

Abstract
Gene transcription is coordinately regulated by the balance between activation and repression mechanisms in response to various external stimuli. Ferritin, composed of H and L subunits, is the major intracellular iron storage protein involved in iron homeostasis. We previously identified an enhancer, termed antioxidant-responsive element (ARE), in the human ferritin H gene and its respective transcriptional activators including Nrf2 and JunD. Here we found that ATF1 (activating transcription factor 1) is a transcriptional repressor of the ferritin H ARE. Subsequent yeast two-hybrid screening identified PIAS3 (protein inhibitor of activated STAT3) as an ATF1-binding protein. Further investigation of the human ferritin H ARE regulation showed that 1) PIAS3 reversed ATF1-mediated repression of the ferritin H ARE; 2) ATF1 was sumoylated, but PIAS3, a SUMO E3 ligase, did not appear to play a major role in SUMO1-mediated ATF1 sumoylation or ATF1 transcription activating function; 3) PIAS3 decreased ATF1 binding to the ARE; and 4) ATF1 knockdown with siRNA increased ferritin H expression, whereas PIAS3 knockdown decreased basal expression and oxidative stress-mediated induction of ferritin H. These results suggest that PIAS3 antagonizes the repressor function of ATF1, at least in part by blocking its DNA binding, and ultimately activates the ARE. Collectively our results suggest that PIAS3 is a new regulator of ATF1 that regulates the ARE-mediated transcription of the ferritin H gene.
Iron is a vital element that participates in numerous metabolic functions in all organisms; however, excess iron is harmful because it catalyzes formation of hydroxyl radicals by reacting with hydrogen peroxide in the Fenton chemistry (1). Hydroxyl radicals as well as other reactive oxygen species potentially damage cellular components such as proteins, membrane lipids, and nucleic acids, which have been implicated in the etiology of many human diseases and disorders including neurode-generative disease, cancer, and aging (2-4). Therefore, cells must tightly control cellular iron levels under various physiological conditions by storing excess iron in a nontoxic but bio-available form.
Ferritin is the major intracellular iron-binding protein comprised of 24 subunits of the heavy (H) and light (L) chains (5). The importance of ferritin is supported by its ubiquitous expression, highly conserved protein structure (6, 7), and embryonic lethality in ferritin H knockout mice (8). The H sub-unit has ferroxidase activity, which is involved in the oxidation and incorporation of ferrous iron into the ferritin shell, whereas the L subunit plays a role in the formation of the iron core (6, 7). Post-transcriptional regulation of ferritin and the transferrin receptor by iron was elegantly characterized by experiments that showed iron-regulated interactions between trans-acting iron regulatory proteins and iron-responsive elements in these mRNAs (9-11). Ferritin expression is also regulated at the transcriptional level in an iron-independent manner during inflam- mation (12), cell differentiation (13, 14), and oxidative stress (15).
We previously characterized and identified an antioxidant-responsive element (ARE)3 involved in the transcriptional activation of the mouse (16) and human (17) ferritin H genes. The ARE is an enhancer element that contains an AP1 - and/or Mafbinding sequence (18-20), to which Nrf2 (NF-E2-related factor-2) (21) binds and activates transcription of a variety of phase II detoxification genes including glutathione S-transferases, NAD(P)H quinone oxidoreductase 1, and heme oxygenase 1 (22-25). A similar ARE element was also identified in the human ferritin L gene (26). Nrf2, a Cap‘n’Collar basic region leucine zipper (b-Zip) family member, is also important for transcriptional activation of the ferritin H gene (27) and ferritin L gene (28) by chemopreventive agents. In addition, we demonstrated that other b-Zip transcription factors, such as JunD and JunB, are involved in activation of the ferritin H ARE (17, 29, 30). Ferritin up-regulation under these stress conditions renders cells more resistant to the stress-mediated toxicity (31, 32) by reducing the labile iron pool (33), which ultimately limits production of reactive oxygen species (34).
ATF1 is a member of the b-Zip transcription factor family that includes CREB and cAMP-response element modulator (35). These b-Zip transcription factors regulate target genes by forming homodimers or heterodimers within the family, as well as with other b-Zip transcription factors such as Jun and Fos family members, on an AP1-binding sequence or a cAMP response element (35, 36). ATF1 expression is increased in transformed lymphocytes (37) and metastatic melanoma cells (38) and may contribute to growth potential of these tumor cells. ATF1 appears to act as either a transcriptional activator (39- 45) or repressor (46-48) of genes involved in cell proliferation; however, the molecular mechanism controlling the diversity of ATF1 function on target genes remains unknown.
Extensive studies that focused on characterization of transcription factors that directly bind to an ARE identified Nrf2 as the major ARE-activating proteins (22). By contrast, little is known about ARE repressors or involvement of transcriptional coregulators. Interaction of a cytoplasmic inhibitory protein Keap1 with Nrf2 was first reported by Yamamoto and co-workers (49) and is the best characterized mechanism of ARE regulation (22). Recruitment of coactivators, corepressors, and post-translational regulation of transcription factors are critical determinants of expression of target genes. One such regula- tory protein, PIAS3 (protein inhibitor of activated STAT3), was originally identified as a specific inhibitor of the STAT3 signaling pathway (50). A growing body of evidence has shown that the PIAS family (PIAS1, PIAS3, PIASx, and PIASy) regulates a variety of cytokine/growth factor signaling pathways and transcription factors, including NF-κB and SMADs, through protein-protein interactions (51). It has also been demonstrated that the PIAS family has a conserved RING finger-like zinc binding domain that is involved in SUMO-E3-ligase activity and regulates activity of transcription factors such as SMAD4 (52) through sumoylation.
We previously identified ATF1 as a binding protein of the mouse ferritin H ARE (30); however, the role of ATF1 and an ATF1 regulatory protein in ARE enhancer activity remained uncharacterized. In this study, we report that ATF1 is a repressor of the human ferritin H ARE and that PIAS3 is an ATF1-binding protein, which counteracts ATF1 repressor function by diminishing its DNA binding in vivo in a sumoylation-independent manner. These results suggest a new role for PIAS3 in the transcriptional regulation of ferritin and in iron metabolism.
EXPERIMENTAL PROCEDURES
Cell Culture
K562 human erythroleukemia cells, purchased from ATCC, were cultured in RPMI 1640 medium supplemented with 25 mm Hepes, 0.3g/liter l-glutamine, and 10% fetal bovine serum (FBS, 35-010-CV; Mediatech). 293 cells (ATCC) were cultured in minimum essential medium containing Earle’s balanced salt solution, 0.1 mm nonessential amino acids, 1 mm sodium pyruvate, and 2 mm l-glutamine and 10% FBS. These cells were incubated at 37 °C in a humidified 5% CO2 condition.
