)]2+ (4b) reveals that 4b is 100-fold more reactive than 2b in oxidizing weak C H bonds. This surprising observation may shed further light on how intermediate Q carries out the hydroxylation of methane.
H bonds. This surprising observation may shed further light on how intermediate Q carries out the hydroxylation of methane.
Keywords: diiron(IV), iron-oxo, nonheme, oxygen activation, electrochemistry
The conversion of methane to methanol is carried out by methane monooxygenases (MMO) that are isolated from methanotrophic bacteria (1). The soluble enzyme has a well characterized nonheme diiron active site (2) that reacts with O2 to yield a diiron(IV) intermediate, Q, responsible for oxidizing CH4 (3–5). Extended x-ray absorption fine structure (EXAFS) analysis of Q has found a short FeFe distance of 2.5 Å, which has led us to propose an [FeIV2(μ-O)2] diamond core structure (6). Density functional theory (DFT) calculations from a number of groups also support the viability of such an intermediate (7–11). Although several mononuclear oxoiron(IV) complexes have been structurally characterized (12), thus providing valuable information regarding the catalytic mechanism of mononuclear nonheme iron enzymes (13), to date there is only one structurally characterized high-valent model complex with an [Fe2(μ-O)2] diamond core, namely [Fe2(μ-O)2(Lc)2](ClO4)3 [Lc, tris(5-ethyl-2-pyridylmethyl)amine] (1c, Scheme 1) (14). However, this complex has an FeIIIFeIV oxidation state, which is one-electron reduced relative to that of MMO-Q.
Scheme. 1.

Ligands used in this study to support the high-valent Fe2(μ-O)2 core.
Thus far, there is no synthetic complex with an FeIV2(μ-O)2 core. In 2001, we claimed to have generated such a complex (15); however, additional data obtained in our laboratories strongly suggested that the compound should be reformulated as an FeIV(OH)(OOR) species (16). In 2005, Collins and coworkers (17) reported the crystallographic and spectroscopic characterization of the first example of a (μ-oxo)diiron(IV) complex; in this case, the high oxidation state was stabilized by the tetra anionic TAML ligand. Here we report a diiron(IV) complex with an [Fe2(μ-O)2] diamond core, obtained by one-electron oxidation of its [FeIIIFeIV(μ-O)2] precursor by using bulk electrolysis. This technique has been demonstrated to be an efficient way for generating high-valent iron and manganese complexes from corresponding lower-valent precursors (18–20). The FeIV2(μ-O)2 complex thus formed has been characterized by electronic, Mössbauer, resonance Raman, and x-ray absorption spectroscopies. The oxidative reactivity of this complex has been compared with those of the FeIIIFeIV precursor and the corresponding mononuclear oxoiron(IV) complex. Our results shed light on the factors that control the oxidative behavior of Fe2(μ-O)2 species.
Results and Discussion
Cyclic Voltammetry.
The valence-delocalized complex 1a (Scheme 1) is the first well characterized high-valent diiron complex possessing an [Fe2(μ-O)2] diamond core (21). As its oxidation state is only one-electron reduced relative to that of intermediate Q, we explored the electrochemistry of 1a to oxidize it to the diiron(IV) form. As shown in Fig. 1A, an oxidative scan of 1a by cyclic voltammetry in MeCN at −40°C displays a reversible redox wave at E1/2 = 900 mV versus Fc+/0 (ΔE = 60 mV at a scan rate 100 mV·s−1).§ This result reveals that it is possible to achieve the one-electron oxidation of 1a to form a new species, 2a. Unfortunately, attempts to oxidize 1a on a preparative scale by bulk electrolysis gave low yields of 2a because of its high self-decay rate.
Fig. 1.

Cyclic voltammetry of 1a (A) and 1b (B) in MeCN at −40°C with 100 mM KPF6 as supporting electrolyte obtained at a scan rate 100 mV/s. Potentials are referenced to Fc+/0.
We then prepared complex 1b (Scheme 1) using a ligand with more electron donating groups on the pyridine rings to stabilize the oxidized complex 2b. The electronic spectrum of 1b is shown in Fig. 2A (dashed line) and resembles that of 1a. Complex 1b shows a quasi-reversible wave at E1/2 = 760 mV (ΔE = 200 mV, Fig. 1B), which is 140 mV lower than that for 1a. The lower potential results in an increased stability for 2b, thereby allowing for its spectroscopic characterization as described below.
Fig. 2.

Interconversion of 1b and 2b. (A) Controlled potential spectroelectrochemistry of the one-electron oxidation of 0.08 mM 1b in MeCN at −40°C with 100 mM KPF6 as supporting electrolyte. (B) Reduction of 0.25 mM 2b to 1b by 0.1-equivalent aliquots of Fc. (Inset) Fc titration plot for the conversion of 2b (absorbance at 485 nm and 875 nm) to 1b (absorbance at 620 nm). Arrows indicate the spectral changes during the oxidation and reduction.
