

Abstract
With the quantity of genomic data increasing at an exponential rate, it is imperative that these data be captured electronically, in a standard format. Standardization activities must proceed within the auspices of open-access and international working bodies. To tackle the issues surrounding the development of better descriptions of genomic investigations, we have formed the Genomic Standards Consortium (GSC). Here, we introduce the minimum information about a genome sequence (MIGS) specification with the intent of promoting participation in its development and discussing the resources that will be required to develop improved mechanisms of metadata capture and exchange. As part of its wider goals, the GSC also supports improving the ‘transparency’ of the information contained in existing genomic databases.
A wealth of genomic and metagenomic sequences
By the end of next year, there will be complete genome sequences of at least draft quality for more than 1,000 bacteria and archaea and 100 eukaryotes1,2 and for even larger numbers of viruses, organelles and plasmids. With the rapid pace at which new genome sequences are appearing, the need to consider how best to ensure stewardship of these data for the long term has never been more pressing.
Our genome collection: more than the sum of its parts
The analysis of genomic information is having an impact on every area of the life sciences and beyond. A genome sequence is a prerequisite to understanding the molecular basis of phenotype, how it evolves over time and how we can manipulate it to provide new solutions to critical problems. Such solutions include therapies and cures for disease, industrial products, approaches for biodegradation of xenobiotic compounds and renewable energy sources. With improvements in sequencing technologies, the growing interest in metagenomic approaches and the proven power of comparative analysis of groups of related genomes, we can envision the day when it will be commonplace to sequence tens to hundreds of genomes or more as part of a single study. At current rates of genome sequencing, it has been estimated that >4,000 bacterial genomes will be available soon after 2010 (ref. 1).
Given the importance of the growing genome collection, the capital investment in its creation and the benefits of leveraging its value through diverse comparative analyses, every effort should be made to describe it as accurately and comprehensively as possible. There is an increasing interest from the community in doing so, for three main reasons. The first is the interest in testing hypotheses about the features observed in genomes using comparative evo- and eco-genomic approaches3. The second is the need to supplement the content of a variety of databases with high-level descriptions of genomes that allow useful grouping, sorting and searching of the underlying data. The third is the growth in genome sequence data from environmental isolates and metagenomes—vast data sets of DNA fragments from environmental samples4-6. The data generated by such studies will dwarf current stores of genomic information, making improved descriptions of genomes even more important.
At present, both top-level descriptors and genome descriptions are incomplete for many reasons. First and foremost, in hindsight we now know the minimum quality and quantity of information that is required to make each description precise, accurate and useful. For example, even for bacterial and archaeal species with validly published names, strain names were not routinely captured in genome annotation documents before the sequencing of large numbers of genomes from the same species7, but such information is now considered essential. Through empirical observations, we are expanding our view of the types of information that are important for testing particular hypotheses, exploring new patterns and quantifying inherent sampling biases3,8.
As the number of habitats and communities sampled using metagenomic approaches increases, we are also being forced to rethink our understanding of the minimum information required to adequately describe a genome sequence. Without adequate description of the environmental context and the experimental methods used, such data sets will be of less value for researchers wishing to conduct comparative genomic studies or link genetic potential with the diversity and abundance of organisms. In fact, given the vast number of uncultivated microbes, it may be that a DNA-centric approach, in which genes are linked to habitats (locations), is more useful than the species-centric view9,10. Finally, sequencing technology is advancing rapidly, and the adoption of new methods11,13 will force the adoption of additional descriptors (e.g., the depth of sequence coverage, quality and whether any ‘finishing’ was used) to be able to distinguish among these methods.
Most often, metadata about genome sequences are found only in the primary literature or in reference works, such as Bergey's Manual14 for bacteria and archaea, rather than in sequence databases. The distributed and patchy nature of this information and the difficulties of curating even a few pieces of information for what are now very large collections of genomes make the vision of a single definitive source of rich genomic descriptions highly desirable.
The need for coordinated efforts
Facilitating and accelerating the process of collecting relevant metadata would clearly reduce ongoing replication of efforts and maximize the ability to share and integrate data within the genomics community. The obvious solution is to develop a consensus-based approach.
