

Abstract
To study the pathogenesis of central nervous system abnormalities in Down syndrome (DS), we have analyzed a new genetic model of DS, the partial trisomy 16 (Ts65Dn) mouse. Ts65Dn mice have an extra copy of the distal aspect of mouse chromosome 16, a segment homologous to human chromosome 21 that contains much of the genetic material responsible for the DS phenotype. Ts65Dn mice show developmental delay during the postnatal period as well as abnormal behaviors in both young and adult animals that may be analogous to mental retardation. Though the Ts65Dn brain is normal on gross examination, there is age-related degeneration of septohippocampal cholinergic neurons and astrocytic hypertrophy, markers of the Alzheimer disease pathology that is present in elderly DS individuals. These findings suggest that Ts65Dn mice may be used to study certain developmental and degenerative abnormalities in the DS brain.
Down syndrome (DS) or trisomy 21 is the most frequent genetic cause of mental retardation with a prevalence of 1:800 live births (1, 2). Abnormal central nervous system (CNS) function is universal in individuals with DS and is noted during development and adult life (3). Hypotonia in the newborn period is followed by developmental delay and mental retardation. In addition, all individuals with DS demonstrate the neuropathology of Alzheimer disease (AD) by age 30–40 years (4). To explore pathogenetic mechanisms in DS, investigators have produced and characterized animal models. Until recently, trisomy 16 (Ts16) mice served as the best model of DS. Mouse chromosome 16 (MMU-16) is the chromosome most homologous to human chromosome 21 (HSA-21), and fetal Ts16 mice demonstrate both systemic and neurological phenotypes similar to those found in DS (5). However, there are two important limitations in examining the Ts16 model. (i) Ts16 is not a perfect genetic model for trisomy of HSA-21. MMU-16 is larger than HSA-21 and contains genes located on HSA-21 as well as other human chromosomes (3). (ii) Ts16 mice do not survive birth (3). The latter prevents analysis of many features of brain development and maturation. Because of these limitations, attempts have been made to develop new genetic models in which only the region syntenic between MMU-16 and HSA-21 is triplicated. Candidate animal models for DS should meet two criteria: (i) subjects possess three copies of all or most of the genetic material present on HSA-21 responsible for the DS phenotype but not genetic material outside this region and (ii) animals survive into adulthood.
The “DS region” of HSA-21 has been defined as the subset of genes that when present in three copies results in the major phenotypic features of DS, including the characteristic facial appearance, congenital heart disease, and mental retardation (6). Interestingly, though most DS cases are caused by the presence of an entire extra copy of HSA-21, a small number have a “partial trisomy,” resulting from an unbalanced translocation or partial duplication of the long arm of HSA-21. The critical DS region appears to reside within the q22 region (6). The distal end of MMU-16 is genetically homologous to the q22 region of HSA-21; at least 14 defined genes have been localized on both the human and mouse chromosomes in this region (7).
Segmentally trisomic mice, Ts(1716)65Dn [herein referred to as Ts65Dn (8)] appear to fulfill the criteria for a candidate DS model. These mice are trisomic for a segment of MMU-16 that is homologous to HSA-21; the segment includes material just proximal to App extending to Mx (7). Thus much of the distal end of MMU-16, encoding most of the MMU-16 segment shared with the long arm of HSA-21 in the q22 region, is translocated to <10% of the centromeric end of MMU-17 to form a small translocation chromosome (8). Importantly, these mice survive into adulthood. Reports regarding Ts65Dn mice have documented behavioral abnormalities in adult animals (7, 9, 10). However, there are no reports documenting the developmental and age-related features expected for the CNS DS phenotype.
To characterize the nervous system of Ts65Dn mice, we have carried out detailed behavioral, morphological, and molecular analyses in both developing and mature subjects. We discovered similarities and differences with respect to DS. Like DS, Ts65Dn animals have (i) abnormal behavior during early postnatal development as well as in adulthood, (ii) normal brain weight in the early postnatal period, (iii) initial normal development of basal forebrain cholinergic neurons (BFCNs), (iv) marked loss of BFCNs with age, (v) astrocytic hypertrophy in the adult, and (vi) abnormal expression of genes both on and off the trisomic segment. Unlike aged DS subjects, there was no evidence of β-amyloid (Aβ)-containing plaques or neurofibrillary tangles in older mice. These findings suggest that Ts65Dn mice may be used to study the pathogenesis of both developmental and neurodegenerative CNS phenotypes in DS.
MATERIALS AND METHODS
Animals.
All mice were maintained on a B6C3HF1 outbred background by mating Ts65Dn female mice (obtained from M. Davisson, Jackson Laboratory; ref. 8) with B6C3HF1 male mice (Jackson Laboratory). Lymphocytes were karyotyped from all animals and one to three Ts65Dn mice were matched and housed with 2n (diploid) littermates of the same sex. Many of the behavioral tests assessed in our analysis require intact vision. While the outbreeding paradigm maintains the mice on a variable genetic background, a recessive gene, rd, is also inherited. The presence of this mutation in the homozygous state results in retinal degeneration and blindness (11). Therefore, we histologically examined the retina of each animal at the completion of our behavioral studies. In all cases, ≈25% of both Ts65Dn and 2n mice had retinal degeneration. Behavioral results from these animals were eliminated from our analyses. Investigators carrying out behavioral as well as histological studies and analysis were blind to animal genotype.