Generation of 2b for Spectroscopic Studies.
Controlled potential bulk electrolysis of 1b carried out at an applied potential of 900 mV in MeCN at −40°C converts green 1b within 1 h into red 2b in near-quantitative yield with absorption maxima at 875 nm (ε = 2,200 M−1·cm−1) and 485 nm (ε = 9,800 M−1·cm−1) (Fig. 2A, solid line). The spectral changes during the conversion of 1b to 2b, recorded in situ and shown in Fig. 2A, give rise to two isosbestic points at 777 nm and 588 nm, indicating the absence of side reactions or decomposition. Complex 2b is quite stable at −40°C (t1/2 ≈ 10 h) and can be rereduced to recover 1b quantitatively by the addition of one equivalent of ferrocene (Fc) (Fig. 2B). Coulometric analysis revealed the extraction of one electron from 1b in the oxidation to 2b, in excellent agreement with the Fc titration experiment. Therefore 2b can be assigned as the one-electron oxidation product of 1b.
Mössbauer Spectroscopy.
In 1995, we reported a detailed Mössbauer study of the mixed-valence complex 1a; this S = 3/2 complex is valence-delocalized, and its equivalent iron sites have quadrupole splittings ΔEQ = 0.49 mm·s−1 and isomer shifts δ = 0.12 mm·s−1 at 100 K (21). Complex 1b (also S = 3/2) yields very similar parameters, namely, ΔEQ = 0.44(2) mm·s−1 and δ = 0.11(1) mm·s−1 at 100 K. Fig. 3 shows Mössbauer spectra of 2b in MeCN recorded at 4.2 K in zero field (A) and in a field of 8.0 T (B) applied parallel to the observed γ-radiation. The zero-field spectrum exhibits a well defined doublet (representing 86–90% of the Fe) with ΔEQ = 2.09(2) mm·s−1 and δ = −0.04(1) mm·s−1. The decrease in δ by 0.15 mm·s−1 in going from 1b to 2b demonstrates that both Fe sites of 2b are now in the FeIV state, in accord with the electrochemical data. The 8.0 T spectrum lacks paramagnetic hyperfine structure, indicating that the two Fe sites of 2b are antiferromagnetically coupled to provide a ground state with S = 0. Given that the iron sites of the FeIIIFeIV species 1a and 1b are low-spin (formally S1 = 1/2 and S2 = 1), it is reasonable to assume that the two local sites of 2b have S1 = S2 = 1. This assignment is in accord with observations by us and others that mononuclear and dinuclear FeIV complexes with N4O2 and N3O3 ligation have S = 1 ground states (12, 13, 16, 18). To date, no mononuclear octahedral FeIV complex with an S = 0 ground state has been reported. Given that all mononuclear FeIV complexes with (octahedral) ligand sets studied in our laboratories have S = 1 ground state, the observation of a diamagnetic ground state for 2b strongly supports the assignment that 2b is a dinuclear species.
Fig. 3.

Shown are the 4.2 K Mössbauer spectra of 2b in acetonitrile, recorded in zero field (A) and a parallel applied field of 8.0 T (B). The solid lines are the sums of spectral simulations for the diamagnetic FeIVFeIV complex 2b (86%) and an 8% FeIIIFeIII contaminant. For the simulations of 2b we used ΔEQ = +2.09 mm·s−1, η = 0, and δ = −0.04 mm·s−1. Simulated spectra of the diiron(III) contaminant, drawn to scale, are shown above the data. An 8.0 T spectrum taken at 100 K with simulations for various J values is shown in supporting information (SI) Fig. 6.
The quadrupole splitting of 2b decreases at temperatures above 90 K, indicating that an excited orbital state is thermally accessible; the observed for ΔEQ(T) (values in mm·s−1) are: 2.02 (100 K), 1.89 (140 K), 1.68 (170 K), and 1.50 (190 K). Currently, we do not have enough information about 2b to identify this orbital state (To determine the energy of the excited state, one needs information about its electric field gradient tensor relative to that of the ground state).
It has been shown for antiferromagnetically coupled FeIIIFeIII complexes that the exchange coupling constant J (ℋ = JS1·S2 with S1 = S2) can be determined by Mössbauer spectroscopy (22, 23), provided J < 100 cm−1. Population of excited states in the spin ladder yields nonzero thermal expectation values of the electronic spin, 〈S〉th, inducing an internal magnetic field at the 57Fe nucleus, Bint(i) = −〈Si〉th·Ai/gnβn, where Ai is the magnetic hyperfine tensor of site i. The presence of Bint(i) reduces the magnetic splitting, yielding the value of J. We have recorded 8.0 T spectra of 2b at 4.2 K (Fig. 3B) and 100 K (SI Fig. 6). The 4.2 K spectrum can be simulated, as shown in Fig. 3B, with the assumption that only the S = 0 ground state is populated. Using the 2SPIN option of WMOSS, we have simulated the 8.0 T spectrum recorded at 100 K for various J values, finding that the exchange coupling constant must be larger than 80 cm−1 (illustrated in SI Fig. 6).