The Genomic Standards Consortium
The GSC is an open-membership, international working body formed in September 2005 (ref. 15). Its goal is to promote mechanisms that standardize the description of genomes and the exchange and integration of genomic data. The GSC community brings together (i) evolutionists, ecologists, molecular biologists and other researchers analyzing collections of genomes, (ii) bio-informaticians producing genomic databases, (iii) those who sequence genomes and (iv) computer scientists, ontology experts and members of other standardization initiatives, such as the International Nucleotide Sequence Database Collaboration (INSDC), which is responsible for the DNA Data Bank of Japan (DDBJ), European Molecular Biology Laboratory (EMBL) and GenBank databases (http://www.insdc.org/). The guidance of DDBJ, EMBL and GenBank will be critical to the success of the GSC initiative, both because they are the official stewards of the public collection of genomes and because of their interest in fulfilling community needs.
Minimum information about genomes and metagenomes
The GSC is working to define a set of core descriptors for genomes and metagenomes in the form of a MIGS specification (Fig. 1). MIGS extends the minimum information already captured by the INSDC. The MIGS checklist is given in Box 1, and the most up-to-date version is available from the consortium's website (http://gensc.sf.net). Examples of MIGS-compliant reports are given in Supplementary Table 1 online. The information required to comply with MIGS is routinely included in primary genome publications (or is referenced therein). However, this information needs to be formalized and made available in electronic form to improve its accessibility16 (Box 2).
Figure 1.
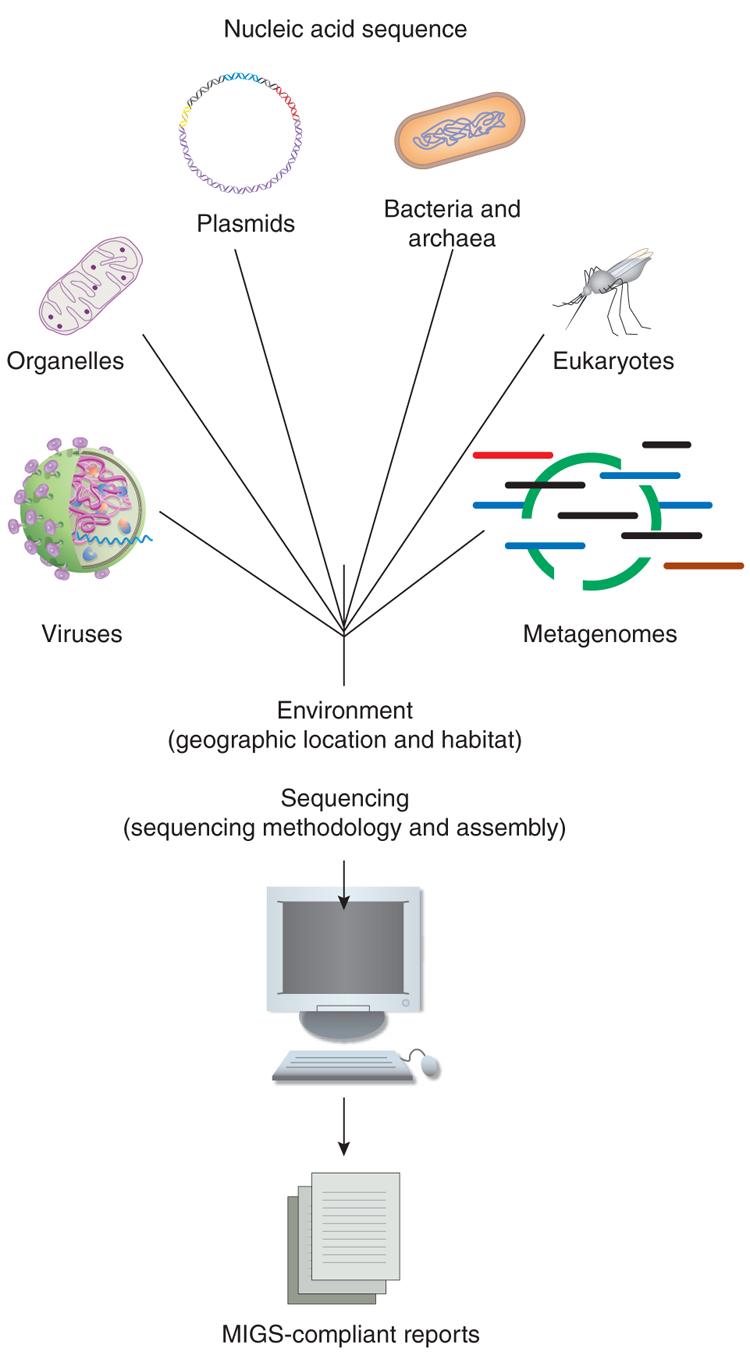
The scope of MIGS. The MIGS specification enables description of the complete range of possible genomes (eukaryotes, bacteria, archaea, plasmids, viruses, organelles) and metagenomes. Core descriptors include information about the origins of the nucleic acid sequence (genome), its environment (latitude and longitude, date and time of sampling and habitat) and sequence processing (sequencing and assembly methods). MIGS-compliant reports can be rendered into an electronic format using the MIGS XML schema and controlled vocabularies through the GSC's Genome Catalogue (http://gensc.sf.net).
