

Abstract
Fifty years since their introduction, thiazide diuretics are established as first-line therapy in the treatment of hypertension. Because the dosing profile for thiazide agents lessened, the mechanism responsible for the blood pressure lowering effect may lie outside their diuretic properties. We evaluated the mechanism driving blood pressure reductions in spontaneously hypertensive rats (SHR) and normotensive WKY by examining the effects of low-dose hydrochlorothiazide (HCTZ) administration on renin-angiotensin system (RAS) components. The 7-day, 1.5 mg/kg/day HCTZ did not change systolic pressure in WKY, but decreased SBP by 41 ± 2 mm Hg (p < 0.0001) in SHR. This reduction was independent of increases in water intake, urine output, or alterations in electrolyte excretion. HCTZ significantly increased the plasma concentrations of angiotensin I (Ang I) and angiotensin II (Ang II) in both WKY and SHR while reducing angiotensin converting enzyme (ACE) activity and the Ang II/Ang I ratio (17.1 ± 2.9 before versus 10.3 ± 2.9 after, p < 0.05) only in SHR. HCTZ increased cardiac ACE2 mRNA and activity, and neprilysin mRNA in WKY, but not SHR. Conversely in SHR, ACE2 activity was decreased and aside from a 75% increase in AT1 mRNA in the HCTZ-treated SHR, the expression of the other variables remained unaltered. Measures of cardiac mas receptor mRNA showed no changes in response to treatment in both strains, although cardiac mas mRNA was significantly lower in untreated SHR. These data, which document for the first time the effect of low-dose thiazide on the activity of the ACE2/Ang-(1–7)/mas-receptor axis, suggest that the opposing arm of the system does not substantially contribute to the antihypertensive effect of low-dose thiazides in SHR.
Keywords: hydrochlorothiazide, angiotensin II, AT1 receptor, angiotensin converting enzyme 2, angiotensin-(1-7), hypertension
Introduction
Thiazide diuretics, the cornerstone of hypertensive and heart failure drug therapy, are purported to exert their antihypertensive and cardiac unloading effects through increased sodium and water excretion. Their mechanistic action was identified when thiazides were prescribed at doses exceeding 50 mg/day. To avoid side effects, the doses were progressively reduced to less than half of the amount considered to exert diuresis when first introduced into the therapeutic armamentarium.
Acting at the cortical diluting tubular segment, high doses of thiazide diuretics induce negative changes in sodium and fluid balance while decreasing circulating blood volume and cardiac output (1). Initially, peripheral vascular resistance increases to partially compensate for the reduction in cardiac output and arterial pressure. Long term, both plasma volume and cardiac output are partly restored, keeping the blood pressure low by decreasing peripheral vascular resistance (2). Since the biphasic nature of the hemodynamic response to thiazide administration was not fully understood (1), additional studies explored an effect of these drugs on vascular reactivity. In 1992, Calder et al (3) investigated direct effects HCTZ on human, guinea pig, and rat arterioles. This report and work by Pickkers et. al (4), showed that HCTZ induced vascular relaxation via stimulation of Ca(2+)-activated K+ channels secondary to a direct effect of HCTZ on vascular carbonic anhydrase. Additional studies suggested the vascular actions of HCTZ were mediated by a vascular endothelium-dependent mechanism (5) whereas, a detailed study in SHR showed that HCTZ inhibits agonist-induced vasoconstriction by calcium desensitization in smooth muscle cells linked to the Rho-Rho kinase pathway (6).
Earlier studies demonstrated that the saluretic action of diuretics evoked heightened renal renin release from the attendant decrease in blood volume and renal perfusion pressure (7). As the decreased HCTZ doses (< 25 mg/day) were administered to patients, the increase in PRA seemed necessary to augment the Ang II blockade effect, and this rationale remained unchallenged to-date (8–10).
There is no systematic analysis of the mechanism of action for low-dose thiazides as the majority of the early investigations utilized doses exceeding 50 mg/day. Another intricacy to ponder is the paradoxical acceptance of the tenet that amplifying the effect of ACE inhibitors and Ang II receptor blockers required the use of low-dose thiazide diuretics as this dogma insinuates that further increasing the activity of the RAS is obligatory toward lower pressure through blockade of this pressor system.
Accumulating evidence that within the RAS, two distinct biochemical pathways yielding effector peptides conveying opposing actions led us to explore the hypothesis that low-dose thiazides may veer the balance between these two axises comprised of ACE/Ang II/AT1 and angiotensin converting enzyme 2 (ACE2)/Ang-(1–7)/mas receptor. Accordingly, we investigated the effect of an equivalent low-dose HCTZ administration in SHR and WKY on hemodynamics, fluid balance, and RAS activity.
Methods
Animals
Ten week old male SHR (n = 24) and WKY rats (n = 24) from Charles River Laboratories (Wilmington, MA) were housed in individual cages with free access to water and rat chow (Purina Mills, Richmond VA) providing 17 and 28 mEq of sodium and potassium per 100 g/day, respectively. The AAALAC approved facility was maintained on a 12-hour light/dark cycle at constant temperature and humidity. All experiments were performed in accordance with protocols of the WFUSM Institutional Animal Care and Use Committee.
Experimental protocol
The rats were randomly divided into 4 groups (WKY-vehicle, SHR-vehicle, WKY-HCTZ, and SHR-HCTZ, n=12/group). The vehicle-treated rats served as controls and drank tap water while the HCTZ groups received 3 mg/kg/day HCTZ dissolved in water on treatment day-1, which slightly increased urine output. Therefore, the dose was decreased to 1.5 mg/kg/day for the remaining 6 treatment days, a dose that decreased blood pressure, but did not induce polyuria. The rats were individually housed in metabolic cages to measure water intake, food consumption, and urine production. The HCTZ solution was adjusted daily to accommodate water intake and body weight variation.