The sample of Fig. 3 also contains a diamagnetic diiron(III) contaminant (≈8%) with ΔEQ = 1.40 mm·s−1 and δ = 0.48 mm·s−1. In a related project we have observed this contaminant as a decay product of 1b. The sample also may contain an as-yet-unidentified contaminant (<5%).
Resonance Raman Spectroscopy.
Resonance Raman spectroscopy has proven to be a valuable tool for the characterization of species with [Fe2(μ-O)2] diamond core structures. A characteristic of such complexes is a band in the 660–700 cm−1 region, which downshifts ≈30 cm−1 upon 18O labeling and has been assigned to a deformation of the [Fe2(μ-O)2] core (24–26). The Raman spectrum of 2b obtained with 514.5-nm excitation shows one intense band at 674 cm−1 (Fig. 4) and additional weaker features at 650 cm−1 and 705 cm−1. When 2b is prepared from 18O-labeled 1b, the 674-cm−1 band shifts to 644 cm−1 (Fig. 4), its 30-cm−1 downshift matching those associated with other [Fe2(μ-O)2] cores (24, 25). This feature therefore is assigned to an Fe2O2 core deformation of 2b. The 705-cm−1 band is not 18O-sensitive, whereas the 650-cm−1 band becomes obscured by the downshift of the 674-cm−1 peak upon 18O labeling and cannot be assigned. The Fe2O2 vibration found in 2b is similar to those of lower-valent complexes with Fe2O2 cores, 1b (661 cm−1) and [FeIII2(μ-O)2(6-Me3-TPA)2]2+ [692 cm−1; 6-Me3-TPA, Tris(6-methyl-2-pyridylmethyl)amine] (24). Indeed, despite differences in iron oxidation and spin state, all three values fit well into the linear correlation between νasym(FeOFe) and ∠FeOFe originally developed for diiron(III) complexes (25, 27), demonstrating that the FeOFe frequency observed reflects only the FeOFe angle. (For 2b, we calculated an FeOFe angle of 101° from rFeFe = 2.73 Å and rFeO = 1.77 Å, see below.)
Fig. 4.

Resonance Raman spectra obtained with 514.5-nm excitation of 2b (A) and 18O-labeled 2b (B; prepared from oxidation of 18O-labeled 1b) in frozen MeCN (77 K) in the presence of 100 mM KPF6. Solvent bands are labeled with “s.”
Although exchange of 1b with 300 equivalents H218O at −30°C is complete within 10 min, 2b becomes labeled with H218O under the same conditions at a much slower rate, taking more than 3 h (SI Fig. 7). Exchange of water into the bridging oxo groups of the [Fe2(μ-O)2] core likely requires breaking of an FeO bond to provide a site for the water to bind (28, 29). So the slower exchange rate observed for 2b suggests a higher kinetic stability for the [FeIV2(μ-O)2] core, consistent with its higher charge and the shorter FeO bond lengths in 2b obtained from EXAFS analysis (see below).
X-Ray Absorption Spectroscopy (XAS).
XAS can be used to gain further insight into the structures of 1b and 2b. Both 1b and 2b in frozen MeCN solution exhibit intense 1s → 3d pre-edge transitions with total areas of 19.7 and 19.0 units, respectively (SI Table 3). The similarity of the pre-edge features suggests that no large structural changes occur upon oxidation of 1b to 2b. However, 2b has a higher Fe edge energy of 7,130.1 eV, compared with 7,129.0 for 1b, reflecting its higher oxidation state.
The Fourier-transformed EXAFS data of 2b exhibit three intense features at r′ = 1.28, 1.64, and 2.26 Å (Fig. 5), where r′ is the actual metal-scatterer distance minus phase-shift corrections of ≈0.3–0.5 Å. The best fit to the EXAFS data reveals a first-coordination sphere consisting of two O/N atoms at 1.78 Å and four N/O atoms at 1.97 Å (Table 1). In agreement with the more oxidized nature of the diiron(IV) complex, these distances are ≈0.05-Å shorter than the average values determined crystallographically for 1c (14). As shown in Table 1, inclusion of a single Fe scatterer at ≈2.7 Å dramatically improves the quality of the EXAFS fit and provides conclusive evidence that 2b is a dinuclear complex with a short FeFe distance. Indeed, the FeFe distance of 2b is comparable to the value of 2.68 Å found in the crystal structure of 1c (14). Finally, as with previous EXAFS studies of Fe complexes with pyridine ligands (14, 16), the fit also is improved by inclusion of an additional shell of six C atoms at 2.93 Å, accounting for the shoulder at r′ = 2.5 Å and corresponding to the pyridine α-carbon atoms. Thus, the XAS analysis provides strong support for the formulation of 2b as [FeIV2(μ-O)2(Lb)2]4+.