Box 1 Minimum Information about a Genome Sequence (MIGS) checklist version 2.0.
| Report type |
||||||
|---|---|---|---|---|---|---|
| Investigation | EU | BA | PL | VI | OR | ME |
| • Submit to trace archives and INSDC | M | M | M | M | M | M |
| • Investigation type (i.e., report type) | M | M | M | M | M | M |
| • Project name2 | M | M | M | M | M | M |
| • Study | ||||||
| • Environment | ||||||
| • Geographic location (latitude and longitudefloat (point, transect and region), depth and altitude of sample)(integer) | M | M | M | M | M | M |
| • Time of sample collection(UCT) | M | M | M | M | M | M |
| • HabitatEnvO | M | M | M | M | M | M |
| MIMS extension: select to report a set of uniform measurements for a given habitat: | M | |||||
| • Water body: (temperature, pH, salinity, pressure, chlorophyll, conductivity, light intensity, dissolved organic carbon (DOC), current, atmospheric data, density, alkalinity, dissolved oxygen, particulate organic carbon (POC), phosphate, nitrate, sulfates, sulfides, primary production)(integer, unit) | ||||||
| • Nucleic acid sequence source | ||||||
| • Subspecific genetic lineage (below lowest rank of NCBI taxonomy, which is subspecies) (e.g., serovar, biotype, ecotype)(CABRI) | M | M | M | M | M | – |
| • Ploidy (e.g., allopolyploid, polyploid)(PATO) | M | |||||
| • Number of replicons (EU, BA: chromosomes (haploid count); VI: segments)(integer) | M | M | – | M | – | – |
| • Extrachromosomal elements(integer) | X | M | ||||
| • Estimated size (before sequencing; to apply to all draft genomes)(integer; base pairs) | M | X | X | X | X | – |
| • Reference for biomaterial (primary publication if isolated before genome publication; otherwise, primary genome report)(PMID or DOI) | X | M | X | X | X | X |
| • Source material identifiers: (cultures of microorganisms: identifiers(alphanumeric) for two culture collections(OBI); specimens (e.g., organelles and Eukarya): voucher condition and location(CV)) | M | M | M | M | M | M |
| • Known pathogenicity | M | M | ||||
| • Biotic relationship (e.g., free-living, parasite, commensal, symbiont)(OBI) | X | M | X | |||
| • Specific host (e.g., host taxid, unknown, environmental)EnvO | X | M | M | M | ||
| • Host specificity or range(taxid) | X | X | X | M | ||
| • Health or disease status of specific host at time of collection (e.g., alive, asymptomatic)PATO | M | M | ||||
| • Trophic level (e.g., autotroph, heterotroph)PATO | M | M | – | – | – | – |
| • Propagation (phage: lytic or lysogenic; plasmid: incompatibility group)(CV) | M | M | M | – | – | |
| • Encoded traits (e.g., plasmid: antibiotic resistance; phage: converting genes)(CV; see caption) | X | M | M | X | ||
| • Relationship to oxygen (e.g., aerobic, anaerobic)PATO | M | – | – | – | – | |
| • Isolation and growth conditions(PMID or DOI) | M | M | M | M | M | M |
| • Biomaterial treatment (e.g., filtering of sea water)(OBI) | M | |||||
| • Volume of sample(integer) | M | |||||
| • Sampling strategy (enriched, screened, normalized)(CV) | M | |||||
| • Assay | ||||||
| • Sequencing | ||||||
| • Nucleic acid preparation (extraction method(CV); amplification(CV) | M | M | M | M | M | M |
| • Library construction (library size(integer), number of reads sequenced(integer), vector(CV) | M | |||||
| • Sequencing method (e.g., dideoxysequencing, pyrosequencing, polony)(OBI) | M | M | M | M | M | M |
| • Assembly (assembly method(CV), estimated error rate(unit) and method of calculation(CV)) | M | M | M | M | M | M |
| • Finishing strategy (status—e.g., complete or draft(CV), coverage(integer), contigs(integer)) | M | M | X | X | X | X |