Behavioral Testing.
Neurobehavioral development on postnatal days (PD) 1–23 was assessed by daily testing of the pups according to the procedure of Fox (12) and was supplemented by a tactile stimulation test and vertical screen test as described (13, 14). The homing test (on PD12) and ultrasonic vocalization test were performed as described (13, 14). Pups were weighed daily, and measures of somatic growth included eyelid and ear opening, incisor eruption, and body and tail length. Open field testing was performed as described (15). Passive avoidance apparatus and testing were similar to that described (15). Adult animals were tested in the Morris water maze adapted for mice (16). For statistical analysis of behavior, an ANOVA for repeated measures was used for processing behavioral data rearranged in blocks of repeated trials. Post hoc comparisons were performed using paired t tests with Bonferroni’s correction. Nonparametric data were analyzed using the Kruskal–Wallis test.
Tissue Processing, Immunocytochemistry, and Northern Blot Analysis.
Tissue processing and immunocytochemistry were performed exactly as described (17). The antibodies utilized for immunostaining were as follows: REX (anti-p75NGFR) (18), RTA (anti-trkA) (19), anti-glial fibrillary acidic protein (GFAP; Chemicon), 1280 (anti-Aβ, a gift of D. Selkoe, Boston), and Alz-50 (gift of P. Davies, Bronx, NY). RNA preparation, Northern blot analysis, and densitometry were performed as described (17).
Assessment of Brain Region Volumes, Cell Number, and Profile Area.
The volumes of brain regions were measured in an unbiased manner using Cavalieri’s method (20). Every third coronal section throughout each brain was stained with Cresyl violet and visualized with an MCID image analysis system (Imaging Research, St. Catherine’s, ON, Canada) linked to a charge-coupled device (CCD) camera with a 35-mm lens. In 20-month-old mice, hippocampus and cortex were measured from their incipience rostrally to the end of the interpeduncular nucleus caudally. All other structures at all ages were measured in their entirety. Cell numbers were estimated in an unbiased manner using the optical disector method (21, 22) in combination with the Cavalieri method for estimating reference volume (20). The first section in each series was chosen randomly, followed by every third section thereafter, with a total of 7–10 sections per structure. Cells were sampled using a disector frame taped to the monitor screen; cells were counted if they contained a nucleolus that fell within the dissector frame under a ×100 objective (n.a., 1.32). The cross-sectional area of each counted cellular profile was then measured as described (19). The movement of the microscope stage in the x, y, and z planes was accomplished manually. Within-individual variance (coefficient of error, CE) was calculated as described (22). Between-group analysis of age-matched samples was carried out using Student’s t test.
RESULTS
Brain Growth and Gross Morphology in Ts65Dn Mice.
The brain of adult Ts65Dn mice has been reported to be grossly normal (7, 8). We confirmed this for both the developing and adult brain and showed that brain weight in Ts65Dn did not differ from those of 2n subjects at PD10 and 3 months. To determine whether there were regional differences, we quantitated the size of the hippocampus, cortex, striatum, and cerebellum at PD2, PD10, PD22, 20 months, and in adults. No significant differences were noted in the volume of these brain regions between Ts65Dn and 2n mice. No abnormalities of neuronal migration were noted in the cortex of Ts65Dn mice. Thus, gross structural aspects of brain growth and development appeared to be unaffected by the segmental trisomy.
Behavioral Characterization of Ts65Dn Mice.
In DS, developmental and somatic growth delay as well as mental retardation are nearly universal (1, 2). We performed a battery of behavioral tests during both the postnatal and adult periods to determine if analogous abnormalities could be detected in Ts65Dn animals. First, somatic and sensorimotor development of pups was assessed from PD1 through PD23. There was delay in the achievement of certain physical and most sensorimotor milestones. Body weight gain was slower in Ts65Dn pups [F(16,256) = 3.57; P < 0.01)]. In contrast, no gross differences were found for the time of eyelid and ear opening or incisor eruption. To evaluate sensorimotor development, a wide range of reflexes were assessed as were more specific behavioral endpoints such as (i) the pattern of ultrasonic vocalization, (ii) performance on a homing test, and (iii) learning performance and locomotor activity in an open field arena (12–14). In almost all reflexes the emergence of adult-like responses was delayed in Ts65Dn pups (Fig. 1A). Differences were statistically significant for cliff aversion, swift righting, screen climbing, vibrissa placing, and ear twitch response [χ2(1) = 5.48, 7.36, 5.58, 5.31, 4.98, respectively; P < 0.05]. Despite delays, both Ts65Dn and 2n mice reached neurobehavioral maturation for these responses near the time for weaning. Ultrasonic vocalization, a communicative behavior aimed at eliciting maternal care (13, 14), was delayed by ≈4 days; its peak expression was observed at PD9 in Ts65Dn mice (Fig. 1C). Ts65Dn mice also demonstrated more scattered locomotion in association with ultrasonic vocalization with a 20% greater number of “stop and go” events [F(1,16) = 5.35); P < 0.05]. Ts65Dn pups were also quite abnormal in homing (Fig. 1B). This could reflect poor motor coordination or an inability to detect olfactory signals. Almost identical to findings in adults (see below), Ts65Dn pups showed a significant increase in locomotor activity (frequency of crossings and wall rearing) at PD21 (Fig. 2A). However, not all behaviors were abnormal. At PD18, learning performance in the passive avoidance task revealed no significant difference between Ts65Dn and 2n mice [F(1,26) = 0.341; P = 0.56].
