

Abstract
Chronic stress causes dendritic regression and loss of dendritic spines in hippocampal neurons that is accompanied by deficits in synaptic plasticity and memory. However, the responsible mechanisms remain unresolved. Here, we found that within hours of the onset of stress, the density of dendritic spines declined in vulnerable dendritic domains. This rapid, stress-induced spine loss was abolished by blocking the receptor (CRFR1) of corticotropin-releasing hormone (CRH), a hippocampal neuropeptide released during stress. Exposure to CRH provoked spine loss and dendritic regression in hippocampal organotypic cultures, and selective blockade of the CRFR1 receptor had the opposite effect. Live, time-lapse imaging revealed that CRH reduced spine density by altering dendritic spine dynamics: the peptide selectively and reversibly accelerated spine retraction, and this mechanism involved destabilization of spine F-actin. In addition, mice lacking the CRFR1 receptor had augmented spine density. These findings support a mechanistic role for CRH–CRFR1 signaling in stress-evoked spine loss and dendritic remodeling.
Keywords: dendritic spine, stress, CRF, CRH, actin, synaptic plasticity, two photon, hippocampus
Introduction
Chronic stress leads to reversible dendritic dedifferentiation in hippocampal neurons (Magarinos and McEwen, 1995). This effect is important because it is generally accompanied by, and perhaps underlies, loss of synaptic plasticity and cognitive impairment (McEwen, 1999; Sapolsky, 2000; Fenoglio et al., 2006). The mechanisms by which stress causes these structural alterations of hippocampal (and select cortical) (Radley et al., 2006) neurons are not fully understood. Molecules that are activated during stress, including glucocorticoid receptors, have been found to contribute to stress-induced dendritic remodeling, as have NMDA receptors and tissue plasminogen activator (Magarinos and McEwen, 1995; Pawlak et al., 2005).
In addition to glucocorticoids, the prototypic stress hormones, other extracellular factors are activated by stress within hippocampus, and are thus candidate mediators of dendritic changes in hippocampal pyramidal cells. These include corticotropin-releasing hormone (CRH), which is expressed in hippocampal interneurons (Swanson et al., 1983; Chen et al., 2001) and is released into the extracellular space during a variety of stressors (Merali et al., 1998; Chen et al., 2004b, 2006; Khan et al., 2004). CRH interacts primarily with CRH receptor type 1 (CRFR1) expressed on pyramidal cell dendrites (Chalmers et al., 1995; Chen et al., 2000, 2004a,b; Van Pett et al., 2000). Activation of this receptor has recently been shown to be necessary for stress-evoked immediate-early gene expression in populations of CA1 and CA3 pyramidal cells of immature and adult hippocampus (Chen et al., 2006). These facts raised the possibility that extracellular CRH might mediate stress-evoked dendritic regression, a notion further supported by finding that, in mice lacking CRFR1, dendritic length and branching were exuberant in hippocampal neurons (Chen et al., 2004a).
Dendritic integrity may be governed by the presence and functions of dendritic spines. Spines, important for synaptic function and plasticity (Segal, 2005), are specialized structures that perform distinct functions including calcium signaling (Koester and Sakmann, 1998; Mainen et al., 1999; Augustine et al., 2003; Yuste and Bonhoeffer, 2004; Calabrese and Halpain, 2005). Changes in both the number and shape of spines may govern synaptic plasticity (Zhou et al., 2004; Calabrese and Halpain, 2005; Segal, 2005; Chen et al., 2007). The balance of spine formation and retraction (spine dynamics) (Maletic-Savatic et al., 1999; Engert and Bonhoeffer, 1999; Fu et al., 2007) influences dendritic integrity (Nägerl et al., 2004; Zhou et al., 2004). Spines are regulated by extracellular factors including neurotransmitters, growth factors, and hormones, that, in turn, are governed by environmental signals including stress (Calabrese and Halpain, 2005; Segal, 2005). Thus, a derangement of spine dynamics that favors loss of spines is a plausible candidate mechanism for stress-evoked dendritic atrophy and associated synaptic dysfunction (Brunson et al., 2005).
Combining in vivo, organotypic culture and live-imaging experiments, we found that exposure of adult mice to restraint stress reduced spine density in dendrites of CA3 neurons already within hours. This rapid, stress-induced spine loss in selective dendritic domains was blocked by a specific inhibitor of CRFR1. In addition, chronic exposure of hippocampal neurons to CRH reduced spine density in stress-vulnerable domains of pyramidal cells via activation of CRFR1. As expected, mice lacking CRFR1 had increased dendritic spine densities in the same apical dendritic regions. Live imaging of yellow fluorescent protein (YFP)-expressing neurons demonstrated that CRH-mediated activation of CRFR1 caused rapid and reversible acceleration in the rate of spine elimination, without altering the appearance of new spines. This effect was blocked by a selective CRFR1 antagonist and involved enhanced activation of the F-actin regulating protein, cofilin. Together, these findings support a mechanistic role for CRFR1 and its endogenous ligands in stress-evoked dendritic remodeling.
Materials and Methods
Animals.