| • Relevant Standard Operating Procedures (SOPs) | M | M | M | M | M | M |
| • Relevant electronic resources | M | M | M | M | M | M |
All proposed descriptors in MIGS and the reports (groups) to which they apply are listed. EU, eukaryotes; BA, bacteria and archaea; PL, plasmid; VI, virus; OR, organelle; ME, metagenome. Each descriptor has superscripts denoting its ‘type’ (e.g., integer or controlled vocabulary (CV) term). For items marked “CV,” candidate OBO ontologies (http://obofoundry.org), if available, have been selected for use. EnvO, The Environment Ontology; PATO, the Phenotype and Trait Ontology; CABRI, Common Access to Biological Resources and Information. Mixed ontologies may be useful for the “encoded traits” descriptor: the PATO term “resistant” could be used with a ChEBI term—for example, “penicillin”—to note antibiotic resistance to a given compound. Descriptors in shaded rows are common to all report types and are considered the ‘core’ of MIGS. “Source material identifier” is an exception; the GSC recommends this be a core descriptor, but as yet, physical archives are not yet routinely created for all cases or types of biological material subjected to genome sequencing (the recommended deposition in at least two culture collections for viable samples20 and vouchers for specimens). This is due to both cultural and technical issues. The need for universal and unique identifiers for metagenomic samples is an idea recently discussed in an exploratory workshop organized by the MetaFunctions group (http://www.metafunctions.org). In fact, the application of MIGS to our complete genome collection will require the designation of permanent and unique identifiers for all genome projects, something the INSDC is working to implement21. Geographic location is applied in principle to all report types, but we recognize that many isolates, especially eukaryotes, are highly domesticated laboratory organisms distantly separated from an environmental context of relevance. All descriptors deemed to be core are marked “M” (minimum) and others which could be optionally applied to other groups with high priority are marked “X” (extra). Taxonomic groups for which a descriptor cannot be meaningfully applied are marked with a dash. This list of minimal information is recognized by the GSC as just a starting point for the description of genomes and metagenomes. PMID, PubMed identifier; DOI, digital object identifier; float, floating-point decimal; UCT, Coordinated Universal Time (YYYY-MM-DD); unit, a suitable unit of measure. The descriptors isolation and growth conditions take citations as their values because the information can not be contained in a single value (or small set of values) like those of all other fields. This could be given as the PMID or DOI of the publication. It could also be an SOP. In principle, all aspects of the checklist could be substantiated with a reference in addition to a value, and this would be captured at the level of implementation.
Box 2 Frequently asked questions about MIGS.
Below we answer general questions about MIGS, its development and how to use it.
What is MIGS?
MIGS specifies a formal way to describe genomes and metagenomes in more detail than is captured at present in DDBJ, EMBL and GenBank documents.
The information in MIGS is intended to be used in comparative genomic analysis, provide a better understanding of the source of each genome and enable us to situate genomes and metagenomes in their geospatial and temporal contexts (when relevant) through the specification of geographic location and sampling date.