Baseline tail-cuff blood pressures were recorded for three days and on treatment days 1, 3, 5, and 7. Once treatment terminated, the rats were anesthetized with halothane (1.5%; Halocarbon Laboratories, River Edge, NJ) and a catheter was inserted into the abdominal aorta to acquire blood for analysis. The heart was removed by cardio-pulmonary excision. The ventricles were isolated, weighed, submerged in liquid N2, and stored at −80°C.
Biochemistry
The aortic blood samples were collected in an inhibitor cocktail and processed as previously reported (11). The minimum detectable levels of these assays were 0.96 fmol/mL for angiotensin I (Ang I), 0.76 fmol/mL for Ang II, and 2.78 fmol/mL for Ang-(1–7). The intra- and inter-assay coefficients of variation were 18% and 22% for Ang I, 12% and 22% for Ang II, and 8% and 20% for Ang-(1–7). Plasma renin concentration (PRC) and serum ACE activity were measured as reported elsewhere (12) (11). ACE2 activity in cardiac membranes was quantified using the ACE2 fluorescent enzymatic assay as previously described (13).
RNA Isolation and Reverse Transcriptase/Real-time Polymerase Chain Reaction
Cardiac ACE, ACE2, neprilysin, and AT1 and mas receptor mRNAs were measured as previously detailed (11). The results were quantified as Ct values, where Ct was defined as the threshold cycle of PCR at which amplified product is first detected, and expressed as relative gene expression (the ratio of target/18S rRNA control).
Western Blot Analysis
AT1 receptor, mas receptor, and β-actin in the cardiac membranes were measured by Western blot hybridization as previously described (14) using an affinity purified polyclonal anti-AT1 antibody (1:500) (Alpha Diagnostics, San Antonio, TX), a polyclonal anti-mas receptor antibody (1:2000) (developed by M.C. Chappell), or a monoclonal anti-β-actin antibody (1:2000) (Sigma, St. Louis, MO).
Statistical analysis
Values are expressed as mean ± SEM. Normality and homogeneity were assessed prior to conducting a 2-way ANOVA with Bonferroni tests for each variable. Significant interactions were analyzed via one-way ANOVA with Bonferroni or unpaired t-tests. P<0.05 was considered statistically significant.
Results
Hemodynamic Effects of HCTZ Therapy
The WKY baseline systolic blood pressures averaged 128 ± 3 mmHg and 194 ± 2 mmHg in SHR (p<0.0001). HCTZ had no effect in the WKY, but progressively decreased blood pressure in SHR (Figure 1) concluding with a mean pressure reduction of 41 ± 2 mmHg (p<0.0001).
Figure 1.
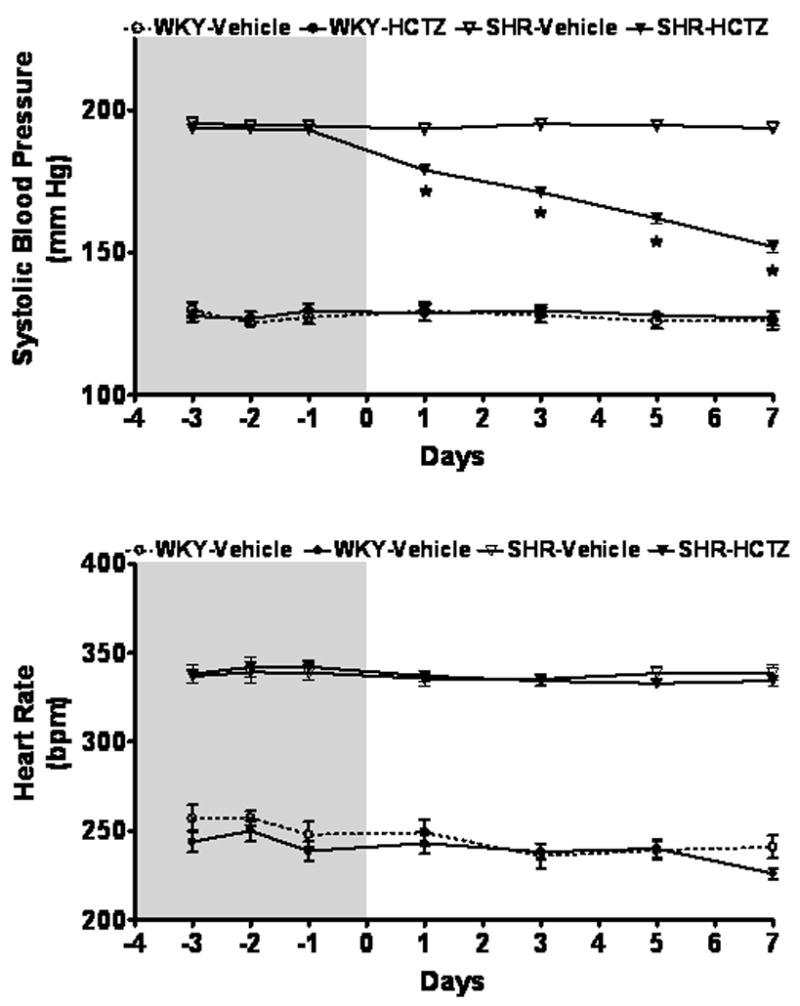
Time course of systolic blood pressure and heart rate measured by tail-cuff plethysmography in WKY and SHR given either vehicle or HCTZ for 7 days. Values are means ± SEM; *P < 0.05 vehicle versus HCTZ
Although age and food consumption did not vary between the strains, SHR were heavier (256 ± 4 g versus 238 ± 7 g) than the WKY prior to initiating treatment and the elevated body mass persisted throughout the experiment (p<0.05).