Fig. 5.

Fourier-transformed Fe K-edge EXAFS data obtained at 10 K of 3 mM 2b in frozen CH3CN solution with 100 mM PF6 electrolyte. (Inset) k3χ′(k) data. Fourier-transform range k = 2–14 Å−1; experimental data (dotted line) and best fit [solid line; consisting of 2 O/N at 1.77 Å (Δσ2, 0.0028), 4 N/O at 1.97 Å (0.0027), 1 Fe at 2.73 Å (0.0007), and 6 C at 2.93 Å (0.0044)].
Table 1.
EXAFS fitting results for 2b
| Complex | FeO/N |
FeN/O |
FeFe |
FeC |
GOF | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | r | Δσ2 | n | r | Δσ2 | n | r | Δσ2 | n | r | Δσ2 | ||
| 2b | 6 | 1.94 | 11.7 | 1,249 | |||||||||
| 2 | 1.78 | 1.7 | 4 | 1.97 | 1.7 | 806 | |||||||
| 2 | 1.76 | 2.0 | 4 | 1.96 | 2.4 | 1 | 2.72 | 1.7 | 364 | ||||
| 2 | 1.77 | 2.8 | 4 | 1.97 | 2.7 | 1 | 2.73 | 0.7 | 6 | 2.93 | 4.4 | 196 | |
| 1c* | 1.83 (av) | 2.02 (av) | 2.683 (1) | 2.86 (av) | |||||||||
Fourier-transformed range k = 2–14 Å−1 (resolution 0.13 Å). r is in unit of Å. Δσ2 is 10−3 Å2. The first shell was fit using scatterers with oxygen parameters, whereas the second employed nitrogen parameters; however, backscatters differing by Z = 1 cannot be distinguished by EXAFS. GOF, goodness of fit proportional to the sum of the squares of the differences between the experimental and fitted data.
*1c, [FeIIIFeIV(μ-O)2(Lc)2] (ClO4)3; crystallographic data from ref. 14.
Reactivity Studies.
Our ability to generate the diiron(IV) complex 2b provides us with an unprecedented opportunity to compare the redox properties of iron(IV) complexes of the same ligand with FeIIIFeIV(μ-O)2, FeIV2(μ-O)2, and FeIV(O) units. To this end, [FeIV(O)(Lb)(NCMe)]2+ (4b) was prepared following procedures reported for the TPA congener (30). (Although it is conceivable for 4b to convert to 2b by initial loss of the MeCN ligand and subsequent dimerization or for 2b to break up into 4b, we have not been able to effect such an interconversion between 2b and 4b.) As noted earlier, the redox potential of the 2b/1b couple is 760 mV versus Fc+/0 by cyclic voltammetry; however, we have not been able to measure the reduction potentials of 1b and 4b by cyclic voltammetry because of kinetic barriers in the electrochemical reduction, as found in the electrochemistry of the related [FeIV(O)(N4Py)]2+ [N4Py, N,N-bis(2-pyridyl methyl)-N-bis(2-pyridyl)methylamine] complex (19). An order of decreasing potential can be deduced by comparing the reactions of 1b and 4b with reductants of known oxidation potential such as Fc (0 mV), acetylferrocene (270 mV), 1,1′-diacetylferrocene (490 mV), and tris(4-bromophenyl)amine (670 mV) (31). Based on these experiments (Table 2), the reduction potentials of the three complexes decrease in the order 2b > 4b > 1b.
Table 2.
Redox properties of iron(IV) complexes of L
| Complex | Reduction potential, mV (vs. Fc+/0)* | kobs (DHA)†, s−1 | KIE |
|---|---|---|---|
| 1b | 270 < E < 490 | 4.0 × 10−5 | 9 |
| 2b | 760 | 5.1 × 10−4 | 10 |
| 4b | 490 < E < 670 | 4.2 × 10−2 | 27 |
*The reduction potentials of 1b and 4b were estimated by titrations with reductants with known potentials, such as acetylferrocene (270 mV), 1,1′-diacetylferrocene (490 mV), and tris(4-bromophenyl)amine (670 mV) (31). Potentials are referenced to Fc+/0.
†Oxidation of 10 mM DHA carried out in MeCN at −30°C under Ar.