Do all genomes and metagenomes fall under the scope of MIGS?
Yes. MIGS has elements describing eukaryotic, bacterial and archaeal, plasmid, viral and organellar genomes as well as metagenomes. Some of the core elements overlap between types of records, and some are unique to one or more groups.
Who has driven the development of MIGS?
MIGS has been developed through a series of GSC workshops involving participants from DDBJ, EMBL, the US National Center for Biotechnology Information (NCBI), European Bioinformatics Institute (EBI), Joint Genome Institute (JGI), Sanger Institute, J. Craig Venter Institute (JCVI, formerly TIGR), Max Planck Institute, the Community Cyberinfrastructure for Advanced Marine Microbial Ecology Research and Analysis (CAMERA) project and a variety of other research institutions.
Who should complete a MIGS report?
Authors of genome and metagenome publications should submit a report after submitting project information to DDBJ, EMBL or Genbank.
Is MIGS very time-consuming to complete?
MIGS is a short specification compared with most other ‘-omic’ checklists (see http://mibbi.sf.net) for three reasons:
MIGS is an extension of the data already captured by DBBJ, EMBL and Genbank to describe genomes and metagenomes and is designed to be complementary to these authoritative sources of metadata. The INSDC genome project database will contain essential administrative information, taxonomy identifiers (taxids) and a genome project identifier (PID).
MIGS was intentionally designed to be ‘minimal’ to encourage its adoption.
Genomic sequences, unlike transcriptomes, proteomes or metabolomes, are ‘state independent’ (a genome sequence is stable with respect to cellular state and environmental factors). In contrast, metagenomic experiments depend on the sampling strategy and the specific habitat of a given microbial community, requiring a further specification (MIMS) to define habitat parameters such as salinity, pH and temperature.
How can I get a unique identifier for my submission for use in my publication?
The Genomes Online Database (http://www.genomesonline.org) is the recognized authority for issuing GCat identifiers for eukaryotes, bacteria and archaea and metagenomes. The Genome Catalogue (GCat) will issue identifiers for other genomes.
Can I submit MIGS-compliant information online?
Yes. The GSC has developed a portal called the ‘Genome Catalogue’ that has been useful in prototyping the MIGS specification. MIGS-compliant information can be submitted through user-friendly web forms with drop-down menus for the selection of appropriate terms; batch uploading functions are being developed (http://gensc.sf.net).
Are sample reports available?
Yes, the Genome Catalogue contains a collection of MIGS-compliant reports. Examples are given in Supplementary Table 1.
How would I report the existence of MIGS-compliant data in my publication?
MIGS-compliant information could be reported as a supplementary table in a publication. Far more beneficial to the wider community would be to submit this information to the Genome Catalogue and report the GCat identifier and the URL of this database.
How can I get involved in the GSC and provide feedback for the development of MIGS?
The GSC has an open call for participation. Further information can be found at http://gensc.sf.net.
Since it was originally proposed16, the MIGS specification has been simplified and changed by the GSC through an iterative revision process to contain (i) only curated information that cannot be calculated from raw genomic sequence and (ii) core descriptors specific to the major taxonomic groups (eukaryotes, bacteria and archaea17, plasmids, viruses, organelles) and metagenomes. MIGS is structured as an ‘Investigation’ composed of a ‘Study’ and an ‘Assay’, according to the Reporting Structures for Biological Investigations (RSBI) working group's recommendation for the modularization of checklists18,19. Under ‘Study’ are the top-level concepts ‘Environment’ and ‘Nucleic Acid Sequence’ and under ‘Assay’ is a description of the sequencing technology.
MIGS aims to support unencumbered access to genomic reagents (such as strains)20, place the complete (meta)genome collection into geospatial and temporal context (latitude, longitude, altitude or depth, date and time of sampling) and provide essential details of the experimental method used (e.g., sequencing method). MIGS also provides a framework for the capture of extra information deemed ‘minimum’ to specific communities. Most importantly, the description of metagenomes in MIGS is being extended in the minimum information about a metagenome sequence (MIMS) specification21. MIMS enables the capture of further measurements that define habitat (such as temperature, salinity, pH, dissolved organic carbon) and extends the original structure of MIGS for describing a single (meta)genomic experiment to allow the capture of information from pooled samples and more than one independent sampling event (e.g., sampling along a transect4).