HCTZ Effect on Renal Excretory Function
At baseline, water intake and urinary output were greater in WKY compared to SHR by an average of 34% (p<0.001) and 52% (p<0.001), respectively. These differences were associated with lower urinary excretion of Na+ (1.6 ± 0.2 mEq/24 h in WKY vs. 1.1 ± 0.1 mEq/24 h in SHR; p<0.001) and K+ (3.3 ± 0.3 mEq/24 h in WKY vs. 2.5 ± 0.1 mEq/24 h in SHR; p<0.001). Urinary osmolality (21.5 ± 0.9 mOsm/24 h vs. 17.6 ± 0.6 mOsm/24 h) was higher and protein concentration lower (1.3 ± 0.1 mg/mL vs. 2.1 ± 0.2 mg/mL) in WKY compared to SHR (p<0.01), but urinary creatinine excretion was similar between the strains.
HCTZ therapy induced a transient increase in urinary volume in SHR that had returned to basal levels by the fifth day of treatment, while HCTZ had no effect on WKY urinary output throughout the treatment period (Figure 2). Concomitantly, there was no effect of HCTZ on urinary electrolyte excretion, creatinine, protein, or osmolality. Following a week of treatment, HCTZ did not change creatinine or K+ excretion rates in either SHR or WKY. Cumulative measurements of water balance and electrolytes did not change.
Figure 2.
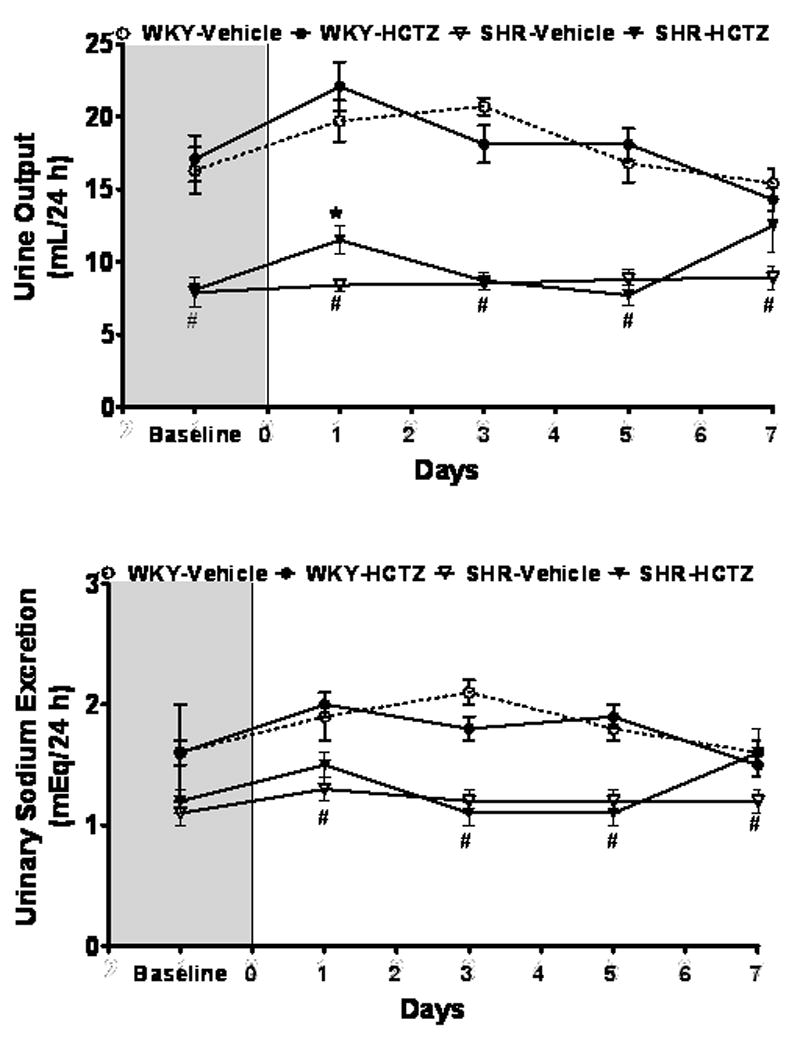
Time course of urine output and urinary sodium (Na+) excretion in WKY and SHR. Values are means ± SEM, *P < 0.05 vehicle versus HCTZ; #P < 0.05 versus WKY-Vehicle
Plasma and Serum RAS Response to Treatment
WKY and SHR baseline plasma concentrations of Ang I and Ang II differed significantly in that the values obtained in the SHR were 1.6- and 1.4-fold less that those in WKY (p<0.05), respectively (Figure 3). Lower basal circulating levels of Ang I and Ang II coincided with a 2.7-fold lower PRC in SHR than in WKY (Figure 4). Baseline plasma Ang-(1–7) levels were comparable in both strains.
Figure 3.