To assess their relative abilities to carry out H-atom abstraction, the reactions of 1b, 2b, and 4b with 9,10-dihydroanthracene (DHA) were investigated in MeCN at −30°C by monitoring the decay of their respective visible or near-IR chromophores (SI Figs. 8–10). In all three cases, DHA was oxidized and kinetic isotope effects (KIEs) > 9 were observed with DHA-d4 as substrate (Table 2), indicating that hydrogen atom abstraction is the rate-determining step for the oxidations (32, 33). Analysis of the oxidation products showed that anthracene was produced in ≈50% yield for 1b and 4b, as deduced from its characteristic UV bands (SI Figs. 8 and 10), indicating that these complexes act as one-electron oxidants. Complex 2b also acts as a one-electron oxidant and afforded 1b quantitatively (SI Fig. 9). The organic products were anthracene and anthraquinone, obtained in respective yields of 14% and 13%, which account for all the iron-oxidizing equivalents consumed. The appearance of anthraquinone as a product derives from the facile oxidation of anthracene [E1/2 = 600 mV versus Fc+/0 (34)], as observed with other high-potential metal-oxo complexes (33, 35).
The transfer of a hydrogen atom from DHA to 1b, 2b, and 4b should result in the reduction of an FeIV(O) fragment to an FeIII(OH) unit (Scheme 2). Complexes 1b and 4b were observed to convert into (μ-oxo)diiron(III) products, [Fe2III(O)(OH)(Lb)2] from 1b (SI Fig. 8) and [Fe2III(O)(O2CR)(Lb)2] from 4b resulting from the facile dimerization of [FeIII(OH)(Lb)] in the presence of a carboxylate (from peracetic acid, the oxidant for generating 4b) (SI Fig. 10). Complex 2b would be expected to convert into the conjugate acid of 1b, which is not very stable. Therefore, a base (5 equivalents of 2,6-lutidine) was added to neutralize this proton. Under these conditions, 1b was obtained quantitatively from 2b.
Scheme. 2.

Hydrogen abstraction by complexes 1, 2, and 4.
As shown in Table 2, the rates of oxidation increase in the order kobs(1b) < kobs(2b) < kobs(4b). The observation that kobs(2b) is ≈10 times faster in oxidizing DHA than kobs(1b) is consistent with its higher potential and oxidation state. However, 4b has a lower potential than 2b but reacts with DHA 100-fold faster than 2b. A simple rationale for this somewhat surprising result is that a terminal oxo is more reactive than a bridging oxo group. This explanation can be scrutinized in light of Mayer's seminal work on hydrogen abstraction reactions of metal-oxo complexes (36). Mayer points out that the hydrogen affinity of a metal-oxo reagent is equivalent to the bond dissociation energy (BDE) of the OH bond that is formed. According to a thermochemical cycle developed by Bordwell (37) and applied by Mayer for metal-oxo systems (38), the OH BDE value is a function of the one-electron reduction potential of the M O oxidant and the pKa of the MOH reduction product. In the case of 4b versus 2b, the 0.2 V estimated difference in redox potential (or ≈4–5 kcal/mol in energy) can be compensated for by a pKa difference of 3–4. The MOH species for 4b is FeIIIOH, and that for 2b can be formulated as FeIIIOHFeIV. We argue that the pKa of FeIIIOHFeIV should be significantly lower than that of FeIIIOH because of the additional coordination of the highly Lewis acidic FeIV to the OH group. Thus the higher oxidative reactivity of the FeIVO complex may be attributed to the stronger basicity of the corresponding FeIIIO− species.
O oxidant and the pKa of the MOH reduction product. In the case of 4b versus 2b, the 0.2 V estimated difference in redox potential (or ≈4–5 kcal/mol in energy) can be compensated for by a pKa difference of 3–4. The MOH species for 4b is FeIIIOH, and that for 2b can be formulated as FeIIIOHFeIV. We argue that the pKa of FeIIIOHFeIV should be significantly lower than that of FeIIIOH because of the additional coordination of the highly Lewis acidic FeIV to the OH group. Thus the higher oxidative reactivity of the FeIVO complex may be attributed to the stronger basicity of the corresponding FeIIIO− species.
Summary Remarks.
We have prepared 2b, the first example of a complex with an FeIV2(μ-O)2 core, by one-electron oxidation of its FeIIIFeIV precursor 1b at −40°C. UV-visible and electrochemical studies show that the interconversion between 1b and 2b is direct, quantitative, and reversible. The presence of an FeIV2(μ-O)2 core in 2b is established by Mössbauer evidence for an antiferromagnetically coupled (J >80 cm−1) diiron(IV) center, the observation by EXAFS of two O scatterers at 1.77 Å and an Fe scatterer at 2.73 Å, and the detection of an 18O-sensitive Raman band at 674 cm−1 that serves as the signature for an [Fe2(μ-O)2] diamond core (24–26). The generation and characterization of 2b as a complex with a significant lifetime at −40°C attests to the chemical viability of an FeIV2O2 diamond core structure and lends credence to the postulation of such a core structure for MMO-Q. There are, however, key differences between 2b and MMO-Q in iron spin state and ligand environment. Unlike 2b, which has a nitrogen-rich ligand environment, the iron(IV) centers of MMO-Q have oxygen-rich ligand sets (FeNO5) (11) and are high spin (4, 5).