How genomes and metagenomes are described in public databases has evolved from how short, simple DNA sequences are described, without special attention to information such as the geographical origin of the sequence. Significant efforts are underway by the INSDC to adapt and extend the infrastructure for describing genomes through the Genome Project Metadata initiative22. The INSDC efforts are open to evolution, albeit at a conservative pace22, and it is the GSC's hope that much, if not all, of the MIGS specification will be included in the Genome Project Metadata initiative. A mapping between INSDC features and MIGS has been developed for the purpose of placing MIGS information into INSDC documents and is available on our website. Any fields that are not already formally defined by the INSDC Feature Table Document (http://www.insdc.org/files/documents/feature_table.html) can be represented within a structured comment block in INSDC records22.
A genome catalog
The development of any checklist must be an open and iterative process that involves a balanced group of participants. Moreover, mechanisms for achieving compliance are needed to facilitate widespread adoption of a checklist. Such mechanisms involve an appropriate reporting structure for capturing and exchanging data (file formats), software, databases and appropriate controlled vocabularies and/or ontologies for defining the terms used in the annotations. The GSC is working toward these combined goals and has created an online system for capturing MIGS-compliant reports (http://gensc.sf.net).
In brief, we have implemented the checklist as an XML schema and built a freely available Genome Catalogue system (GCat) (http://gensc.sf.net). GCat is designed to generate forms automatically and ‘on the fly’ from this schema for the sake of data input. It also allows users to view and search genome descriptions as they accumulate during the process of refining the MIGS checklist. The GCat system is generic and could be applied to the capture of more expressive metadata for subsets of genomes. Indeed, it is flexible enough to support the implementation of any checklist that can be structured as an appropriate XML schema (MIGS.xsd, being developed into the Genomic Contextual Data Markup Language (GCDML)). The GSC is also working in the area of controlled vocabulary and ontology development through the collation of controlled vocabularies already in use in the community and through contributions to the Ontology for Biomedical Investigations (OBI, previously known as the Functional Genomics Investigation Ontology (FuGO)23) and the Environment Ontology (EnvO) project (http://environmentontology.org). As a part of this process, GCat makes use of existing controlled vocabulary terms and accepts new terms.
Improving genomic databases
By design, MIGS contains only primary, curated information. This is because secondary, or derived, information that can be calculated from a genome sequence is subject to frequent change, can be generated using more than one method and should be acquired directly from those producing the calculations. Still, access to computed information (e.g., in the simplest cases, G+C content or total number of predicted proteins) should be made as easy as possible.
Genomic sequences and their initial annotations must be submitted to the INSDC (http://www.insdc.org/) (and subsequent high-quality, curated annotations derived from empirical observations to the Third Party Annotation data set24), but there are an ever increasing number of genomic databases containing a wide range of additional computations. Although GSC does not endorse any particular method of analysis or database, it supports increased transparency of such resources for the sake of accurate data interpretation and integration.
The first issue is that of exchanging calculated information. This could be facilitated in part by widespread adoption of a common exchange format, such as the Generic Feature Format Version 3 (GFF3) file format (http://song.sourceforge.net/gff3.shtml). There are many tools that support the reformatting of a variety of file types into GFF3, so database providers would find it straightforward to generate appropriate files. The availability of a wide suite of tools for downstream analyses of files in GFF3 format also means that users could combine the weight of evidence from many sources when examining a particular genome. This could reveal instances of systemic bias and therefore lead to better genomic annotations, as more composite features would be available and conflicting annotations could be highlighted for resolution.
Exchanging data also relies on common standards for computational analyses, and supporting data downloads is not enough, regardless of format. Data resources should also be expected, within reason, to provide clear specifications for how the data are generated (for example, standard operating procedures (SOPs) that describe computations such as gene prediction and operon and ortholog identification). One example of this type of documentation is provided in AboutIMG, a web-based description of the Integrated Microbial Genomes (IMG) system25.