Yeast Two-hybrid Screening
Yeast two-hybrid screening of a mouse NIH3T3 pAct2 cDNA library (purchased from Clontech) was carried out using a full-length human ATF1 as bait with Saccharomyces cerevisiae strain PJ69-4A (MATa, trp1-901, leu2-3,112, ura3-52, his3-200, gal4Δ, gal80Δ, GAL2- ADE2, LYS2:GAL1-HIS3, met2:GAL7-LacZ) (53). PJ69-4A was transformed with pGBDATF1 (cultured on tryptophan-deficient synthetic drop-out medium), and expression of a GAL4DBD-ATF1 fusion protein was confirmed by Western blotting with anti-ATF1 antibody. A single colony expressing the bait protein was transformed with 20 μg of the NIH3T3 library cDNA by successive treatment of the yeast strain with 0.1 m lithium acetate, 44% polyethylene glycol 3350 for 30 min at 30 °C and 10% Me2SO for 15 min at 42 °C. Yeast cells transformed with the cDNA library were plated on tryptophan/leucine/histidine-deficient synthetic drop-out agar medium containing 2 mm 3-aminotriazole. cDNA plasmids were recovered from 20 positive clones grown on the selection medium by mechanical extraction of plasmid DNA with glass beads (425-600 microns; Sigma) and transformation of Escherichia coli HB101 by electroporation (1300 V in 0.1-cm cuvette, BTX). HB101 transformed with the extracted cDNA were grown on M9/leucine-deficient agar media containing 50 μg/ml ampicillin, followed by isolation of plasmid DNA, characterization with restriction enzymes, and DNA sequencing (SeqWright Co., Houston, TX). Two clones among the positive pool turned out to be identical, and both encoded the C-terminal region of mouse PIAS3.
DNA Transfection, Immunoprecipitation, and Western Blotting
293 cells were transfected with pCMVHA-ATF1, pCMVFLAG-PIAS3, and/or pCMVT7-SUMO1 by electroporation (Bio-Rad X-Cell) using a preoptimized setting developed by Bio-Rad. To detect ATF1-PIAS3 interaction in the cells, cell lysates were immunoprecipitated with anti-FLAG antibody (M2; Sigma) and protein A-agarose, followed by Western blotting with anti-HA antibody (HA11; Covance). For human ferritin H-luciferase reporter assays, cloning of the human ferritin H 5’ upstream enhancer/promoter region and construction of luciferase plasmids were reported previously (17). 0.5-1 μgofa ferritin H luciferase plasmid containing ARE (-4.5 kb) or lacking ARE (-4.4 kb) was transiently transfected into K562 cells (1 × 107 cells/100 μl in a 0.2-cm gap cuvette) by electroporation (Bio-Rad X-Cell) or into 293 cells by JetPEI (according to the manufacturer’s instructions; polyplus transfection) along with ATF1 and/or PIAS3 expression plasmids. As a internal control for transfection, 20 ng of pRL-CMV or pRL-EF (elongation factor promoter) was simultaneously transfected. Transfected K562 cells were then plated at a density of 2-3 × 106 cells/60-mm plate containing 4 ml of the culture medium. After incubation for 36-48 h, the cells were harvested, and luciferase activity was measured using dual luciferase assay reagent (Promega). Firefly luciferase expression driven by the ferritin H gene was normalized to Renilla luciferase expression or, in some experiments, by protein concentration of the cell extracts.
Chromatin Immunoprecipitation (ChIP) Assay
Binding of transfected or endogenous ATF1 to the ferritin H ARE was assessed by ChIP assay as described previously (17). Briefly, 1 × 107 K562 cells were transiently transfected with 1 μg of HA-tagged ATF1 expression plasmid for 40-48 h, followed by cross-linking chromatin with 1% formaldehyde and preparation of cell lysates using the ChIP assay kit (Upstate/Millipore). The cross-linked chromatin-DNA was sonicated with a Sonic Dismembrator (Fisher) for 12 cycles of pulses (10 s) and intervals (20 s). 1/10 aliquots of sonicated DNA were immunoprecipitated with 1 μg of mouse IgG or anti-HA antibody (HA11; Covance), and semiquantitative PCR for the ferritin H ARE or a non-ARE region was performed using a primer set as shown below. Simultaneously 1/50 aliquots of each sonicated DNA were used for quantitative PCR to assess the amount of input DNA. To detect endogenous ATF1 binding to the ferritin H ARE, similar ChIP assays were performed using an ATF1-specific antibody (SC-243x; Santa Cruz Biotechnology). To study the effect of PIAS3 on the binding of ATF1 to the ferritin H ARE, 1 μg of HA-tagged ATF1 expression plasmid was transfected into 1 × 107 K562 cells together with 0, 2, or 9 μg of pCMVFLAGPIAS3 by electroporation, and a HA-ATF1-chromatin complex was immunoprecipitated with anti-HA antibody. Semiquantitative PCR was performed in the presence of 0.1 μCi of [α-32P]dCTP and the following PCR primers specific to the ferritin H ARE or another sequence in the ferritin H gene that does not contain the ARE: ferritin ARE primers: 5’-CCCTCCAGGTCTTATGACTGCTC-3’ and 5’-GTTTCTGGAGGTTCAGCACGTC-3’; ferritin non-ARE primers: 5’-ACATAGTACTTCATTAAACATACAAAT-3’ and 5’-TCTTCTGAGGCTGTCAGAAGC-3’.
Small Interfering RNA (siRNA) Transfection
In ATF1 siRNA experiments, two different transfection methods (electroporation and Lipofectamine 2000) were used in 293 cells. For electroporation, final 20, 100, or 500 nm of siATF1 (J-01004508, sense 5’-GAUCCGAACUACACCUUCAUU-3’, and antisense 5’-UGAAGGUGUAGUUCGGAUCUU-3’; Dharmacon) or nontargeting siControl (D-001210-01, sense 5’-UAGCGACUAAACACAUCAAUU-3’, antisense 5-UUGAUGUGUUUAGUCGCUAUU-3’; Dharmacon) RNA were transfected into 1 × 107 293 cells using Gene Pulser X-Cell (Bio-Rad preoptimized setting for 293 cells) in medium without FBS or antibiotics. After incubation of cells in the cuvette for 5-10 min at room temperature, the cells were suspended in 10 ml of FBS/antibiotic-free OptiMEM (Invitrogen) and incubated for 48 h in a 100-mm dish. For Lipofectamine-mediated transfection, 40, 200, and 400 pmol of siATF1 or siControl RNA (final 40, 200, and 400 nm was mixed with 5 μl of Lipofectamine 2000 (Invitrogen) in 500 μl of FBS/antibiotic-free OptiMEM. The resulting RNA-lipid complexes were incubated for 30 min at room temperature. 2 ml of growth medium (1 × 105 293 cells/well in 6-well plate) was aspirated, and 500 μlof OptiMEM (FBS/antibiotic-free) and 500 μl of the Lipofectamine 2000-siRNA mixture was added to 293 cells and incubated for 48 h. To detect expression of ATF1, ferritin H, and β-actin, 50 μg of whole cell lysates was subjected to Western blotting with anti-ATF1 (sc-243; Santa Cruz Biotechnology), anti-ferritin H (sc-25617; Santa Cruz Biotechnology), and anti-β-actin (A5441; Sigma) antibodies. Total RNA was also isolated using TRIzol (Invitrogen), and 10 μg of RNA was subjected to Northern blotting with 32P-labeled human ferritin H cDNA probe. In PIAS3 siRNA experiments, 100 pmol of two different siRNAs (final concentration, 1 μm) for human PIAS3 (M-004164-05, sense 5’-GGAGCCAAAUGUGAUUAUAUU-3’, and antisense 5’-UAUAAUCACAUUUGGCUCCUU-3’; J-004164-12, sense 5’-GACAGAGAGUCAGCACUAUUU-3’, and antisense 5’-AUAGUGCUGACUCUCUGUCUU-3’; Dharmacon) or siControl RNA (D-001210-01; Dharmacon) was transfected into 1 × 107 K562 cells by electroporation using Gene Pulser X-Cell (Bio-Rad, a preoptimized setting for K562) without FBS or antibiotics. 4 h after transfection, FBS was added to a final 20%, and the cells were incubated for 40 h. The cells were then treated with 50 μm t-BHQ or hemin for 8 h and harvested. The cell lysates were subjected to Western blotting to detect PIAS3, ferritin H, and β-actin expression levels using anti-PIAS3 (AP1245a; Abgent), anti-ferritin H (sc-25617) and anti-β-actin (A5441; Sigma) antibodies. Total RNA was also isolated, and 10 μg of RNA was subjected to Northern blotting for hybridization with 32P-labeled human ferritin H cDNA probe.