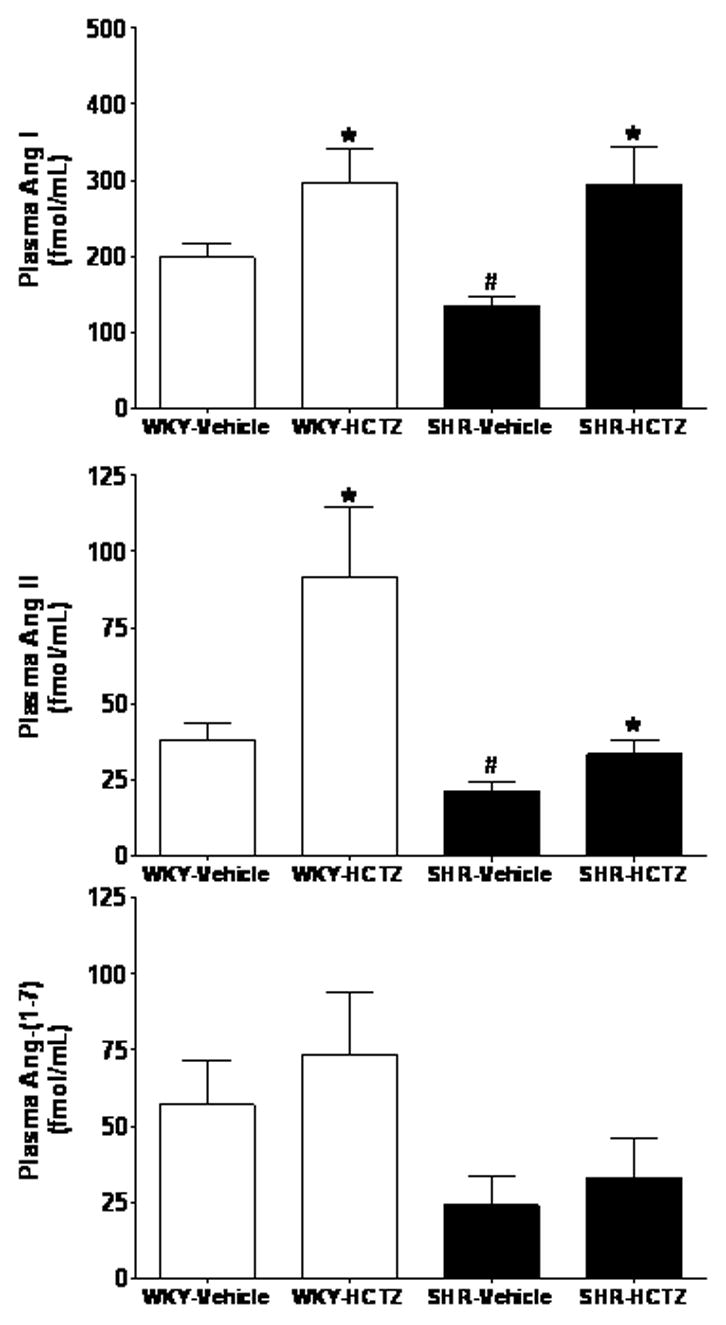
Plasma concentrations of Ang I, Ang II, and Ang-(1–7) in WKY and SHR given either vehicle or HCTZ for 7 days. Values are means ± SEM; *P < 0.05 vehicle versus HCTZ; #P < 0.05 versus WKY-Vehicle.
Figure 4.
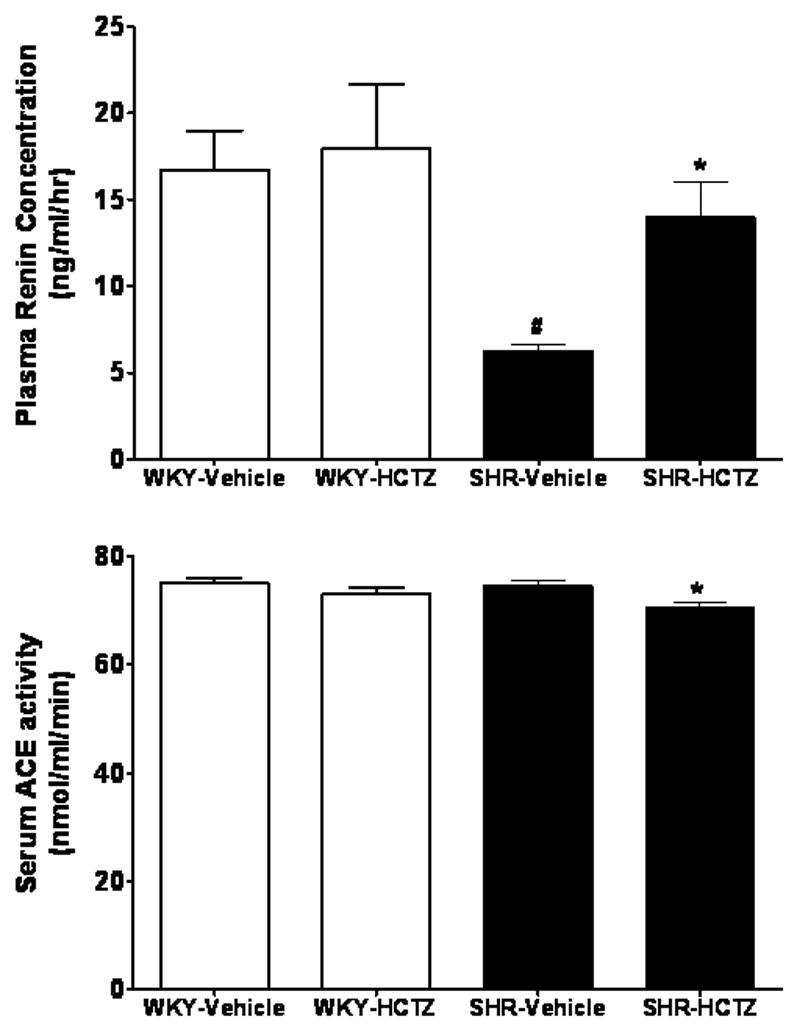
Effects of 7-day administration of HCTZ on plasma renin concentration and serum ACE activity in WKY and SHR. Values are means ± SEM; *P < 0.05 vehicle versus HCTZ; #P < 0.05 versus WKY-Vehicle
Figure 3 shows comparable peak increases in plasma Ang I levels in response to HCTZ treatment in both strains, although the response appears more robust in SHR as their baseline levels were lower compared with WKY controls. In contrast, the response to HCTZ in terms of plasma Ang II is significantly blunted in the SHR compared to WKY. Plasma Ang-(1–7) did not change in either WKY or SHR following HCTZ treatment (Figure 3). HCTZ administration was associated with increases in PRC only in SHR (Figure 4). Although baseline serum ACE activity values were comparable in both WKY and SHR, HCTZ induced a small but significant decrease in ACE activity in SHR (Figure 4). The lower serum ACE activity induced by the treatment in SHR was consistent with a decreased Ang II/Ang I ratio, an index of ACE activity (17.1 ± 2.9 before versus 10.3 ± 2.9 after, p<0.05) (15).
HCTZ Induced Changes in RAS Gene Expression and Activity
HCTZ’s effects on cardiac ACE2 expression and activity of WKY and SHR are illustrated in Figure 5. Basal levels of cardiac ACE2 mRNA were identical in WKY versus SHR. HCTZ increased ACE2 mRNA in WKY, but did not alter ACE2 gene expression in SHR. ACE2 activity paralleled the ACE2 mRNA results in WKY demonstrating augmentation of the activity following HCTZ while in SHR, ACE2 activity decreased in response to HCTZ treatment (Figure 5).