In the future it should be far simpler to combine various genomic features, exact details of how they were generated and enough information about the provenance (origin) of the analyses to be able to transparently share data from different sources. Such interoperability, especially when provided by participating databases in a way that would enable automatic harvesting of the data (e.g., through web service technology), would multiply the individual value of these databases many times over and open up new opportunities to examine genome sequences in unprecedented detail.
Future directions
The effort required to achieve the degree of transparency advocated here is considerable but offers substantial and immediate benefits. We argue that the cost of achieving such standardization is trivial compared with the sums spent generating the data. The capture of MIGS-compliant information will not only facilitate comparative genomic and metagenomic analyses but also enhance the available descriptions of downstream ‘-omic’ experiments based on genomic data. It will also enhance the much larger ‘halos’ of 16S ribosomal RNA sequences that are now available for many sequenced genomes and metagenomes. For example, the genome sequence of the marine bacterium Silicibacter pomeroyi26 is ‘embedded’ in a large number of environmental 16S rRNA sequences affiliated with the Roseobacter lineage, which is accompanied by a fairly extensive literature describing the distribution, ecology and other properties of this group27.
Through its ongoing efforts, the GSC hopes to stimulate discussion of the MIGS specification and solicit further feedback from the community. It therefore has an open call for participation and is eager to solicit MIGS-compliant genome reports (including batch uploads) and collect relevant controlled vocabulary terms useful in the description of genomes and metagenomes. GCat identifiers have been implemented and are available for past or future projects, and MIGS-compliant genome reports are starting to become available online (e.g., refs. 28-31). We expect a production version of MIGS (2.0) to be released by early 2008 with an appropriate set of terms formalized within OBI19 and other relevant Open Biomedical Ontology (http://obofoundry.org/) ontologies. We would hope that this milestone (release of MIGS 2.0) will be accompanied by recognition by journals and implementation by a variety of databases. Beyond this, the MIGS specification should still remain flexible enough to allow it to be revised in accordance with advances in technology and our biological knowledge. It should also be considered for use in combination with other checklists in the context of the Minimum Information about a Biomedical or Biological Investigation (MIBBI) Foundry (http://mibbi.sf.net), of which the GSC is a founding community19. The most up-to-date information about GSC activities is available at our website (http://gensc.sf.net).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank the UK National Institute of Environmental eScience (NIEeS) and the European Bioinformatics Institute (EBI) for hosting GSC workshops and the UK Natural Environmental Research Council for providing funds for coordination (NE/D01252X/1) and infrastructure building activities (NE/E007325/1).
Footnotes
Note: Supplementary information is available on the Nature Biotechnology website.
Publisher's Disclaimer: DISCLAIMER
Opinions, findings and conclusions or recommendations expressed in this paper are those of the authors, and do not necessarily reflect the views of the US National Science Foundation.
References
- 1.Overbeek R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liolios K, Mavromatis K, Tavernarakis N, Kyrpides NC. The Genomes On Line Database (GOLD) in 2007: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2008;36(database issue):D475–D479. doi: 10.1093/nar/gkm884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martiny J, Field D. Ecological perspectives on our complete genome collection. Ecology Letters. 2005;8:1334–1345. [Google Scholar]
- 4.Rusch DB, et al. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol. [online] 2007;5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards RA, et al. Using pyrosequencing to shed light on deep mine microbial ecology under extreme hydrogeologic conditions. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Committee on Metagenomics: Challenges and Functional Applications, National Research Council . The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet. National Academies Press; Washington, DC: 2007. [PubMed] [Google Scholar]