RESULTS
ATF1 Binds to the Ferritin H ARE and Represses Its Transcription
To investigate the role of ATF1 in the regulation of the ferritin H ARE, we first performed ChIP assay for ATF1 binding to the ferritin H ARE in the cells. Transfection of HA-ATF1 into K562 cells, followed by ChIP assay, revealed that transfected HA-ATF1 binds to the ARE of the ferritin H gene in the cells (Fig. 1A). We also detected endogenous ATF1 binding to the ferritin H ARE in K562 cells in ChIP assay using ATF1 antibodies (Fig. 1B). These results indicate that ATF1 is a binding protein of the ferritin H ARE.
FIGURE 1. ATF1 is a binding protein to the ferritin H ARE.
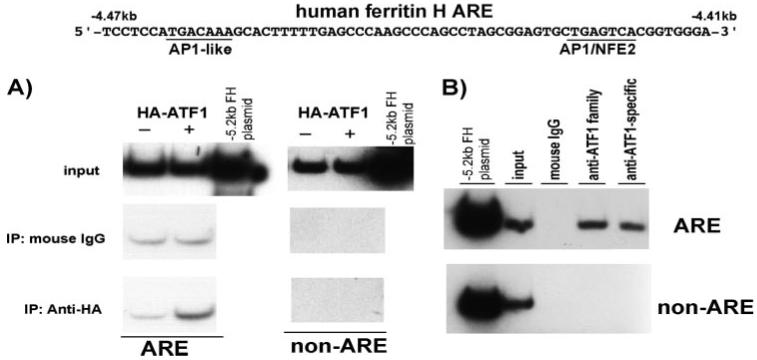
A,1 μg of either pCMVHAATF1 or pCMVHA plasmid was transfected into 1 × 107 K562 cells by electroporation. After 48 h of incubation, chromatin immunoprecipitation (IP) with mouse IgG or anti-HA antibody was performed as described under “Experimental Procedures.” The immunoprecipitates were subjected to quantitative PCR in the presence of [α-32P]dCTP and primer sets for the ferritin H ARE and non-ARE (the primer sequences under “Experimental Procedures”), followed by 8% polyacrylamide gel electrophoresis and autoradiography. 0.5 pg of -5.2-kb ferritin H-luciferase plasmid DNA was used as a template of the PCR positive control and size marker of the PCR-amplified 155-bp DNA for the ferritin H ARE and non-ARE regions. A representative of three independent experiments is shown. B, 1 × 106 K562 cells were subjected to ChIP assay to see the endogenous ATF1 binding to the ferritin H ARE using an anti-ATF1 family or an ATF1-specific antibody. Input DNA as well as ChIP DNA samples were amplified by PCR in the presence of [32P]dCTP, and the amplified 155-bp DNA was visualized by 8% acrylamide gel electrophoresis and autoradiography. A representative of four independent experiments is shown.
We then asked whether ATF1 regulates ferritin H transcription through the ARE. Varied amounts of ATF1 expression plasmid were transfected into K562 cells along with the -4.5-kb human ferritin H luciferase reporter that contains the ARE sequences or the -4.4-kb human ferritin H luciferase reporter that lacks the ARE, and luciferase assays were performed. Unexpectedly, ATF1 repressed expression of the ferritin H luciferase containing the ARE (-4.5-kb ARE (+)), whereas the luciferase construct lacking the ARE (-4.4-kb ARE (-)) showed decreased basal expression and no response to ATF1 (Fig. 2A). To confirm this observation, and to assess the function of endogenous ATF1 on ferritin H expression, we performed knockdown of ATF1 by transfection of siATF1 RNA and examined ferritin H expression by Northern and Western blots. We also attempted treatment with either tert-butylhydroquinone (t-BHQ) or hemin, both of which induced the transcription of the human ferritin H gene through the ARE (17, 56). As shown in Fig. 2 (B and C), ATF1 knockdown increased endogenous ferritin H protein and mRNA expression, both at the steady state and the t-BHQ or hemin-mediated induced levels, suggesting that endogenous ATF1 is a repressor of the ferritin H gene. From these collective results, we concluded that ATF1 is a transcriptional repressor of the human ferritin H gene through the ARE.
FIGURE 2. ATF1 is a transcriptional repressor of the ferritin H ARE.
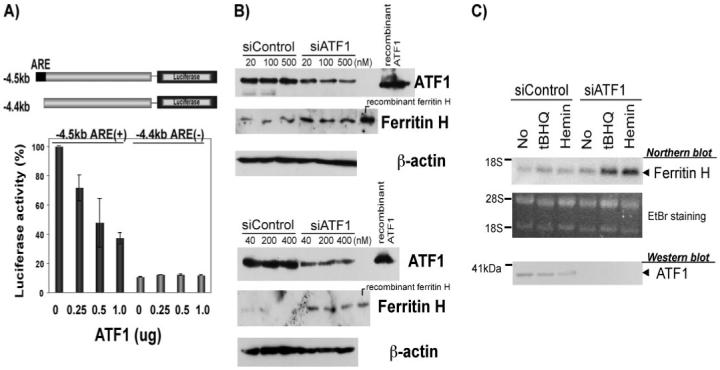
A,1 × 107 K562 cells were electroporated with 1 μg of -4.5-kb (ARE +)or -4.4-kb (ARE-)-ferritin H-luciferase, plus 20 ng of pRL-CMV as an internal control of transfection along with 0.25, 0.5, or 1 μg of ATF1 expression plasmid. Cells were harvested for dual luciferase assay after 36 h of incubation. Expression of firefly luciferase was normalized by that of Renilla luciferase, and the value in -4.5-kb ferritin H-luciferase with no ATF1 was defined as 100%. The results of three independent experiments and standard errors are shown. B, varied amounts (final concentrations are shown) of siRNA Control or siATF1RNA were transfected using electroporation (top) or Lipofectamine 2000 (bottom) into 293 cells and incubated for 48 h. 50 μg of whole cell lysates were subjected to Western blotting with anti-ATF1- and anti-ferritin H-specific antibodies. Recombinant ATF1 and human ferritin H proteins were loaded as positive control of Western blotting. β-Actin Western blots are shown as a loading control. C,5 × 106 K562 cells were transfected with 100 pmol (final, 1 μM) of nontargeting siRNA (siControl) or ATF1siRNA (siATF1). After 48 h of transfection, the cells were treated with 10 μM t-BHQ or 10 μM hemin for 24 h, and total RNA and whole cell lysate were isolated. 5 μg of total RNA was subjected to Northern blotting probed with 32P-labeled ferritin H cDNA. Ethidium bromide staining of total RNA is shown below to verify equal loading and integrity of RNA. The positions of 28 and 18 S ribosomal RNA are indicated. 50 μg of whole cell lysates were used for Western blotting to detect endogenous ATF1 expression levels with anti-ATF1 antibody.