Figure 1.
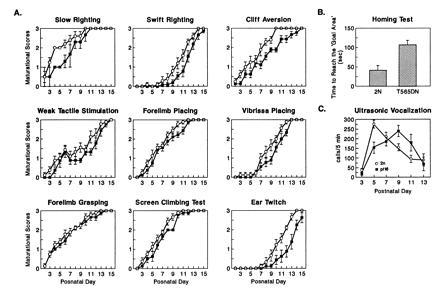
Neurobehavioral data for developing Ts65Dn and 2n mice. In A, sensorimotor reflexes (12) were assessed. Pups were scored on a scale from 0 to 3 with a score of 3 representing an adult-like response. In B, the homing test and the vocalization test were performed (13, 14). In the homing test, the time for each pup to reach the goal area was recorded (cut off time 3 min). (2n, n = 9; Ts65Dn, n = 9).
Figure 2.
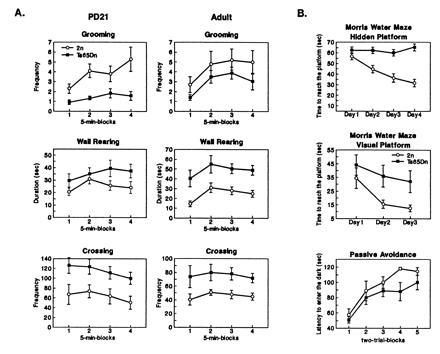
Behavioral data for adult Ts65Dn and 2n mice. (A) Results of open field testing. The following responses were measured: crossing (scored by dividing the floor of the arena in squares and counting the crossing of square limits with both forepaws), wall rearing (alternating forelimb placing movements on the wall), and grooming (wiping, licking, combing, or scratching of any part of the body). (B) Morris water maze. On each day of testing, mice were evaluated in four consecutive trials in the visible platform task or six consecutive trials in the hidden platform task. Different groups of animals were tested in hidden and visible tasks. There was a 10-sec intertrial interval and a 90-sec maximum swim latency per trial. Data collection was automated by a computerized video tracking system (San Diego Instruments). The testing apparatus for passive avoidance was similar to that described (15). The procedure consisted of two phases: acquisition occurred on day 1 and retention occurred on day 2. In the acquistion phase, mice underwent a multitrial session. The sliding door between compartments was raised and the mouse was allowed to cross into a dark chamber. When the mouse crossed thus lowering the tilting door, the door shut, and a 3-sec 0.3-mA footshock was delivered to the grid floor. A trial ended when the mouse gave the step-through response or remained in the start compartment for 120 sec. The acquisition phase ended when the subject either remained in the start compartment for 2 consecutive trials or after 10 trials by stepping through. For the retention phase, the procedure consisted of one trial not punished by footshock. The trial ended when the mouse gave the step-through response or remained in the start compartment for 120 sec.
Ts65Dn animals were also assessed on behavioral tasks at 6–8 months of age. In open field testing, several differences were noted (Fig. 2A). There was an increase in the frequency of crossings (main effect of karyotype [F(1,19) = 5.4; P < 0.05], and in frequency of wall rearing [F(1,19) = 10.3; P < 0.05]. Post hoc comparisons indicated that these differences were more consistent in the last 10 min of the test, particularly in the frequency of crossings (2n versus Ts65Dn; P < 0.05 in blocks 3 and 4). In addition, a trend toward reduction in duration of grooming was also evident [F(1,19) = 3.4; P = 0.07]. No differences were found in the latency to approach a novel object or in lying still.