- 7.Coenye T, Vandamme P. Bacterial whole-genome sequences: minimal information and strain availability. Microbiology. 2004;150:2017–2018. doi: 10.1099/mic.0.27307-0. [DOI] [PubMed] [Google Scholar]
- 8.Haft DH, Selengut JD, Brinkac LM, Zafar N, White O. Genome properties: a system for the investigation of prokaryotic genetic content for microbiology, genome annotation and comparative genomics. Bioinformatics. 2005;21:293–306. doi: 10.1093/bioinformatics/bti015. [DOI] [PubMed] [Google Scholar]
- 9.Lombardot T, et al. Megx.net—database resources for marine ecological genomics. Nucleic Acids Res. 2006;34(database issue):D390–D393. doi: 10.1093/nar/gkj070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tautz D, Arctander P, Minelli A, Thomas E, Vogler AP. A plea for DNA taxonomy. Trends Ecol. Evol. 2003;18:70–74. [Google Scholar]
- 11.Zhang K, et al. Sequencing genomes from single cells by polymerase cloning. Nat. Biotechnol. 2006;24:680–686. doi: 10.1038/nbt1214. [DOI] [PubMed] [Google Scholar]
- 12.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shendure J, Mitra RD, Varma C, Church GM. Advanced sequencing technologies: methods and goals. Nat. Rev. Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- 14.Garrity GM, editor. Bergey's Manual of Systematic Bacteriology. 2nd edn. Vol. 1. Springer; New York: 2001. [Google Scholar]
- 15.Field D, et al. Meeting report: eGenomics: cataloguing our complete genome collection I. Comp. Funct. Genomics. 2006;6:357–362. doi: 10.1002/cfg.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Field D, Hughes J. Cataloguing our current genome collection. Microbiology. 2005;151:1016–1019. doi: 10.1099/mic.0.27914-0. [DOI] [PubMed] [Google Scholar]
- 17.Pace NR. Time for a change. Nature. 2006;441:289. doi: 10.1038/441289a. [DOI] [PubMed] [Google Scholar]
- 18.Sansone SA, et al. A strategy capitalizing on synergies: the Reporting Structure for Biological Investigation (RSBI) working group. OMICS. 2006;10:164–171. doi: 10.1089/omi.2006.10.164. [DOI] [PubMed] [Google Scholar]
- 19.Taylor C, et al. Promoting coherent minimum reporting requirements for biological and biomedical investigations: the MIBBI project. Nat. Biotechnol. doi: 10.1038/nbt.1411. in the press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward N, Eisen J, Fraser C, Stackebrandt E. Sequenced strains must be saved from extinction. Nature. 2001;414:148. doi: 10.1038/35102737. [DOI] [PubMed] [Google Scholar]
- 21.Field D, et al. Meeting report: eGenomics: cataloguing our complete genome collection III. Comp. Funct. Genomics. 2007;2007:47304. doi: 10.1002/cfg.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrison N, et al. Concept of sample in OMICS technology. OMICS. 2006;10:127–137. doi: 10.1089/omi.2006.10.127. [DOI] [PubMed] [Google Scholar]
- 23.Whetzel PL, et al. Development of FuGO: an ontology for functional genomics investigations. OMICS. 2006;10:199–204. doi: 10.1089/omi.2006.10.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cochrane G, et al. Evidence standards in experimental and inferential INSDC Third Party Annotation data. OMICS. 2006;10:105–113. doi: 10.1089/omi.2006.10.105. [DOI] [PubMed] [Google Scholar]
- 25.Markowitz VM, et al. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 2008;36(database issue):D534–D538. doi: 10.1093/nar/gkm869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moran MA, et al. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature. 2004;432:910–913. doi: 10.1038/nature03170. [DOI] [PubMed] [Google Scholar]
- 27.Buchan A, Gonzalez JM, Moran MA. Overview of the marine roseobacter lineage. Appl. Environ. Microbiol. 2005;71:5665–5677. doi: 10.1128/AEM.71.10.5665-5677.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angly FE, et al. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer M, et al. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ. Microbiol. 2006;8:2201–2213. doi: 10.1111/j.1462-2920.2006.01152.x. [DOI] [PubMed] [Google Scholar]
- 30.Glockner FO, et al. Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc. Natl. Acad. Sci. USA. 2003;100:8298–8303. doi: 10.1073/pnas.1431443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabus R, et al. The genome of Desulfotalea psychrophila, a sulfate-reducing bacterium from permanently cold Arctic sediments. Environ. Microbiol. 2004;6:887–902. doi: 10.1111/j.1462-2920.2004.00665.x. [DOI] [PubMed] [Google Scholar]
- 32.Raes J, Foerstner KU, Bork P. Get the most out of your metagenome: com-
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.