PIAS3 Is an ATF1-binding Protein and Reverses ATF1-mediated Transcriptional Repression of the Ferritin H Gene through the ARE
To investigate ATF1-mediated regulation of the ferritin H ARE, we employed yeast two-hybrid screening to identify ATF1-binding proteins that may regulate ATF1 activity through a protein-protein interaction. We screened a NIH3T3 mouse cDNA library using a full-length human ATF1 as bait and identified a cDNA encoding C-terminal 212 amino acids (amino acids 418-630) of PIAS3 (Fig. 3A). Because PIAS3 is highly conserved in its amino acid sequences between mouse and human (97%) including the C-terminal region isolated from our two-hybrid screening, we investigated the role of human PIAS3 in the regulation of human ATF1 transcription factor throughout the rest of this study. First, we confirmed the interaction between PIAS3 and ATF1 in mammalian cells by coimmunoprecipitation assays. Full-length FLAG-PIAS3 and HA-ATF1 expression plasmids were cotransfected into 293 cells. The cell lysates were subjected to immunoprecipitation with anti-FLAG antibody and Western blotting with anti-HA antibody. Under the condition of equal expression levels of HA-ATF1 and FLAG-PIAS3 (Fig. 3B, middle and bottom panels), HA-ATF1 was coprecipitated only when FLAG-PIAS3 was expressed in the cells, although an unidentified nonspecific band with a slightly larger size than the HA-ATF1 band was also detected in all three lanes (Fig. 3B, top panel). Then we examined the endogenous PIAS3 and ATF1 interaction by immunoprecipitation with anti-PIAS3 antibody followed by Western blotting with the anti-ATF1 family antibody (crossreactive to CREB and cAMP-response element modulator). As shown in Fig. 3C, PIAS3 antibody coimmunoprecipitated endogenous ATF1 as well as CREB, whereas control IgG failed to precipitate these proteins. Collectively, these results indicate that human PIAS3 is an ATF1-binding protein.
FIGURE 3. PIAS3 is an ATF1-binding protein.

A, yeast two-hybrid analysis of ATF1-PIAS3 interaction. PJ69-4A yeast cells were transformed with pGBD and pAct2 (-/-), pGBDATF1 and pAct2 (ATF1/-), pGBDATF1 and pAct2PIAS3 (ATF1/PIAS3), and pGBD and pAct2PIAS3 (-/PIAS3), and transformed colonies were tested for their growth on SD-adenine deficient agar plate. pGBDATF1 is a human full-length ATF1, and pAct2 PIAS3 is a mouse C-terminal PIAS3 isolated from pAct2 NIH3T3 cDNA library. B,1 μg of pCMVHA-hATF1 and 9 μg of pCMVFLAG-hPIAS3 were transfected into 293 cells, and cell lysates were subjected to immunoprecipitation (IP) with anti-FLAG antibody followed by Western blotting (WB) with anti-HA antibody (top panel), or whole cell lysates were subjected to Western blotting with anti-HA antibody (middle panel) or anti-FLAG antibody (bottom panel). pCMVHA-ATF1 transfected cell lysate was simultaneously loaded on the IP/WB gel to identify the HA-ATF1 band. A representative of four independent experiments is shown. C, 800 μg of 293 whole cell lysates were immunoprecipitated with 1 μg of rabbit IgG or anti-PIAS3 antibody, followed by Western blotting with anti-ATF1 family antibody. 50 μg of whole cell lysates were used for Western blots of ATF1/CREB and PIAS3 to confirm the positions of these proteins in the SDS-PAGE gel. A representative of three independent experiments is shown.
To investigate role of PIAS3 on ATF1 function and ferritin H transcription, we cotransfected -4.4-kb ARE(-)- or -4.5-kb ARE(+)-ferritin H-luciferase reporter along with PIAS3 into K562 cells to test whether PIAS3 activates or inhibits the ferritin H ARE. As shown in Fig. 4A, PIAS3 specifically activated -4.5-kb ARE(+) reporter expression. Next, we asked whether PIAS3 modulates the repressive effect of ATF1 on the ferritin H ARE. Transfection of -4.5-kb ARE(+)-ferritin H-luciferase reporter together with ATF1 and varied amounts of PIAS3 into K562 cells showed that ATF1 reproducibly repressed ferritin H ARE-luciferase and that the ATF1-mediated ARE repression was reversed by increased amounts of PIAS3 (Fig. 4B). This reversal effect was not due to decreased expression of ATF1 after cotransfection with PIAS3 (Fig. 4B, bottom panel). We obtained similar results in 293 cells, showing that the reversal effect of ATF1 repression by PIAS3 was dependent on the presence of the ferritin H ARE (Fig. 4C). Thus, we concluded that PIAS3 overrides ATF1-mediated transcriptional repression of the human ferritin H gene through the ARE.
FIGURE 4. PIAS3 reverses ATF1-mediated repression of the human ferritin H transcription via the ARE.
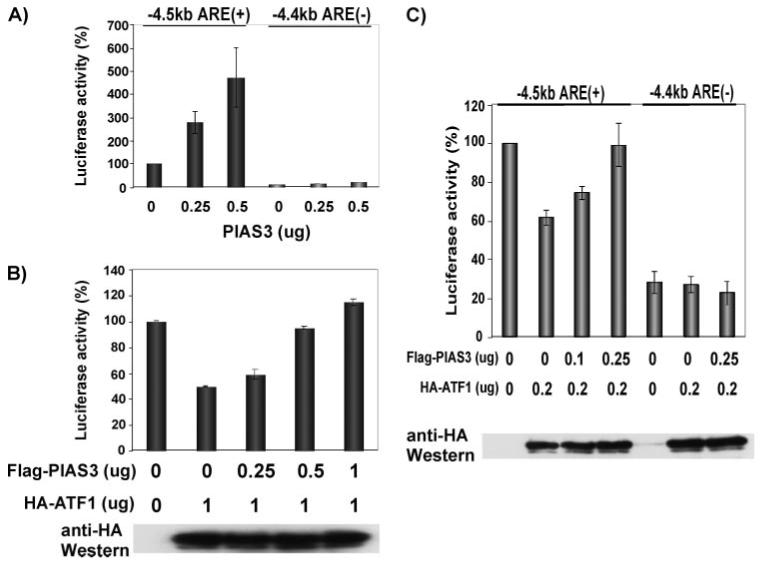
A,1 × 107 K562 cells were electroporated with 0.5 μg of either -4.5-kb (ARE +)or -4.4-kb (ARE-) ferritin H-luciferase plus 0.25 μg or 0.5 μg of pCMVFLAG-PIAS3 (the total amount of plasmid DNA was made up to 1 μg by adding pCMVFLAG empty vector). Cells were harvested after 36 h, and luciferase activity was measured. Expression of firefly luciferase was normalized by that of Renilla luciferase, and the value in -4.5-kb ferritin H-luciferase with no PIAS3 was defined as 100%. The results of three independent experiments and standard errors are shown. B,1 × 107 K562 cells were electroporated with 0.5 μg of -4.5-kb ferritin H-luciferase and 1 μg of pCMV-HAATF1 together with varied amount (0.25, 0.5, and 1 μg) of pCMVFLAG-hPIAS3. The cell lysates were prepared 36 h after transfection and subjected to luciferase assays and Western blotting of HA-ATF1 (bottom). Fold induction of luciferase expression was determined by setting the luciferase reading of no ATF1, no PIAS3 (transfected with the empty vectors) as 100%, and three independent experiments and standard errors are shown. C, 293 cells were transfected with 50 ng of -4.5 (+ARE) or -4.4 (-ARE) ferritin H-luciferase promoter together with indicated amounts of HA-ATF1 and/or FLAG-PIAS3 expression vector using the Jet-PEI transfection reagent. The total amount plasmid DNA in each transfection was equalized to 500 ng by adding pCMV empty vector. 48 h after transfection, the cells were harvested for luciferase assay and Western blotting of HA-ATF1. The luciferase value of the -4.5-kb ARE (+) reporter along with the empty vectors was defined as 100%. The means and standard errors from four independent experiments are shown.