With respect to oxidative reactivity, diiron(IV) complex 2b is 100-fold less effective in carrying out H-atom abstraction of DHA than its mononuclear analog 4b, despite having a higher redox potential. It would appear then that there may be a larger kinetic barrier for oxidation by an [FeIV2(μ-O)2] center than for an FeIVO center. Indeed, related, more thermally stable mononuclear oxoiron(IV) complexes have been shown to attack CH bonds as strong as 99 kcal·mol−1 (39). MMO-Q oxidizes methane and is thus a superior oxidant than any of these synthetic low-spin iron(IV) complexes. Its greater reactivity may arise from the fact that it has high-spin iron(IV) centers, which DFT calculations find to be more reactive than their low-spin counterparts in hydrogen-atom abstraction (40–42). However, our observations suggest in addition that its [FeIV2(μ-O)2] core may be further activated in the course of the reaction with methane by isomerization to a more reactive ring-opened form with a terminal FeVO unit, such as FeIIIOFeVO, as originally proposed in 1997 by Siegbahn and Crabtree (7). This notion would be consistent with nature's general strategy of protecting the enzyme active site by unmasking a powerful oxidant only in the presence of the target substrate.
Materials and Methods
Materials.
2-Chloromethyl-4-methoxy-3,5-dimethylpyridine hydrochloride, KPF6 (99.9+%), tris(4-bromophenyl)amine (98%), peracetic acid (32% in dilute acetic acid solution), 1,1′-diacetylferrocene (97%), acetylferrocene (95%), and Fc (98%) were purchased from Aldrich. H218O (95% 18O) was purchased from ICON Isotopes (Summit, NJ). DHA purchased from Aldrich (97%) was recrystallized from EtOH under Ar, and DHA-d4 was prepared following the procedures described in ref. 32. HPLC grade acetonitrile was dried by distilling from CaH2 under Ar. The synthesis and characterization of tris(4-methoxy-3,5-dimethylpyridyl-2-methyl)amine (Lb), [FeIIIFeIV(μ-O)2(Lb)2](ClO4)3 (1b), and [Fe2(μ-O)(H2O)(OH)(Lb)2](ClO4)3 (3b), [FeIIIFeIV(μ-O)2(La)2](ClO4)3 (1a) [La, tris(5-methyl-2-pyridylmethyl)amine], and [FeIV(O)Lb(MeCN)](OTf)2 (4b) are detailed in SI Text.
Physical Methods.
UV-visible spectra were recorded on a Hewlett-Packard 8453A diode array spectrometer equipped with a cryostat from Unisoku Scientific Instruments, Osaka, Japan. 1H-NMR spectra were recorded on a Varian Inova VXR-500 spectrometer. Chemical shifts (ppm) were referenced to the residual solvent peaks. ESI-MS was performed on a Finnigan-MAT (San Jose, CA) LCQ ion-trap mass spectrometer. Elemental analysis was performed by Atlantic Microlab (Norcross, GA). Cyclic voltammetry and bulk electrolysis were performed on a Cypress CS-1200 with the standard three-electrode system.
Mössbauer spectra were recorded with two spectrometers, using Janis Research (Wilmington, MA) SuperVaritemp dewars that allow studies in applied magnetic fields up to 8.0 T in the temperature range from 1.5 to 200 K. Mössbauer spectral simulations were performed by using the WMOSS software package (WEB Research, Minneapolis). Isomer shifts are quoted relative to Fe metal at 298 K. Resonance Raman spectra were recorded on an Acton AM-506 spectrophotometer, using a Kaiser Optical holographic supernotch filter with a Princeton Instruments LN/CCD-1100-PB/UVAR detector cooled with liquid nitrogen. Laser excitation was provided by a 2065 argon ion laser. The spectra were obtained at 77 K by using a 135°-backscattering geometry, and the Raman frequencies were referenced to indene. XAS data for 1b and 2b were collected at beamlines 7–3 and 9–3 of the Stanford Synchrotron Radiation Laboratory (SSRL), Stanford Linear Accelerator Center. X-ray absorption spectra at the iron K-edge were collected between 6.9 and 8.0 eV at a constant temperature of ≈10 K. To avoid photoreduction, flux intensity was reduced by insertion of Al foil in the beamline. The monochromator was calibrated by using the edge energy of Fe foil at 7,112.0 eV. XAS data analysis was performed with the programs EXAFSPAK (43) and SSExafs (44), as previously described (45).