Figure 5.
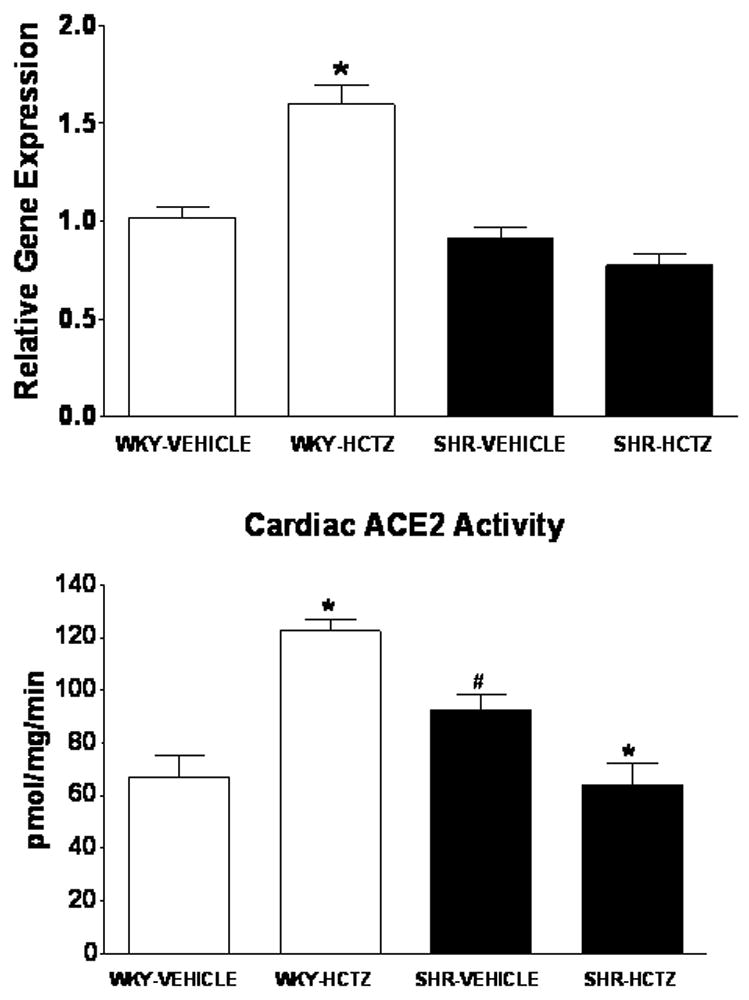
Expression and activity of ACE2 in the left ventricle of WKY and SHR receiving either vehicle or HCTZ. Values are means ± SEM; *P < 0.05 vehicle versus HCTZ; #P < 0.05 versus WKY-Vehicle
Baseline expression of cardiac ACE, neprilysin, and AT1 receptor mRNAs were higher in SHR compared to WKY (Figure 6). HCTZ augmented the expression of ACE, neprilysin, and AT1 receptor mRNAs in WKY, whereas only the AT1 receptor mRNA was elevated following HCTZ in SHR (Figure 6). Contrasting with the differences between WKY and SHR regarding other RAS mRNA components, mas mRNA was 47% lower in SHR compared to WKY (p<0.05) before initiation of treatment. However, HCTZ had no effect on mas mRNA in either strain (Figure 6). Both the cardiac AT1 and mas receptor levels were not different following the HCTZ in either the WKY or SHR.
Figure 6.
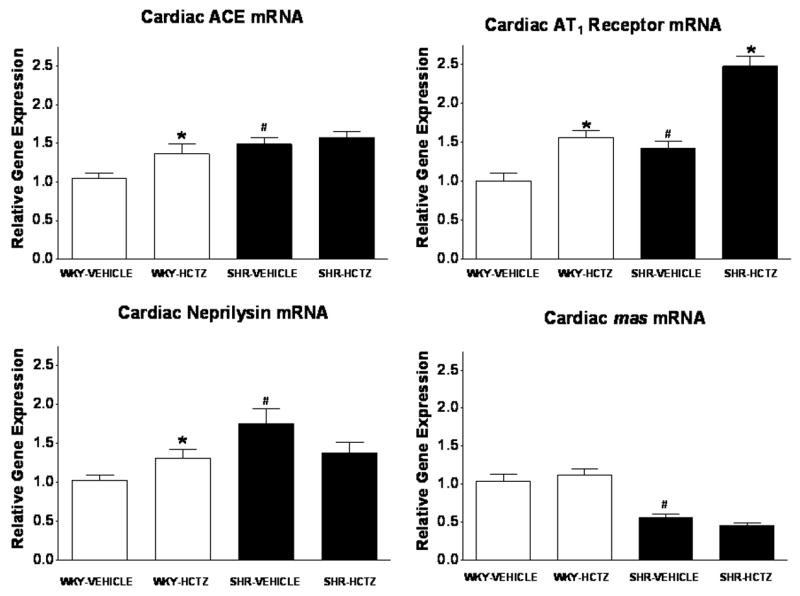
Expression of ACE, neprilysin, AT1 receptor, and mas receptor mRNA in ventricular tissue following vehicle or HCTZ administration. Values are means ± SEM; *P < 0.05 vehicle versus HCTZ; #P < 0.05 versus WKY-Vehicle.
Multiple regression analysis revealed significant associations of ACE and ACE2 mRNAs with AT1 receptor mRNA as well as ACE activity (Figure 7) only in WKY, indicating a 32%, 64%, and 48% relationship of the detectable variation (r2) between these variables. There was no significant relationship present among the AT1 receptor, ACE, and ACE2 mRNAs in the SHR comparable to those accessed in the WKY. Neprilysin mRNA, negatively correlated with plasma Ang-(1–7) (r = 0.595, p < 0.006), while it demonstrated a 46% related variation with the values of ACE2 gene expression (p < 0.0003).