ATF1 Is Sumoylated in a PIAS3-independent Manner
It has recently been reported that CREB is activated by sumoylation at lysine 285 and lysine 304 under hypoxic conditions (54). CREB and ATF1 belong to the b-Zip family of transcription factors with highly conserved amino acids including these two lysine residues; one is Lys215 and another is Lys234 of ATF1 (supplemental Fig. S1B). Because PIAS3 has SUMO E3-ligase activity and modulates functions of various targeted proteins (55), we first tested whether ATF1 is sumoylated at Lys215 or Lys234 and whether PIAS3 is involved in this ATF1 sumoylation. Our results of cotransfection of PIAS3, ATF1, and SUMO1 followed by Western blot analysis suggested that ATF1 can be sumoylated, but PIAS3 does not play a major role in the SUMO1-mediated ATF1 sumoylation at Lys215 and Lys234 (supplemental Fig. S1). In addition, mutations in the two sumoylation sites of ATF1 (K215R, K234R, and K215/234R) did not affect PIAS3 function for the reversal of ATF1-mediated ARE repression (supplemental Fig. S2). To confirm these results, we tested whether the E3-SUMO ligase domain of PIAS3 is important for the reversal effect on the ATF1-mediated ARE repression. PIAS3 1-395, which is composed of the N-terminal 395 amino acids of PIAS3 and contains both the RING finger-like zinc-binding domain and the PINIT motif for the SUMO E3-ligase activity but lacks the C-terminal ATF1 interaction domain (amino acids 418-630 of the mouse PIAS3; Fig. 3A), failed to reverse ATF1-mediated ferritin H repression (Fig. 5). In contrast, PIAS3 395-619, which does not have the SUMO-E3 ligase domain but contains the ATF1 interaction domain, showed a reversal of ATF1-mediated ferritin H repression as efficient as wt PIAS3 (Fig. 5). We therefore concluded that derepression of ATF1 activity on the ferritin H ARE by PIAS3 is not due to sumoylation-mediated activation of ATF1 transcriptional function by PIAS3.
FIGURE 5. SUMO-E3 ligase domain of PIAS3 is not required for the override of ATF1-mediated ferritin H ARE repression.
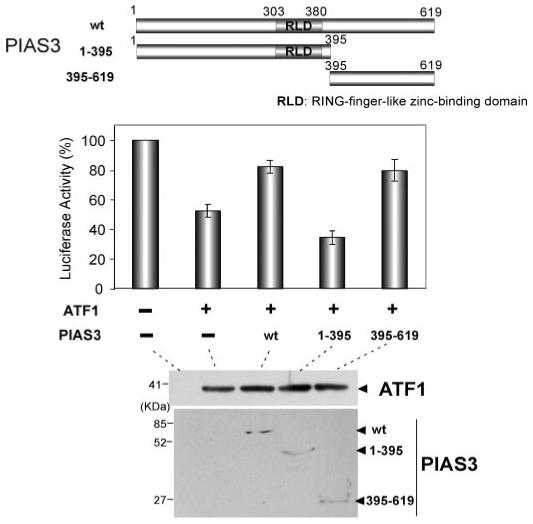
50 ng of -4.5-kb ferritin H-luciferase reporter was transfected into 293 cells together with HA-ATF1 (100 ng) and FLAG-PIAS3 expression plasmid (wt, 250 ng; 1-395, 500 ng; 395-619, 125 ng, in which the varied amounts of wt and mutant PIAS3 plasmids were used to attain comparable expression levels). The total amount plasmid DNA in each transfection was equalized to 650 ng by adding pCMV empty vector. 48 h after transfection, the cells were harvested for luciferase assay. The luciferase value of the -4.5-kb ARE (+) reporter along with the empty vectors was defined as 100%. The means and standard errors from four independent experiments are shown. Comparable expression levels of transfected ATF1 and PIAS3 (wt and mutants) were verified by Western blots (bottom). RLD, RING finger-like zinc-binding domain.
PIAS3 Decreases DNA Binding of ATF1 to the Ferritin H ARE
Because PIAS3 did not enhance ATF1 transcriptional activity, we examined the possibility of inhibition of ATF1 binding to the ferritin H ARE through interaction with PIAS3. To address this, K562 cells were transfected with HA-ATF1 plasmid along with increasing amounts of FLAG-PIAS3 plasmid and detected HA-ATF1 binding to the ferritin H ARE in ChIP assays. Without PIAS3 transfection, specific interaction between HA-ATF1 and the ferritin H ARE was confirmed by increased amounts of the 155-bp PCR product derived from ChIP DNA precipitated with anti-HA antibody, compared with control IgG (Fig. 6). This ATF1-ferritin H ARE interaction was diminished by increased expression of PIAS3 (Fig. 6). PIAS3 expression did not alter ATF1 expression levels (Fig. 6, bottom panels), suggesting that PIAS3 inhibits binding of ATF1 to the ARE. These results also suggest that PIAS3 ultimately serves as an activator of basal ferritin H expression (Fig. 4) at least in part through blocking binding of ATF1 to the ferritin H ARE.
FIGURE 6. PIAS3 blocks ATF1 binding to the ARE in vivo.
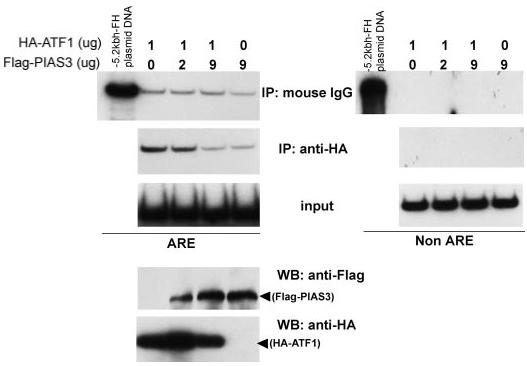
1 × 107 K562 cells were transfected with 1 μg of pCMV-ATF1 together with 0, 2, or 9 μg of pCMVFLAG-PIAS3 for 48 h, and ChIP assay was carried out using antimouse IgG or anti-HA antibody. Input DNA as well as ChIP DNA samples were PCR-amplified in the presence of [32P]dCTP and a set of primer to amplify 155 bp containing the ferritin H ARE (left) or a non-ARE region (right). 0.5 pg of pBluescript-5.2-kb ferritin H-luciferase plasmid DNA was simultaneously amplified in the same PCR condition for a positive control of PCR and a size marker. The samples were subjected to 8% acrylamide gel electrophoresis and autoradiography. The transfected K562 cell lysates were also used for Western blotting (WB) with anti-HA and antiFLAG antibodies to confirm the expression of HA-ATF1 and FLAG-PIAS3, respectively. The experiment was repeated three times, and the representative results are shown. IP, immunoprecipitation.