Mice were also tested in the Morris water maze. In the hidden platform task, subjects must learn the location of a submerged, invisible platform to escape the water. While the latency to reach the platform decreased by ≈50% in 2n animals over 4 days of testing, Ts65Dn animals showed no significant improvement in performance (Fig. 2B). On testing days 2 through 4, the latency to reach the platform was significantly greater when comparing Ts65Dn and 2n animals: Ts65Dn (n = 13) versus 2n (n = 17): day 1, P = 0.312; day 2, P = 0.01; day 3, P = 0.007; day 4, P < 0.001. A second group of Ts65Dn and 2n littermates underwent testing on the visible platform task in the Morris water maze. On all days of testing, Ts65Dn animals performed more poorly in this task with significant differences recorded on both day 2 and day 3 (P < 0.05) (Fig. 2B). Of note, there was greater variability in the performance of Ts65Dn animals. Some Ts65Dn animals were able to learn the task while others never appeared to improve. Significantly, there were no differences in swim speed between Ts65Dn and 2n mice.
Adult mice were also assessed in the passive avoidance task. No statistical differences were detected between the groups at any time. However, in the second part of the acquisition phase, step-through latencies from the Ts65Dn mice were shortened (Fig. 2B). As in the visible platform task, some Ts65Dn animals had scores comparable to 2n mice, whereas others were clearly impaired throughout the acquisition session. In fact, analysis performed on the standard deviation values revealed a significant difference between groups (Wilcoxon, P < 0.05), observations from Ts65Dn mice being more scattered. Of note, similar variability in performance was found in both young and older Ts65Dn animals; whereas variability in performance decreased somewhat with age in 2n animals (data not shown).
Age-Related Abnormalities in the Septohippocampal Cholinergic System in Ts65Dn Mice.
Given the presence of AD pathological markers in elderly DS patients, these were evaluated in Ts65Dn and 2n animals at 6 months and 20 months of age. Brains were examined for the presence of plaques and tangles with both Bielshowsky silver staining as well as with Aβ and Alz-50 immunostaining. There was no evidence for abnormal staining resembling that seen in DS and AD.
In both DS and in AD, BFCNs are selectively vulnerable to degeneration (5, 23). These neurons supply the major cholinergic input to both the hippocampus and neocortex, and their degeneration may contribute to the dementia of DS and AD. Interestingly, studies on DS patients suggest that while BFCNs are normal at birth, during infancy, and in the early adult period (24, 25), they degenerate in older adults (23). Using unbiased stereological techniques for neuronal counting (the optical disector method), we assessed the number of BFCNs in the medial septal nucleus of both Ts65Dn mice and 2n age-matched control littermates at PD2, PD10, PD22, 6 months, and 20 months of age. BFCNs were identified by immunostaining with an antibody to p75NGFR, a protein which in this region is localized specifically to cholinergic neurons. During the first weeks of postnatal development, there was no difference in either the number or size of BFCNs in Ts65Dn and 2n control subjects (Fig. 3). The situation in adult animals was very different; age-related neuronal atrophy and loss of BFCNs was detected (Fig. 3). At 6 months of age, the number of p75NGFR-immunoreactive (IR) neurons in the Ts65Dn mice was decreased by 30% and at 20 months there was 40% decrease. In contrast, even in 20-month-old 2n animals there was no decrease in the number of p75NGFR-IR neuronal profiles. Significantly, the loss of cholinergic neurons in Ts65Dn mice appears to be somewhat selective since there was no difference in the number of caudate–putamen cholinergic neurons when comparing 6-month-old Ts65Dn and 2n mice (mean ± SEM: Ts65Dn = 5150 ± 328, n = 5 versus 2n = 4142 ± 390, n = 5; P = 0.083). The neuronal atrophy of remaining Ts65Dn BFCNs with age is analogous to that seen in remaining BFCN neurons in AD (26).
Figure 3.
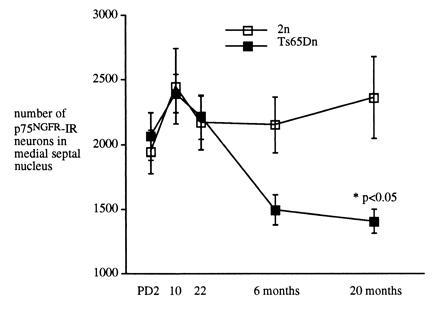
Analysis of the number and size of p75NGFR-IR neurons in the medial septal nucleus of Ts65Dn and 2n mice during development and in the adult. The mean cross-sectional profile area of p75NGFR-IR neurons with a nucleolus were assessed in Ts65Dn and 2n mice. No significant differences were found at PD2, PD10, or PD22. At later times, areas were as follows: (mean ± SEM) 6 months, Ts65Dn, 149 ± 2.2 (n = 5, n = 300) and 2n, 156 ± 2.3 (n = 5, n = 300), P = 0.04; 20 months, Ts65Dn, 147 ± 0.5 (n = 5, n = 506) and 2n, 153 ± 0.3 (n = 5, n = 506), P < 0.001.
Glial Abnormalities in Ts65Dn Mice.