Reactivity Studies.
The reactivity studies were carried out at −30°C in dry MeCN under Ar unless otherwise stated. For a typical redox potential titration, 1 equivalent of reducant was added to a solution of the appropriate iron(IV) complex in a cuvette, and the reaction was monitored by following the characteristic absorption band of the complex (620 nm for 1b, 485 nm for 2b, and 720 nm for 4b). For DHA oxidation studies, 10 mM DHA or DHA-d4 was added to solution of the appropriate iron complex, and the progress of the reaction was monitored after the decay of the characteristic absorption of the complex. The time traces were fitted with a pseudo-first-order model. After removing the iron complexes by filtration through silica gel columns, anthracene product was quantified by its characteristic UV-visible absorption (377 nm, 7,700 M−1·cm−1) and anthraquinone product was quantified by GC. In the DHA oxidation by 2b, 5 equivalents of 2,6-lutidine was added, and the reaction was quenched by 1 equivalent of Fc when 2b was completely converted to 1b.
Supplementary Material
ACKNOWLEDGMENTS.
This work was supported by National Institutes of Health Grants GM38767 (to L.Q.) and EB-001475 (to E.M.) and Postdoctoral Fellowship FGM079839 (to A.T.F.). XAS data were collected on beamlines 7–3 and 9–3 at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, and Biomedical Technology Program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 20641.
This article contains supporting information online at www.pnas.org/cgi/content/full/0708516105/DC1.
The previously reported reversible redox wave at 560 mV versus Fc+/0 (ref. 21) was irreproducible and probably artifactual.
References
- 1.Dalton H. Adv Appl Microbiol. 1980;26:71–87. [Google Scholar]
- 2.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 3.Lee S-K, Nesheim JC, Lipscomb JD. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 4.Lee S-K, Fox BG, Froland WA, Lipscomb JD, Münck E. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 5.Liu KE, Valentine AM, Wang D, Huynh BH, Edmondson DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1995;117:10174–10185. [Google Scholar]
- 6.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 7.Siegbahn PEM, Crabtree RH. J Am Chem Soc. 1997;119:3103–3113. [Google Scholar]
- 8.Siegbahn PEM. Inorg Chem. 1999;38:2880–2889. doi: 10.1021/ic981332w. [DOI] [PubMed] [Google Scholar]
- 9.Basch H, Mogi K, Musaev DG, Morokuma K. J Am Chem Soc. 1999;121:7249–7256. [Google Scholar]
- 10.Dunietz BD, Beachy MD, Cao Y, Whittington DA, Lippard SJ, Friesner RA. J Am Chem Soc. 2000;122:2828–2839. [Google Scholar]
- 11.Baik M-H, Gherman BF, Friesner RA, Lippard SJ. J Am Chem Soc. 2002;124:14608–14615. doi: 10.1021/ja026794u. [DOI] [PubMed] [Google Scholar]
- 12.Shan X, Que L., Jr J Inorg Biochem. 2006;100:421–433. doi: 10.1016/j.jinorgbio.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Costas M, Mehn MP, Jensen MP, Que L., Jr Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 14.Hsu H-F, Dong Y, Shu L, Young VG, Jr, Que L., Jr J Am Chem Soc. 1999;121:5230–5237. [Google Scholar]
- 15.Costas M, Rohde J-U, Stubna A, Ho RYN, Quaroni L, Münck E, Que L., Jr J Am Chem Soc. 2001;123:12931–12932. doi: 10.1021/ja017204f. [DOI] [PubMed] [Google Scholar]
- 16.Jensen MP, Costas M, Ho RYN, Kaizer J, Mairata i, Payeras A, Münck E, Que L, Jr, Rohde J-U, Stubna A. J Am Chem Soc. 2005;127:10512–10525. doi: 10.1021/ja0438765. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh A, Tiago de Oliveira F, Yano T, Nishioka T, Beach ES, Kinoshita I, Münck E, Ryabov AD, Horwitz CP, Collins TJ. J Am Chem Soc. 2005;127:2505–2513. doi: 10.1021/ja0460458. [DOI] [PubMed] [Google Scholar]