We previously demonstrated that induction of oxidative stress by either t-BHQ or hemin treatment induces the transcription of the human ferritin H gene through the ARE (17, 56). To investigate the role of endogenous PIAS3 in AREmediated ferritin H gene expression, PIAS3 expression was transiently blocked by transfection of PIAS3 siRNA into K562 cells, and t-BHQ- or hemin-mediated induction of ferritin H was examined by Western and Northern blots. As shown in Fig. 7, PIAS3 knockdown decreased both basal expression and t-BHQ- or hemin-mediated induction of ferritin H mRNA and protein. This is consistent with the fact that an ARE regulates both basal transcription and oxidative stress-mediated activation of ferritin H (56). Taken together with the results in Fig. 6 showing the inhibition of ATF1 binding to the ferritin H ARE by PIAS3, these results suggest that PIAS3 ultimately serves as an activator of the ferritin H expression at least in part by abrogation of the ATF1 repressor function on the ferritin H ARE.
FIGURE 7. PIAS3 knockdown decreases basal expression and oxidative stress-mediated induction of ferritin H.

A,1 × 107 K562 cells were transfected with 100 pmol (final, 1 μm) of nontargeting siRNA (siControl) or PIAS3-targeting siRNA (siPIAS3, M-004164-05; Dharmacon). After 40 h of transfection, the cells were treated with 50 μm t-BHQ or 50 μm hemin for 8 h. 50 μg of each cell lysate was subjected to Western blotting with antibodies for PIAS3, ferritin H, and β-actin. The experiment was repeated three times, and the representative results of Western blots are shown. B,5 × 106 K562 cells were transfected with 100 pmol (final, 1 μm) of no siRNA (no siRNA), nontargeting siRNA (siControl) or PIAS3 siRNA (siPIAS3, J-004164-12; Dharmacon). After 48 h of transfection, the cells were treated with 10 μm t-BHQ or 10 μm hemin for 24 h. 5 μg of total RNA was subjected to Northern blotting probed with 32P-labeled ferritin H cDNA. Ethidium bromide staining of total RNA is shown below to verify equal loading and integrity of RNA. The positions of 28 and 18 S ribosomal RNA are indicated. 50 μg of whole cell lysates were analyzed for expression of PIAS3 and β-actin proteins by Western blotting.
DISCUSSION
Tight regulation of iron homeostasis in erythroid cells is essential not only for supply of sufficient iron during heme synthesis but also for cell defense against oxidative stress (6, 57). Ferritin plays a pivotal role in storage of surplus of intracellular free iron and belongs to a group of antioxidant genes that are transcriptionally regulated through the ARE (16, 17, 26, 56). Studies of transcription factors that activate an ARE demonstrated that Nrf2 is an important transcription factor (22-24). In addition to Nrf2, we and others reported that some other AP1/Maf family members, such as JunD (17) and Nrf1 (58, 59), are transcriptional activators of the ferritin gene and other antioxidant genes that are regulated through the ARE.
In this study, we identified ATF1 as a binding protein to the ferritin H ARE that serves as a transcriptional repressor (Figs. 1 and 2). ATF1 is involved in regulation of cell growth and differentiation through transcriptional activation of hypoxia-inducible genes, such as p21waf1/cip1, enolase, and erythropoietin in cortical neuronal cells (43), CCAAT/enhancer-binding proteins-β and -δ (C/EBP-β and -δ) in preadipocytes (40, 44), egr-1 (the early growth response gene-1) (42), transforming growth factor-β2 (41), histone H4 (60), and a breast cancer susceptibility gene, BRCA1 (39). ATF1 is also one of the transcription factors that induces the type II hexokinase gene associated with high glucose metabolism in many cancer cells (45).
In addition to ATF1-mediated transcriptional activation, ATF1 also represses several genes with tumor-suppressive functions such as retinoblastoma, gelsolin, and thrombospondin-I through a CREB/ATF1-binding site (46-48). Therefore, ATF1 can function as a transcriptional activator or a repressor. The function of ATF1 in regulating target gene expression may depend on the particular target genes regulated by ATF1-binding enhancer sequences, ATF1-binding partners, and phosphorylation status of ATF1; however, the molecular mechanism that determines transcriptional activation or repression by ATF1 remains unknown.
To better understand ATF1-mediated regulation of the ferritin H transcription via the ARE enhancer, we carried out yeast two-hybrid screening and identified PIAS3 as an ATF1-binding protein (Fig. 3). Proposed or demonstrated mechanisms of PIAS-mediated transcriptional regulation are: 1) PIAS represses or activates transcription of target genes by sumoylation of transcription factors (55); 2) PIAS blocks DNA binding ability of transcription factors through protein-protein interactions (50); and 3) PIAS proteins recruit transcriptional coregulators, such as histone deacetylases (61) or p300/CREB-binding protein (62).
Our results in this study suggested that PIAS3-mediated activation of the human ferritin H gene via the ARE was not due to PIAS3-mediated ATF1 sumoylation of Lys215 and Lys234 (supplemental Figs. S1 and S2) but rather due to PIAS3-mediated blockade of ATF1 binding to the ferritin H ARE in cells (Fig. 6). To test whether PIAS3 inhibits ATF1 binding to the ferritin H ARE in vitro, we performed gel shift assays using purified recombinant ATF1 (His-tagged ATF1 or GSTATF1) and recombinant PIAS3 (His-tagged PIAS3). Our repeated experiments showed that recombinant PIAS3 did not inhibit ATF1 binding to the ferritin H ARE in vitro (data not shown); presumably, the ATF1-PIAS3 complex might be very unstable in in vitro binding assay conditions.
PIAS3-mediated regulation of ATF1 may be similar or related to PIASx-mediated inhibition of Stat4 or PIASy-mediated inhibition of Stat1, in which these PIAS proteins inhibit Stat1 and Stat4 transcription factors but do not inhibit in vitro DNA binding activity in gel retardation assays (63, 64). Our ChIP assay results showing decreased ATF1 binding to the ferritin H ARE by PIAS3 (Fig. 6) suggest that, for instance, PIAS3 may sequester ATF1 prior to the ferritin H ARE binding, or PIAS3 may need an additional cofactor(s) to inhibit ATF1 binding to the ferritin H ARE in vivo. Because ATF1 is a repressor of the ferritin H ARE (Fig. 2), sequestering ATF1 from the ARE by PIAS3 may ultimately allow other activators, such as Nrf2 and JunD (17, 56), to bind and activate the ARE. It is also likely that PIAS3 may have a direct activation function with regard to the ARE through chromatin remodeling. It has recently been shown that PIAS family members alleviate chromatin compaction, leading to transcriptional activation of target genes (65, 66). Because PIAS3 has a zinc finger-like motif in its sequence (51), we also tested whether PIAS3 directly binds to the ferritin H ARE sequence by gel mobility shift assays. However, we did not observe a retarded PIAS3-ferritin H ARE band (data not shown). These results suggest that the competition of the ferritin H ARE binding between PIAS3 and ATF1 does not appear to be the mechanism by which PIAS3 overrides ATF1-mediated transcriptional repression of the ferritin H ARE.