Figure 7.
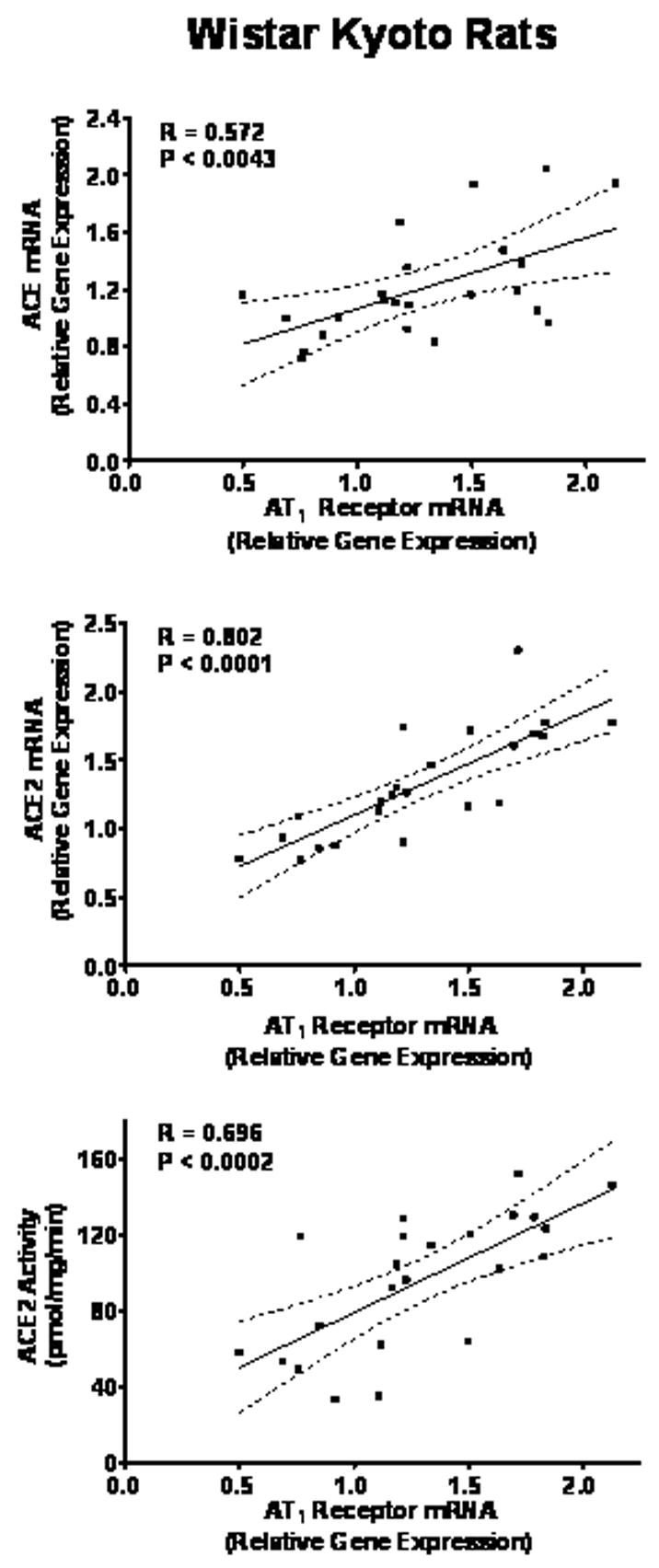
Multiple regression analysis of ACE and ACE2 gene expression, AT1 mRNA, and ACE2 activity in vehicle and HCTZ treated WKY. Regression coefficients are significant for p < 0.01.
Discussion
These studies are the first to evaluate directly low-dose HCTZ effects on the RAS endocrine response. The picture emerging from these experiments suggest that the mechanistic action of low-dose diuretics surpasses in complexity what was originally accepted in terms of the effect of the treatment on circulatory hemodynamics. The activation of the renin angiotensin system in response to HCTZ treatment appears to appropriately compensate for a loss of fluid volume in WKY rats, whereas the same does not occur in SHR. In this strain (SHR), the antihypertensive effect of low dose HCTZ was associated with blunted increases in plasma Ang II concentrations, decreased plasma ACE activity, and larger increases in PRC. The differential response to low dose HCTZ in the two strains is further documented in the contrasting effects of the diuretic on cardiac gene transcripts whereby in the WKY, HCTZ increased cardiac ACE, NEP, ACE2, and AT1 mRNAs while only the AT1 mRNA was elevated in the SHR. These contrasting observations implicate a significant derangement in the regulatory sensitivity of the renin angiotensin system axis in SHR to the application of the stimulus. The current results both reinforce and extend knowledge of the mechanism by which HCTZ acts as an antihypertensive agent by demonstrating a reduced plasma Ang II increases in the SHR in the face of a robust blood pressure reduction. The decreased plasma Ang II, accompanied by increased AT1 receptor mRNA is further evidence of an effect of HCTZ on reducing the pressor activity of the ACE/AngII/AT1 axis. Further work will be required to understand the nature of the signaling mechanism by which this antihypertensive dose of HCTZ silences the response on cardiac ACE, ACE2, NEP, and AT1 message.
In these studies we also report for the first time differences in the expression of the mas receptor, a site that binds Ang-(1–7), which was significantly reduced in SHR. Because our data shows that the effects of low-dose diuretics cannot be merely explained by their impact on diuresis or Na+ excretion, the low-dose HCTZ antihypertensive effect must reside elsewhere.
The chosen dose had no significant effect on the arterial blood pressure of normotensive rats, but elicited a 41 mm Hg decrease in systolic pressure in SHR. The low-dose of HCTZ had minimal effects on fluid intake and output. At the furthest extreme, this dose induced a transient increase in urine volume, a diuretic effect occurring only at day-1 of treatment. There were no significant alterations in urinary sodium and potassium excretion. Therefore, we suggest that activation of the RAS induced by low-dose thiazide in SHR may compensate for the arterial pressure decrease, but the response is inadequate partly because HCTZ attenuates Ang II induced vasoconstriction at AT1 receptors (6).