Glial abnormalities have been described in both the developing and adult DS brain. These include astrocytic hypertrophy and an increase in astrocyte number (27, 28). GFAP is an astrocyte-specific protein. There was evidence of a small but statistically significant hypertrophy of GFAP-IR astrocytes in the hippocampus of Ts65Dn animals (mean ± SEM, Ts65Dn = 62.5 ± 0.88 μm2, n = 5, n = 2500, versus 2n = 59.8 ± 0.82 μm2, n = 5, n = 2500; P = 0.02). We also counted the number of GFAP-IR astrocytes in the right hippocampus of Ts65Dn and 2n mice at 3 months of age. There were 27% more GFAP-IR astrocytes in the Ts65Dn hippocampus. This change was not statistically significant (mean ± SEM, Ts65Dn = 10, 316 ± 1414, n = 5, versus 2n = 8150 ± 1134, n = 5, P = 0.24).
Abnormal Gene Expression in Ts65Dn Mice.
Most studies in DS and other aneuploid organisms have shown that gene expression is proportional to gene dose (1). Nevertheless, there are examples in both DS and in Ts16 mice to indicate that expression of some genes cannot be predicted on the basis of gene dosage (17, 29). To examine the situation in Ts65Dn, we measured brain mRNA levels for genes present in either two or three copies. Genes present in three copies in Ts65Dn mice include App and Sod-1. On the basis of gene dose, mRNA levels would be predicted to be 1.5-fold the 2n level. Examination of gene expression by Northern blot analysis revealed that APP mRNA was present at levels 2-fold and SOD-1 mRNA was at 1.5-fold that seen in 2n mice (Fig. 4). Both values were significantly greater than 1.0, suggesting that genes on the segmental chromosome were expressed. Though APP mRNA levels were increased beyond that expected for gene dose versus 1.5), the increase was not significantly greater than 1.5. We also examined forebrain mRNA levels of genes present in two copies in Ts65Dn animals: (i) trkB [which is encoded on MMU-13 (30)], (ii) Gap-43 [which is encoded on the proximal arm of MMU-16 (31) but is not present on the translocation chromosome in Ts65Dn mice], and (iii) ApoE [which is encoded on MMU-7 (32)]. While trkB and GAP-43 mRNA levels were similar in Ts65Dn and 2n littermates, apoE mRNA levels in the Ts65Dn brain were 1.5-fold the level found in the 2n brain (P < 0.05) (Fig. 4).
Figure 4.
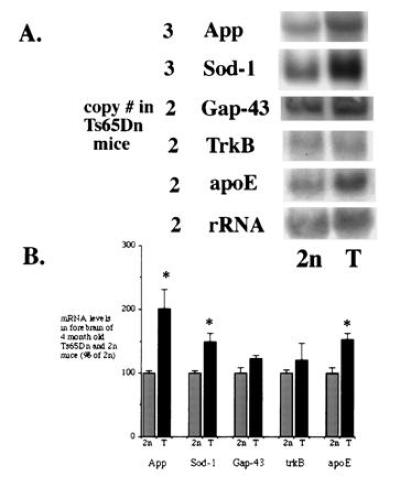
Northern blot analysis of mRNA levels from the forebrain of 4-month-old Ts65Dn and 2n mice. (A) The level of several mRNA species was assessed for genes that were either localized to the 1716 translocation chromosome (present in three copies) or to another chromosomal region (present in two copies). (B) Densitometric scanning of x-ray film revealed significant increases in mRNA levels for genes localized to the translocation chromosome in Ts65Dn mice. In addition, there was significant overexpression of apoE mRNA in Ts65Dn mice (∗, P < 0.05, Ts65Dn versus 2n).
DISCUSSION
The development of segmental (Ts65Dn) mice (8) has provided a genetic model of DS in which much of the MMU-16 region that is homologous with the long arm of HSA-21 is present in three copies. Importantly, there is very little genetic information from MMU-17, which is also in dosage imbalance (7). Determining whether the genetic abnormality present in these animals leads to the hallmark nervous system abnormalities seen in developing and mature DS individuals is critical to define the contribution of specific genetic elements to particular phenotypes. Similar to earlier reports (7, 9, 33), we demonstrate that there are behavioral abnormalities in adult Ts65Dn mice. Significantly, we now show that like DS subjects, Ts65Dn mice have behavioral evidence of developmental delay and cognitive impairment in young subjects and have, in older subjects, a selective age-dependent loss and cellular atrophy of BFCNs. We conclude that Ts65Dn mice are a model for examining certain developmental and age-related DS CNS phenotypes.
The behavior of Ts65Dn animals was abnormal during the entire lifespan. Developmental delay and hyperactivity were noted during early development. These findings parallel what is observed in infants and children with DS (1, 2). Indeed, like DS children (34), Ts65Dn mice did achieve developmental sensorimotor milestones, albeit later than normal. As was present at PD21, Ts65Dn adults also exhibited spontaneous hyperactivity in open field testing; moreover, adult subjects showed evidence of impaired learning in the Morris water maze and in some there was poor performance in passive avoidance testing. While there was no significant difference between the performance of Ts65Dn and 2n mice on passive avoidance, we did note greater variability between animals in the Ts65Dn group. In fact, Ts65Dn mice failed to show a normal decrease in variability on this task with age. This may have been due to an increase in biological variability. It is also possible that it represents a delay or arrest in behavioral development. If so, it would be analogous to what has been observed in DS. While DS children show continuous but gradual improvement in mental age throughout childhood; intelligence quotients generally decline from early in the first year to late childhood (35).