Currently, we do not have evidence showing how ATF1 represses transcription of the ferritin H ARE. However, in our yeast two-hybrid screening with ATF1 as bait, we isolated several uncharacterized cDNA clones in addition to PIAS3, some of which appear to show transcriptional corepressor activities in our preliminary luciferase reporter experiments.4 Further investigation will be necessary to delineate the molecular mechanism by which ATF1 represses ARE enhancer activity.
What is the biological significance of ATF1-mediated ferritin H repression? ATF1 is overexpressed in many transformed lymphocytes (37) and in metastatic melanoma cells (67). In addition, a gene fusion between EWS (Ewing’s sarcoma protein) and ATF1 by t(12;22) chromosomal translocation was found in clear cell sarcoma (68). The EWS/ATF1 chimeric transcription factor plays a role in maintaining tumor cell viability because anti-ATF1 single chain antibody inhibiting the EWS/ATF reduced viability of clear cell sarcoma (69) and also suppressed tumorigenicity and metastatipotential of melanoma cells (38). According to the results presented in this study, we speculate that some of these cancer cells with increased ATF1 activity may have decreased expression of the ferritin H gene through the ARE-mediated transcriptional repression. This appears to be advantageous to the tumor cell growth because several lines of evidence indicated that down-regulation of ferritin H increases the labile iron pool in K562 erythroleukemic cells and other cancer cells, which allows enhanced cell growth and tumorigenicity (70, 71). This is also consistent with our previous finding that NIH3T3 mouse fibroblasts transformed with the adenovirus E1A oncogene showed transcriptional repression of the ferritin H gene (29, 72).
In summary, ATF1 is a transcriptional repressor of the human ferritin H gene through the ARE. PIAS3 binds ATF1, but did not play a major role in SUMO1-mediated ATF1 sumoylation or induction of ATF1 transcription activity. PIAS3-mediated sequestration of ATF1 from the ARE appears to be the mechanism by which ferritin H expression is up-regulated. Our study proposes that ATF1 and PIAS3 are new regulatory proteins of the ferritin ARE and perhaps other phase II detoxification genes regulated by AREs.
Supplementary Material
Acknowledgements
We are grateful to Dr. Satoshi Kishida for superb technical advice and help in the yeast two-hybrid assay. We thank Dr. Elizabeth L. MacKenzie for reading this manuscript thoroughly.
Footnotes
- ARE
- antioxidant-responsive element
- CREB
- cAMP response element-binding protein
- STAT
- signal transducers and activators of transcription
- SUMO
- small ubiquitin-like modifier
- FBS
- fetal bovine serum
- ChIP
- chromatin immunoprecipitation
- HA
- hemagglutinin
- siRNA
- small interfering RNA
- t-BHQ
- tert-butylhydroquinone
- wt
- wild type
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
This work was supported in part by National Institutes of Health Grant DK-60007 (to Y. T.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Papanikolaou G, Pantopoulos K. Toxicol. Appl. Pharmacol. 2005;202:199–211. doi: 10.1016/j.taap.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 2.Andersen JK. Nat. Med. 2004;10(suppl):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 3.Hussain SP, Hofseth LJ, Harris CC. Nat. Rev. Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 4.Balaban RS, Nemoto S, Finkel T. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Harrison PM, Arosio P. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 6.Arosio P, Levi S. Free Radic. Biol. Med. 2002;33:457–463. doi: 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 7.Theil EC. J. Nutr. 2003;133:1549S–1553S. doi: 10.1093/jn/133.5.1549S. [DOI] [PubMed] [Google Scholar]
- 8.Ferreira C, Santambrogio P, Martin ME, Andrieu V, Feldmann G, Henin D, Beaumont C. Blood. 2001;98:525–532. doi: 10.1182/blood.v98.3.525. [DOI] [PubMed] [Google Scholar]
- 9.Klausner RD, Rouault TA, Harford JB. Cell. 1993;72:19–28. doi: 10.1016/0092-8674(93)90046-s. [DOI] [PubMed] [Google Scholar]
- 10.Hentze MW, Kuhn LC. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hentze MW, Muckenthaler MU, Andrews NC. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 12.Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Proc. Natl. Acad. Sci., U. S. A. 1991;88:4946–4950. doi: 10.1073/pnas.88.11.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marziali G, Perrotti E, Ilari R, Testa U, Coccia EM, Battistini A. Mol. Cell. Biol. 1997;17:1387–1395. doi: 10.1128/mcb.17.3.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beaumont C, Seyhan A, Yachou A-K, Grandchamp B, Jones R. J. Biol. Chem. 1994;269:20281–20288. [PubMed] [Google Scholar]
- 15.Torti FM, Torti SV. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 16.Tsuji Y, Ayaki H, Whitman SP, Morrow CS, Torti SV, Torti FM. Mol. Cell. Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsuji Y. Oncogene. 2005;24:7567–7578. doi: 10.1038/sj.onc.1208901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rushmore TH, Morton MR, Pickett CB. J. Biol. Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 19.Prestera T, Holtzclaw WD, Zhang Y, Talalay P. Proc. Natl. Acad. Sci., U. S. A. 1993;90:2965–2969. doi: 10.1073/pnas.90.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wasserman WW, Fahl WE. Proc. Natl. Acad. Sci. U. S. A. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moi P, Chan K, Asunis I, Cao A, Kan YW. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Motohashi H, Yamamoto M. Trends Mol. Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Li J, Lee JM, Johnson DA, Johnson JA. Methods Enzymol. 2004;378:238–258. doi: 10.1016/S0076-6879(04)78019-2. [DOI] [PubMed] [Google Scholar]
- 24.Jaiswal AK. Methods Enzymol. 2004;378:221–238. doi: 10.1016/S0076-6879(04)78018-0. [DOI] [PubMed] [Google Scholar]
- 25.Alam J, Camhi S, Choi AMK. J. Biol. Chem. 1995;270:11977–11984. doi: 10.1074/jbc.270.20.11977. [DOI] [PubMed] [Google Scholar]
- 26.Hintze KJ, Theil EC. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15048–15052. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietsch EC, Chan JY, Torti FM, Torti SV. J. Biol. Chem. 2003;278:2361–2369. doi: 10.1074/jbc.M210664200. [DOI] [PubMed] [Google Scholar]