While exposure of WKY and SHR to the low-dose diuretic yielded comparable directional effects in plasma Ang I and Ang II, the antihypertensive response was associated with increases in PRC and reduced serum ACE activity in the SHR. HCTZ’s effect on serum ACE activity in SHR is noteworthy as it was first inferred by us from changes in the Ang II/Ang I ratio in hypertensive patients given HCTZ at 25 mg/day (16). The current findings are the first to document directly an effect of thiazides on serum ACE activity and the Ang II/Ang I ratios. While the mechanism for the effect of thiazides on ACE activity remains to be fully investigated, Ackerman et al (17) showed that both nitric oxide (NO) and NO-donors inhibited ACE activity in a concentration-dependent and competitive way both in vitro and in vivo.
Divergent responses in cardiac ACE2 mRNA and activity occurred in response to treatment with HCTZ in WKY and SHR. In WKY the thiazide induced increases in both cardiac ACE2 mRNA and activity, whereas in SHR the unchanged cardiac ACE2 mRNA was associated with decreased ACE2 activity. A critical participation of ACE2 in forming Ang-(1–7) from Ang II in vitro is well documented (18; 19) and we showed that Ang II negatively regulates ACE2 expression in cultured astrocytes (20). Baseline differences in ACE2 gene expression in WKY and SHR agree with our suggestion that in hypertension the ACE2/Ang-(1–7)/mas receptor pathway does not compensate for RAS activation (18). In keeping with this interpretation, HCTZ did not change blood pressure while evoking increases in ACE2 and neprilysin mRNA and ACE2 activity in WKY rats. Failure to detect comparable changes in plasma Ang-(1–7) do not negate its contribution as measurements of tissue Ang-(1–7) levels were not obtained in these experiments. We showed previously that Ang-(1–7) plays a critical role in the maintenance of blood pressure as administration of either a selective Ang-(1–7)-antibody (21) or the Ang-(1–7) receptor antagonist, [(D-Ala7)Ang-(1–7)] (22) elevated blood pressure in SHR and transgenic hypertensive rats given a low-salt-diet and a diuretic.
Detection of significant correlations among cardiac ACE mRNA, ACE2 mRNA, ACE2 activity, and cardiac AT1 receptor mRNA suggests that the drug may alter AT1 receptor expression and responsiveness as suggested by Zhu et al (6). Experiments utilizing alternative thiazides complement these data by illustrating similar inhibition of the thiazide on the contractile response in SHR, but not in their WKY counterparts (5). Furthermore, the reduction in cardiac mas receptor mRNA in untreated SHR may be an expression of the altered counterbalancing action of this receptor on AT1 receptor mediated effects since Ang-(1–7) stimulates eNOS release by coupling to this receptor (23; 24). Further studies will be required to ascertain the mechanism that accounts for this difference.
In summary, we show for the first time the complex effects of low-dose thiazide diuretic on the endocrine response mediated by the opposing arms of the renin angiotensin system. In WKY the effects of the low-dose administration of HCTZ on blood pressure may entail a compensatory increase in the Ang-(1–7) forming enzymes activities to buffer the drug’s effect on increased Ang II formation and activity. Blunting of this mechanism in SHR may occur from activation of other vasodilator mechanisms that desensitize or reduce AT1 receptor-mediated agonist vasoconstriction including NO release.
Acknowledgments
Support was provided by NIH (HL-51952, HL-56973), Unifi Inc., Greensboro, NC, and Farley-Hudson Foundation, Jacksonville, NC.
Footnotes
There are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Freis ED. How diuretics lower blood pressure. Am Heart J. 1983;106:185–187. doi: 10.1016/0002-8703(83)90116-3. [DOI] [PubMed] [Google Scholar]
- 2.Birkenhager WH. Diuretics and blood pressure reduction: physiologic aspects. J Hypertens Suppl. 1990;8:S3–S7. [PubMed] [Google Scholar]
- 3.Calder JA, Schachter M, Sever PS. Direct vascular actions of hydrochlorothiazide and indapamide in isolated small vessels. Eur J Pharmacol. 1992;220:19–26. doi: 10.1016/0014-2999(92)90006-p. [DOI] [PubMed] [Google Scholar]
- 4.Pickkers P, Garcha RS, Schachter M, Smits P, Hughes AD. Inhibition of carbonic anhydrase accounts for the direct vascular effects of hydrochlorothiazide. Hypertension. 1999;33:1043–1048. doi: 10.1161/01.hyp.33.4.1043. [DOI] [PubMed] [Google Scholar]
- 5.Colas B, Slama M, Masson H, Colas JL, Collin T, Arnould ML, Hary L, Safar M, Andrejak M. Direct vascular actions of methyclothiazide and indapamide in aorta of spontaneously hypertensive rats. Fundam Clin Pharmacol. 2000;14:363–368. doi: 10.1111/j.1472-8206.2000.tb00417.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Z, Zhu S, Liu D, Cao T, Wang L, Tepel M. Thiazide-like diuretics attenuate agonist-induced vasoconstriction by calcium desensitization linked to Rho kinase. Hypertension. 2005;45:233–239. doi: 10.1161/01.HYP.0000152701.97426.5f. [DOI] [PubMed] [Google Scholar]
- 7.Chan PS, Ronsberg MA, Cervoni P. Studies on the mechanism of the synergistic antihypertensive activity of captopril and hydrochlorothiazide following acute administration in spontaneously hypertensive rats. Clin Exp Hypertens A. 1982;4:1019–1034. doi: 10.3109/10641968209060769. [DOI] [PubMed] [Google Scholar]