The neural substrate for the abnormal behaviors seen in Ts65Dn mice have yet to be defined. It is conceivable that the loss and dysfunction of BFCNs contribute to the abnormalities found on testing in the hidden platform task (36); however, the behavioral abnormalities observed in Ts65Dn suggest a more general cognitive impairment analogous to the mental retardation seen in human trisomy 21 and in translocation DS (6). For example, while it is possible that functional abnormalities involving the hippocampus or its afferent and efferent projections would explain the failure on the hidden platform task, such lesions alone would not account for impaired performance in the visual platform task (16).
The most striking neuropathological phenotype in Ts65Dn was encountered in mature but not developing DS subjects. The number and size of BFCNs was similar in Ts65Dn and 2n subjects during development. It was only in adult subjects that atrophy and progressive loss of BFCNs was detected. These data suggest that triplication of genetic material in the distal arm of MMU-16 does not affect neurogenesis or early development and differentiation of BFCNs. This is not the case in fetal Ts16 fetal animals in which there is a decrease in the number of BFCNs generated (37). Thus, the presence of an extra copy of genetic material in MMU-16 that is not triplicated in DS results in a different cholinergic neuronal phenotype. It is interesting to note that, similar to Ts65Dn mice, young persons with DS appear to be born with a normal septohippocampal cholinergic system (24) and that degeneration of this system only appears with aging (23, 25). Our findings suggest that as in DS, an extra copy of normal genetic material produces BFCN degeneration in mature subjects. That the Ts65Dn brain does not show a generalized late-onset loss of neurons is suggested in the data for regional brain volumes in mature subjects. In regard to regional volumes, it appears that the mature Ts65Dn brain differs from that in DS, where some but not all brain regions are smaller than normal age-matched subjects (38). It is possible that development abnormalities of other neuronal populations in Ts65Dn mice escaped detection in our studies. It will now be important to further evaluate this possibility looking specifically for other abnormalities seen in the DS brain.
The age-related loss of cholinergic neurons in basal forebrain but not the striatum of Ts65Dn mice mimics the specific age-related decrease in cholinergic function seen in DS and AD (23, 25, 39). These findings raise the possibility that this model may be used further to understand mechanisms underlying selective neuronal vulnerability seen in DS and AD. Disturbed trophic support is a possible mechanism. It is noteworthy that nerve growth factor (NGF) is required for the normal differentiation of BFCNs (19). Equally intriguing are recent data suggesting that the BFCN cell loss and atrophy that occurs in aged rats is accompanied by a decrease in retrograde transport of NGF from its site of synthesis in the hippocampus to BFCN cell bodies in the septum (40). It will be important to determine whether there are abnormalities in NGF levels or signaling in Ts65Dn mice.
Similar to what has been described in Ts16 and Ts65Dn mice (5, 7, 17), it appears that for some genes expressed in the young adult Ts65Dn brain, gene dose and gene expression are directly correlated. This was the case for Sod-1, which is located on the trisomic segment, and for trkB and Gap-43, which are located on non-trisomic segments. However, for other genes, this was not the case. App (MMU-16) and apoE (MMU-7) were overexpressed more than predicted on the basis of gene dose. Overexpression of these genes may have significance for AD-related neuropathology. One isoform of apoE (apoE4) has been shown to be a major risk factor for AD (41). Its presence is associated with earlier onset of dementia in subjects with DS (42). Since, apoE is selectively expressed in astrocytes (43), the increase in apoE mRNA in the Ts65Dn mice and in AD may be associated with the activation of astrocytes (hypertrophy) (44). Hypertrophy of astrocytes has been described even in younger DS subjects before gross AD changes have occurred (27, 28). There may be similar changes in Ts65Dn mice. It has been postulated that overexpression of APP in DS leads to Aβ deposition (45). Over the 2-year lifespan of the Ts65Dn mouse, App overexpression did not lead to Aβ deposition. In fact, the only reports to date showing extensive Aβ deposition in the brain of mice are in animals in which a mutant form of human amyloid precursor protein (APP) has been overexpressed (46). Data showing that overexpression of normal human APP in mice also fails to result in plaque formation (47) suggests that the occurrence of AD pathology in DS may be due to more than overexpression of APP.
In summary, our analysis of the developing and adult CNS of Ts65Dn mice documents functional and morphological changes in Ts65Dn animals analogous to some of those seen in DS. Our findings suggest that further studies with Ts65Dn mice may be particularly useful to understand the biological basis for some of the cognitive abnormalities seen in infants, children, and young adults with DS. It is possible that this model can also be used to provide insight into some of the AD-like pathology seen in DS such as selective neuronal vulnerability and glial abnormalities. While the model does not currently allow us to study Aβ deposition and neurofibrillary tangle formation, it is conceivable that through genetic modifications of the model, these markers can be produced thus providing this opportunity.