- 28.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 29.Tsuji Y, Akebi N, Lam TK, Nakabeppu Y, Torti SV, Torti FM. Mol. Cell. Biol. 1995;15:5152–5164. doi: 10.1128/mcb.15.9.5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsuji Y, Torti SV, Torti FM. J. Biol. Chem. 1998;273:2984–2992. doi: 10.1074/jbc.273.5.2984. [DOI] [PubMed] [Google Scholar]
- 31.Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P. J. Biol. Chem. 2000;275:25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 32.Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Biochem. J. 2001;357:241–247. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picard V, Epsztejn S, Santambrogio P, Cabantchik ZI, Beaumont C. J. Biol. Chem. 1998;273:15382–15386. doi: 10.1074/jbc.273.25.15382. [DOI] [PubMed] [Google Scholar]
- 34.Epsztejn S, Glickstein H, Picard V, Slotki IN, Breuer W, Beaumont C, Cabantchik ZI. Blood. 1999;94:3593–3603. [PubMed] [Google Scholar]
- 35.Mayr B, Montminy M. Nat. Rev. Mol. Cell. Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 36.Hai T, Curran T. Proc. Natl. Acad. Sci. U. S. A. 1991;88:3720–3724. doi: 10.1073/pnas.88.9.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsueh YP, Lai MZ. J. Immunol. 1995;154:5675–5683. [PubMed] [Google Scholar]
- 38.Jean D, Tellez C, Huang S, Davis DW, Bruns CJ, McConkey DJ, Hinrichs SH, Bar-Eli M. Oncogene. 2000;19:2721–2730. doi: 10.1038/sj.onc.1203569. [DOI] [PubMed] [Google Scholar]
- 39.Atlas E, Stramwasser M, Mueller CR. Oncogene. 2001;20:7110–7114. doi: 10.1038/sj.onc.1204890. [DOI] [PubMed] [Google Scholar]
- 40.Belmonte N, Phillips BW, Massiera F, Villageois P, Wdziekonski B, Saint-Marc P, Nichols J, Aubert J, Saeki K, Yuo A, Narumiya S, Ailhaud G, Dani C. Mol. Endocrinol. 2001;15:2037–2049. doi: 10.1210/mend.15.11.0721. [DOI] [PubMed] [Google Scholar]
- 41.Kingsley-Kallesen ML, Kelly D, Rizzino A. J. Biol. Chem. 1999;274:34020–34028. doi: 10.1074/jbc.274.48.34020. [DOI] [PubMed] [Google Scholar]
- 42.Rolli M, Kotlyarov A, Sakamoto KM, Gaestel M, Neininger A. J. Biol. Chem. 1999;274:19559–19564. doi: 10.1074/jbc.274.28.19559. [DOI] [PubMed] [Google Scholar]
- 43.Zaman K, Ryu H, Hall D, O’Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. J. Neurosci. 1999;19:9821–9830. doi: 10.1523/JNEUROSCI.19-22-09821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang JW, Klemm DJ, Vinson C, Lane MD. J. Biol. Chem. 2004;279:4471–4478. doi: 10.1074/jbc.M311327200. [DOI] [PubMed] [Google Scholar]
- 45.Lee MG, Pedersen PL. J. Biol. Chem. 2003;278:41047–41058. doi: 10.1074/jbc.M307031200. [DOI] [PubMed] [Google Scholar]
- 46.Dong Y, Asch HL, Ying A, Asch BB. Exp. Cell Res. 2002;276:328–336. doi: 10.1006/excr.2002.5534. [DOI] [PubMed] [Google Scholar]
- 47.Salnikow K, Wang S, Costa M. Cancer Res. 1997;57:5060–5066. [PubMed] [Google Scholar]
- 48.Okuyama Y, Sowa Y, Fujita T, Mizuno T, Nomura H, Nikaido T, Endo T, Sakai T. FEBS Lett. 1996;397:219–224. doi: 10.1016/s0014-5793(96)01178-7. [DOI] [PubMed] [Google Scholar]
- 49.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 51.Shuai K, Liu B. Nat. Rev. Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 52.Ohshima T, Shimotohno K. J. Biol. Chem. 2003;278:50833–50842. doi: 10.1074/jbc.M307533200. [DOI] [PubMed] [Google Scholar]
- 53.James P, Halladay J, Craig EA. Genetics. 1996;144:1425–1436. doi: 10.1093/genetics/144.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Comerford KM, Leonard MO, Karhausen J, Carey R, Colgan SP, Taylor CT. Proc. Natl. Acad. Sci. U. S. A. 2003;100:986–991. doi: 10.1073/pnas.0337412100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kotaja N, Karvonen U, Janne OA, Palvimo JJ. Mol. Cell. Biol. 2002;22:5222–5234. doi: 10.1128/MCB.22.14.5222-5234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwasaki K, Mackenzie EL, Hailemariam K, Sakamoto K, Tsuji Y. Mol. Cell. Biol. 2006;26:2845–2856. doi: 10.1128/MCB.26.7.2845-2856.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ganz T, Nemeth E. Biochim Biophys Acta. 2006;1763:690–699. doi: 10.1016/j.bbamcr.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 58.Venugopal R, Jaiswal AK. Proc. Natl. Acad. Sci., U. S. A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Venugopal R, Jaiswal AK. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 60.Guo B, Stein JL, van Wijnen AJ, Stein GS. Biochemistry. 1997;36:14447–14455. doi: 10.1021/bi971781s. [DOI] [PubMed] [Google Scholar]
- 61.Long J, Matsuura I, He D, Wang G, Shuai K, Liu F. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9791–9796. doi: 10.1073/pnas.1733973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Long J, Wang G, Matsuura I, He D, Liu F. Proc. Natl. Acad. Sci. U. S. A. 2004;101:99–104. doi: 10.1073/pnas.0307598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu B, Gross M, ten Hoeve J, Shuai K. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3203–3207. doi: 10.1073/pnas.051489598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arora T, Liu B, He H, Kim J, Murphy TL, Murphy KM, Modlin RL, Shuai K. J. Biol. Chem. 2003;278:21327–21330. doi: 10.1074/jbc.C300119200. [DOI] [PubMed] [Google Scholar]
- 65.Lyst MJ, Nan X, Stancheva I. EMBO J. 2006;25:5317–5328. doi: 10.1038/sj.emboj.7601404. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Huang CY, Beliakoff J, Li X, Lee J, Sharma M, Lim B, Sun Z. Mol. Endocrinol. 2005;19:2915–2929. doi: 10.1210/me.2005-0097. [DOI] [PubMed] [Google Scholar]
- 67.Chenais B, Andriollo M, Guiraud P, Belhoussine R, Jeannesson P. Free Radic. Biol. Med. 2000;28:18–27. doi: 10.1016/s0891-5849(99)00195-1. [DOI] [PubMed] [Google Scholar]
- 68.Zucman J, Delattre O, Desmaze C, Epstein AL, Stenman G, Speleman F, Fletchers CD, Aurias A, Thomas G. Nat. Genet. 1993;4:341–345. doi: 10.1038/ng0893-341. [DOI] [PubMed] [Google Scholar]
- 69.Bosilevac JM, Olsen RJ, Bridge JA, Hinrichs SH. J. Biol. Chem. 1999;274:34811–34818. doi: 10.1074/jbc.274.49.34811. [DOI] [PubMed] [Google Scholar]
- 70.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Biochem. J. 2001;356:311–316. doi: 10.1042/0264-6021:3560311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kakhlon O, Gruenbaum Y, Cabantchik ZI. Biochem. J. 2002;363:431–436. doi: 10.1042/0264-6021:3630431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsuji Y, Kwak E, Saika T, Torti SV, Torti FM. J. Biol. Chem. 1993;268:7270–7275. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.