- 8.Carter BL, Ernst ME, Cohen JD. Hydrochlorothiazide versus chlorthalidone: evidence supporting their interchangeability. Hypertension. 2004;43:4–9. doi: 10.1161/01.HYP.0000103632.19915.0E. [DOI] [PubMed] [Google Scholar]
- 9.Freis ED. Studies in hemodynamics and hypertension. Hypertension. 2001;38:1–5. doi: 10.1161/01.hyp.38.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Hughes AD. How do thiazide and thiazide-like diuretics lower blood pressure? J Renin Angiotensin Aldosterone Syst. 2004;5:155–160. doi: 10.3317/jraas.2004.034. [DOI] [PubMed] [Google Scholar]
- 11.Ferrario CM, Jessup J, Gallagher PE, Averill DB, Brosnihan KB, Ann TE, Smith RD, Chappell MC. Effects of renin-angiotensin system blockade on renal angiotensin-(1–7) forming enzymes and receptors. Kidney Int. 2005;68:2189–2196. doi: 10.1111/j.1523-1755.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- 12.Chappell MC, Gallagher PE, Averill DB, Ferrario CM, Brosnihan KB. Estrogen or the AT1 antagonist olmesartan reverses the development of profound hypertension in the congenic mRen2.Lewis rat. Hypertension. 2003;42:781–786. doi: 10.1161/01.HYP.0000085210.66399.A3. [DOI] [PubMed] [Google Scholar]
- 13.Joyner J, Neves LA, Granger JP, Alexander BT, Merrill DC, Chappell MC, Ferrario CM, Davis WP, Brosnihan KB. Temporal-spatial expression of angiotensin-(1–7) and angiotensin converting enzyme 2 in the kidney of normal and hypertensive pregnant rats. Am J Physiol Regul Integr Comp Physiol. 2007 doi: 10.1152/ajpregu.00387.2006. [DOI] [PubMed] [Google Scholar]
- 14.Shaltout HA, Westwood BM, Averill DB, Ferrario CM, Figueroa JP, Diz DI, Rose JC, Chappell MC. Angiotensin metabolism in renal proximal tubules, urine, and serum of sheep: evidence for ACE2-dependent processing of angiotensin II. Am J Physiol Renal Physiol. 2007;292:F82–F91. doi: 10.1152/ajprenal.00139.2006. [DOI] [PubMed] [Google Scholar]
- 15.Kohara K, Brosnihan KB, Ferrario CM. Angiotensin(1–7) in the spontaneously hypertensive rat. Peptides. 1993;14:883–891. doi: 10.1016/0196-9781(93)90063-m. [DOI] [PubMed] [Google Scholar]
- 16.Lee D, DeQuattro V, Brosnihan KB, Ferrario CM. Benazepril therapy in hypertension: LVH regression related to neural tone, blood pressure non responders have reflex increase in angiotensin (1–7) peptide. Journal of Hypertension. 1992;10:S64. [Google Scholar]
- 17.Ackermann A, Fernandez-Alfonso MS, Sanchez de RR, Ortega T, Paul M, Gonzalez C. Modulation of angiotensin-converting enzyme by nitric oxide. Br J Pharmacol. 1998;124:291–298. doi: 10.1038/sj.bjp.0701836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrario CM, Trask AJ, Jessup JA. Advances in the Biochemical and Functional Roles of Angiotensin Converting Enzyme 2 and Angiotensin-(1–7) in the Regulation of Cardiovascular Function. Am J Physiol Heart Circ Physiol. 2005 doi: 10.1152/ajpheart.00618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383:45–51. doi: 10.1042/BJ20040634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallagher PE, Chappell MC, Ferrario CM, Tallant EA. Distinct roles for ANG II and ANG-(1–7) in the regulation of angiotensin-converting enzyme 2 in rat astrocytes. Am J Physiol Cell Physiol. 2006;290:C420–C426. doi: 10.1152/ajpcell.00409.2004. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura S, Averill DB, Chappell MC, Diz DI, Brosnihan KB, Ferrario CM. Angiotensin receptors contribute to blood pressure homeostasis in salt-depleted SHR. Am J Physiol Regul Integr Comp Physiol. 2003;284:R164–R173. doi: 10.1152/ajpregu.00210.2002. [DOI] [PubMed] [Google Scholar]
- 22.Iyer SN, Averill DB, Chappell MC, Yamada K, Allred AJ, Ferrario CM. Contribution of angiotensin-(1–7) to blood pressure regulation in salt-depleted hypertensive rats. Hypertension. 2000;36:417–422. doi: 10.1161/01.hyp.36.3.417. [DOI] [PubMed] [Google Scholar]
- 23.Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1–7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. 2007;49:185–192. doi: 10.1161/01.HYP.0000251865.35728.2f. [DOI] [PubMed] [Google Scholar]
- 24.Su Z, Zimpelmann J, Burns KD. Angiotensin-(1–7) inhibits angiotensin II-stimulated phosphorylation of MAP kinases in proximal tubular cells. Kidney Int. 2006;69:2212–2218. doi: 10.1038/sj.ki.5001509. [DOI] [PubMed] [Google Scholar]
- 25.Lanctot PM, Leclerc PC, Clement M, uger-Messier M, Escher E, Leduc R, Guillemette G. Importance of N-glycosylation positioning for cell-surface expression, targeting, affinity and quality control of the human AT1 receptor. Biochem J. 2005;390:367–376. doi: 10.1042/BJ20050189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leclerc PC, Lanctot PM, uger-Messier M, Escher E, Leduc R, Guillemette G. S-nitrosylation of cysteine 289 of the AT1 receptor decreases its binding affinity for angiotensin II. Br J Pharmacol. 2006;148:306–313. doi: 10.1038/sj.bjp.0706725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kedziora-Kornatowska K, Czuczejko J, Szewczyk-Golec K, Motyl J, Szadujkis-Szadurski L, Kornatowski T, Pawluk H, Kedziora J. Effects of perindopril and hydrochlorothiazide on selected indices of oxidative stress in the blood of elderly patients with essential hypertension. Clin Exp Pharmacol Physiol. 2006;33:751–756. doi: 10.1111/j.1440-1681.2006.04436.x. [DOI] [PubMed] [Google Scholar]