Acknowledgments
We thank Dr. Muriel Davisson for initially providing Ts65Dn mice and Dr. Peter Mouton for advice with unbiased stereological procedures. This work was supported by National Institutes of Health Grants AG00445 (D.M.H.), AG08938 (C.J.E.), and AG08938 and AG10672 (W.C.M.). D.M.H. was supported by the National Down Syndrome Society and by a Paul Beeson Physician Faculty Scholar Award from the American Federation for Aging Research. W.C.M. was supported by the Adler Foundation and the McGowan Charitable Trust. D.S. was supported by TELETHON and INPHP “Prevention of risk factors in maternal and child health.”
Footnotes
Abbreviations: CNS, central nervous system, DS, Down syndrome; AD, Alzheimer disease; Ts16, trisomy 16; BFCN, basal forebrain cholinergic neuron; PD, postnatal day; Aβ, β-amyloid; IR, immunoreactive; GFAP, glial fibrillary acidic protein.
References
- 1.Epstein C J. Consequences of Chromosome Imbalance: Principals, Mechanisms, and Models. New York: Cambridge Univ. Press; 1986. [Google Scholar]
- 2.Hayes A, Batshaw M L. Pediatr Clin North Am. 1994;40:523–529. doi: 10.1016/s0031-3955(16)38548-0. [DOI] [PubMed] [Google Scholar]
- 3.Holtzman D M, Epstein C J, Mobley W C. Ment Retard Dev Disabil Rev. 1996;2:66–72. [Google Scholar]
- 4.Wisniewski K E, Wisniewski H M, Wen G Y. Ann Neurol. 1985;17:278–282. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 5.Coyle J T, Oster-Granite M L, Reeves R H, Gearhart J D. Trends Neurosci. 1988;11:390–394. doi: 10.1016/0166-2236(88)90075-6. [DOI] [PubMed] [Google Scholar]
- 6.Korenberg J R. Nat Genet. 1995;11:109–111. doi: 10.1038/ng1095-109. [DOI] [PubMed] [Google Scholar]
- 7.Reeves R H, Irving N G, Moran T H, Wohn A, Kitt C, Sisodia S S, Schmidt C, Bronson R T, Davisson M T. Nat Genet. 1995;11:177–183. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 8.Davisson M T, Schmidt C, Reeves R H, Irving N G, Akeson E C, Harris B S, Bronson R T. In: Segmental Trisomy as a Mouse Model for Down Syndrome. Epstein C J, editor. New York: Wiley–Liss; 1993. pp. 117–133. [PubMed] [Google Scholar]
- 9.Escorihuela R M, Fernandez-Teruel A, Vallina I F, Baamonde C, Lumbreras M A, Tobena A, Florez J. Neurosci Lett. 1995;199:143–146. doi: 10.1016/0304-3940(95)12052-6. [DOI] [PubMed] [Google Scholar]
- 10.Coussonsread M E, Crnic L S. Behav Genet. 1996;26:7–13. doi: 10.1007/BF02361154. [DOI] [PubMed] [Google Scholar]
- 11.Keeler C. J Hered. 1966;57:47–50. doi: 10.1093/oxfordjournals.jhered.a107462. [DOI] [PubMed] [Google Scholar]
- 12.Fox M. Anim Behav. 1965;13:234–241. doi: 10.1016/0003-3472(65)90041-2. [DOI] [PubMed] [Google Scholar]
- 13.Alleva E, Laviola G, Tirelli E, Bignami G. Psychopharmacology. 1985;87:434–441. doi: 10.1007/BF00432509. [DOI] [PubMed] [Google Scholar]
- 14.Santucci D, Calamandrei G, Alleva E. Neurotoxicol Teratol. 1993;15:131–137. doi: 10.1016/0892-0362(93)90071-u. [DOI] [PubMed] [Google Scholar]
- 15.Calamandrei, G., Ricceri, L. & Valanzano, A. (1996) Brain Res., in press. [DOI] [PubMed]
- 16.Morris R G M. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 17.Holtzman D M, Bayney R M, Li Y, Khosrovi H, Berger C N, Epstein C J, Mobley W C. EMBO J. 1992;11:619–627. doi: 10.1002/j.1460-2075.1992.tb05094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weskamp G, Reichardt L F. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Holtzman D M, Kromer L F, Kaplan D R, Chua-Couzens J, Clary D O, Knusel B, Mobley W C. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavalieri B. Geometria degli indivisibili. Torino, Italy: Unione Tipografico; 1966. [Google Scholar]
- 21.Gunderson H J G. J Microsc (Paris) 1986;143:3–45. [Google Scholar]
- 22.West M J. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- 23.Yates C M, Simpson J, Gordon A, Maloney A F J, Allison Y, Ritchie I M, Urquhart A. Brain Res. 1983;280:119–126. doi: 10.1016/0006-8993(83)91179-4. [DOI] [PubMed] [Google Scholar]