- 18.Slep LD, Mijovilovich A, Meyer-Klaucke W, Weyhermuller T, Bill E, Bothe E, Neese F, Wieghardt K. J Am Chem Soc. 2003;125:15554–15570. doi: 10.1021/ja030377f. [DOI] [PubMed] [Google Scholar]
- 19.Collins MJ, Ray K, Que L. Inorg Chem. 2006;45:8009–8011. doi: 10.1021/ic061263i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collomb MN, Mantel C, Romain S, Duboc C, Lepretre JC, Pecaut J, Deronzier A. Eur J Inorg Chem. 2007:3179–3188. [Google Scholar]
- 21.Dong Y, Fujii H, Hendrich MP, Leising RA, Pan G, Randall CR, Wilkinson EC, Zang Y, Que L, Jr, Fox BG, et al. J Am Chem Soc. 1995;117:2778–2792. [Google Scholar]
- 22.Kauffmann KE, Popescu CV, Dong Y, Lipscomb JD, Que L, Jr, Münck E. J Am Chem Soc. 1998;120:8739–8746. [Google Scholar]
- 23.Krebs C, Bollinger JM, Jr, Theil EC, Huynh BH. J Biol Inorg Chem. 2002;7:863–869. doi: 10.1007/s00775-002-0371-1. [DOI] [PubMed] [Google Scholar]
- 24.Wilkinson EC, Dong Y, Zang Y, Fujii H, Fraczkiewicz R, Fraczkiewicz G, Czernuszewicz RS, Que L., Jr J Am Chem Soc. 1998;120:955–962. [Google Scholar]
- 25.Zheng H, Zang Y, Dong Y, Young VG, Jr, Que L., Jr J Am Chem Soc. 1999;121:2226–2235. [Google Scholar]
- 26.Skulan AJ, Hanson MA, Hsu H-f, Que L, Jr, Solomon EI. J Am Chem Soc. 2003;125:7344–7356. doi: 10.1021/ja021137n. [DOI] [PubMed] [Google Scholar]
- 27.Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]
- 28.Tagore R, Chen HY, Crabtree RH, Brudvig GW. J Am Chem Soc. 2006;128:9457–9465. doi: 10.1021/ja061348i. [DOI] [PubMed] [Google Scholar]
- 29.Tagore R, Crabtree RH, Brudvig GW. Inorg Chem. 2007;46:2193–2203. doi: 10.1021/ic061968k. [DOI] [PubMed] [Google Scholar]
- 30.Lim MH, Rohde J-U, Stubna A, Bukowski MR, Costas M, Ho RYN, Münck E, Nam W, Que L., Jr Proc Natl Acad Sci USA. 2003;100:3665–3670. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 32.Goldsmith CR, Jonas RT, Stack TDP. J Am Chem Soc. 2002;124:83–96. doi: 10.1021/ja016451g. [DOI] [PubMed] [Google Scholar]
- 33.Larsen AS, Wang K, Lockwood MA, Rice GL, Won T-J, Lovell S, Sadflek M, Turecek F, Mayer JB. J Am Chem Soc. 2002;124:10112–10123. doi: 10.1021/ja020204a. [DOI] [PubMed] [Google Scholar]
- 34.Neikam WC, Desmond MM. J Am Chem Soc. 1964;86:4811–4814. [Google Scholar]
- 35.de Visser SP, Oh K, Han A-R, Nam W. Inorg Chem. 2007;46:4632–4641. doi: 10.1021/ic700462h. [DOI] [PubMed] [Google Scholar]
- 36.Mayer JM. Acc Chem Res. 1998;31:441–450. [Google Scholar]
- 37.Bordwell FG, Cheng J-P, Ji G-Z, Satish AV, Zhang X. J Am Chem Soc. 1991;113:9790–9795. [Google Scholar]
- 38.Gardner KA, Mayer JM. Science. 1995;269:1849–1851. doi: 10.1126/science.7569922. [DOI] [PubMed] [Google Scholar]
- 39.Kaizer J, Klinker EJ, Oh NY, Rohde J-U, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L., Jr J Am Chem Soc. 2004;126:472–473. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 40.Hirao H, Kumar D, Que L, Jr, Shaik S. J Am Chem Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- 41.Shaik S, Hirao H, Kumar D. Acc Chem Res. 2007;40:523–542. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]
- 42.Bernasconi L, Louwerse MJ, Baerends EJ. Eur J Inorg Chem. 2007:3023–3033. [Google Scholar]
- 43.George GN, Pickering IJ. EXAFSPAK. Stanford, CA: Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center; 2000. [Google Scholar]
- 44.Scarrow RC, Trimitsis MG, Buck CP, Grove GN, Cowling RA, Nelson MJ. Biochemistry. 1994;33:15023–15035. doi: 10.1021/bi00254a011. [DOI] [PubMed] [Google Scholar]
- 45.Rohde J-U, Torelli S, Shan X, Lim MH, Klinker EJ, Kaizer J, Chen K, Nam W, Que L., Jr J Am Chem Soc. 2004;126:16750–16761. doi: 10.1021/ja047667w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
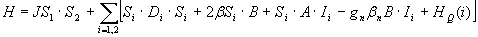{kind=link}