- 24.Kish S, Karlinsky H, Becker L, Gilbert J, Rebbetoy M, Chang L-J, DiStefano L, Hornykiewicz O. J Neurochem. 1989;52:1183–1187. doi: 10.1111/j.1471-4159.1989.tb01864.x. [DOI] [PubMed] [Google Scholar]
- 25.Casanova M F, Walker L C, Whitehouse P J, Price D. Ann Neurol. 1985;18:310–313. doi: 10.1002/ana.410180306. [DOI] [PubMed] [Google Scholar]
- 26.Vogels O J M, Broere A J, Ter Laak H J, Ten Donkelaar H J, Nieuwenhuys R, Schulte B P M. Neurobiol Aging. 1990;11:3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- 27.Griffin W S T, Stanley L C, Ling C, White L, MacLeod V, Perrot L J, White C L, Araoz C. Proc Natl Acad Sci USA. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mito T, Becker L E. Exp Neurol. 1993;120:170–176. doi: 10.1006/exnr.1993.1052. [DOI] [PubMed] [Google Scholar]
- 29.Neve R L, Finch E A, Dawes L R. Neuron. 1988;1:669–677. doi: 10.1016/0896-6273(88)90166-3. [DOI] [PubMed] [Google Scholar]
- 30.Barbacid M, Lamballe F, Pulido D, Klein R. Biochim Biophys Acta. 1991;1072:115–127. doi: 10.1016/0304-419x(91)90010-i. [DOI] [PubMed] [Google Scholar]
- 31.Reeves R H, Citron M P. Mamm Genome. 1994;5:S229–S237. [PubMed] [Google Scholar]
- 32.Hoffer M J, Hofker M H, van Eck M M, Havekes L M, Frants R R. Genomics. 1993;15:62–67. doi: 10.1006/geno.1993.1010. [DOI] [PubMed] [Google Scholar]
- 33.Coussonsread M E, Crnic L S. Behav Genet. 1996;26:7–13. doi: 10.1007/BF02361154. [DOI] [PubMed] [Google Scholar]
- 34.Wishart J G. In: Early Learning in Infants and Young Children with Down Syndrome. Nadel L, editor. Cambridge, MA: MIT Press; 1988. pp. 7–50. [Google Scholar]
- 35.Wang P P. Ment Retard Dev Disabil Rev. 1996;2:102–108. [Google Scholar]
- 36.Olton D S, Givens B S, Markowska A L, Shapiro M, Golski S. In: Mnemonic Functions of the Cholinergic Septohippocampal System. Squire L R, Weinberger N M, McGaugh J L, editors. London: Oxford Univ. Press; 1991. [Google Scholar]
- 37.Sweeney J E, Hohmann C F, Oster-Granite M L, Coyle J T. Neuroscience. 1989;31:413–425. doi: 10.1016/0306-4522(89)90384-9. [DOI] [PubMed] [Google Scholar]
- 38.Kesslak J P, Negata S F, Lott I, Nalcioglu O. Neurology. 1994;44:1039–1045. doi: 10.1212/wnl.44.6.1039. [DOI] [PubMed] [Google Scholar]
- 39.Mann D M A, Yates P O, Marcyniuk B. Neuropathol Appl Neurobiol. 1984;10:185–207. doi: 10.1111/j.1365-2990.1984.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 40.Cooper J D, Lindholm D, Sofroniew M V. Neuroscience. 1994;62:625–629. doi: 10.1016/0306-4522(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 41.Strittmatter W J, Weisgraber K H, Huang D Y, Dong L-Y, Salvesen G S, Pericak-Vance M, Schmechel D, Saunders A M, Goldgaber D, Roses A D. Proc Natl Acad Sci USA. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardy J, Crook R, Perry R, Raghavan R, Roberts G. Lancet. 1994;343:979–980. doi: 10.1016/s0140-6736(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 43.Pitas R E, Boyles J K, Lee S H, Foss D, Mahley R W. Biochim Biophys Acta. 1987;917:148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 44.Diedrich J F, Minnigan H, Carp R I, Whitaker J N, Race R, Frey W, Haase A T. J Virol. 1991;65:4759–4768. doi: 10.1128/jvi.65.9.4759-4768.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rumble B, Retalack R, Hilbich C, Simms G, Multhaup G, Martins R, Hockey A, Montgomery P, Beyreuther K, Masters C L. N Engl J Med. 1989;320:1446–1452. doi: 10.1056/NEJM198906013202203. [DOI] [PubMed] [Google Scholar]
- 46.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, et al. Nature (London) 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 47.Lamb B T, Sisodia S S, Lawler A M, Slunt H H, Kitt C A, Kearns W G, Pearson P L, Price D L, Gearhart J D. Nat Genet. 1993;5:22–30. doi: 10.1038/ng0993-22. [DOI] [PubMed] [Google Scholar]