B6.Cg-TgN transgenic mice expressing YFP under the Thy1 promoter (Thy1-YFP; The Jackson Laboratory, Bar Harbor, ME) were used on postnatal day 0 (P0) to P14 or as adults (3 months of age). In addition, YFP-expressing CRFR1 mutant mice (YFP/CRFR1−/− mice) were generated by crossing C57BL background CRFR1−/− mice [courtesy of M. Stenzel-Poore (Oregon Health & Science University, Portland, OR), F. Holsboer, and W. Wurst (Max Planck Institute of Psychiatry, Munich, Germany)] with B6.Cg-TgN mice to generate YFP/CRFR1+/− breeding pairs. Progeny of the breeding pairs were genotyped by PCR analysis (Preil et al., 2001), and YFP/CRFR1−/− mice and their wild-type YFP-expressing littermates were compared on P7, P14, or at 3 months of age. Organotypic slice cultures were derived from these strains. Adult males only were used to avoid cyclic fluctuations of spine densities that occur in females (Woolley et al. 1990).
Animals were born and maintained in a quiet, uncrowded, National Institutes of Health (NIH)-approved facility on a 12 h light/dark cycle, with ad libitum access to lab chow and water. Efforts were made to minimize the number of animals used and their suffering. All experiments were approved by University Animal Care Committees and conformed to NIH guidelines.
Organotypic slice cultures.
Hippocampal slice cultures were prepared from P1 mice as described previously (Chen et al., 2004a). Briefly, after decapitation, mouse brains were removed and placed in minimal essential medium (1× MEM; Invitrogen, Carlsbad, CA), and a block of tissue including entorhinal cortex and hippocampus was dissected from adhering tissue. Transverse slices (300 μm) were cut (McIlwain tissue chopper; Mickle Lab, Gomshall, UK). YFP-expressing sections were placed onto Millicell-CM culture membranes (Millipore, Bedford, MA), and then transferred to six-well plates filled with 1.2 ml of culture medium (0.5× MEM, 25% horse serum, 25% HBSS, 30 mm glucose, 7.5% NaHCO3, and 0.01% ascorbic acid, pH 7.2). Cultures were incubated in a humidified, 5% CO2 atmosphere at 36°C for 4–14 d in vitro (DIV), with medium changes every 2–3 d.
Application of CRH and CRFR1 antagonist.
To study the effects of chronic exposure to CRH on dendritic spines and the role of CRFR1 in these CRH actions, synthetic rat/human CRH (100 nm; Bachem, King of Prussia, PA) or the CRFR1 antagonist 2,5-dimethyl-3-(6-dimethyl-4-methylpyridin-3-yl)-7-dipropylamino-pyrazolo[1,5-a]pyrimidine (NBI 30775) (1 μm; previously R121919; a generous gift from Dr. D. E. Grigoriadis, Neurocrine Biosciences Inc., San Diego, CA) were applied to the culture medium on DIV 2 as well as to refreshed medium. After 6 or 13 d, cultures were fixed in fresh 4% paraformaldehyde for 30 min and processed for fluorescent immunocytochemistry. To examine whether degradation of F-actin was involved in the effects of CRH on spine retraction, CRH was applied on DIV 6, and cultures were harvested 5, 10, or 30 min later. Each experiment included equal numbers of cultures from control and CRH-treated groups, derived from the same mouse pups, and the data are described per mouse (n = 5 mice per group).
Fixed cultures were imaged using a Zeiss (Oberkochen, Germany) LSM510 Meta or Bio-Rad (Hercules, CA) Radiance 2000 confocal scanning systems. Dendritic trees and spines of CA3 pyramidal neurons were captured using “z-stack” three-dimensional projections (12–16 optical slices; 2 μm each) and files were converted to Adobe Photoshop. Microscope objectives used were a dry 20×, numerical aperture (NA) 0.75 (Nikon), and an oil-immersion 40×, NA 1.3 (Olympus, Tokyo, Japan) for capturing dendritic branches and spines, respectively. The 2 μm optical sections were usually collected at an additional electronic zoom factor of 1.6× for dendrites and 3.2× for spines.
Live imaging studies.
To study the dynamic effects of CRH on dendritic spines, cultures (DIV 5–6) were transferred to a superfusion chamber maintained at 36°C via a heating platform (Warner Instruments, Hamden, CT) to avoid hypothermia-related spine loss (Roelandse and Matus, 2004). The chamber was mounted on an upright Zeiss microscope, and slices were superfused with culture medium saturated with 95% O2 and 5% CO2. CRH (100 nm) or vehicle was infused for 30 min (1 ml/min) followed by a normal medium washout. When used, the CRFR1 antagonist NBI 30775 (1 μm) was infused 5–10 min before CRH application. In addition, the potential effects of NBI 30775 itself on spines was evaluated by infusing the compound for 30 min. To avoid phototoxicity, dendrites of YFP-expressing CA3 pyramidal neurons were imaged using a Zeiss LSM510 Meta Mai-Tai two-photon scanning system with excitation wavelength of 920 nm. Under these conditions, repetitive imaging did not affect the morphology or viability of YFP-expressing neurons, dendrites, and spines (see Fig. 4). Based on the established localization of the effects of stress and CRH (Chen et al., 2004a; Brunson et al., 2005), we focused on the apical dendrites of CA3 pyramidal cells and obtained z-stacks (12–20 optical slices; 2 μm/slice) of 100-μm-long dendritic segments. Stress had little effect on the most proximal segments (0–50 μm from stratum lucidum). Therefore, we focused on the dendritic domain of the third- and fourth-order dendritic branches (∼50–100 μm within stratum radiatum).
Figure 4.
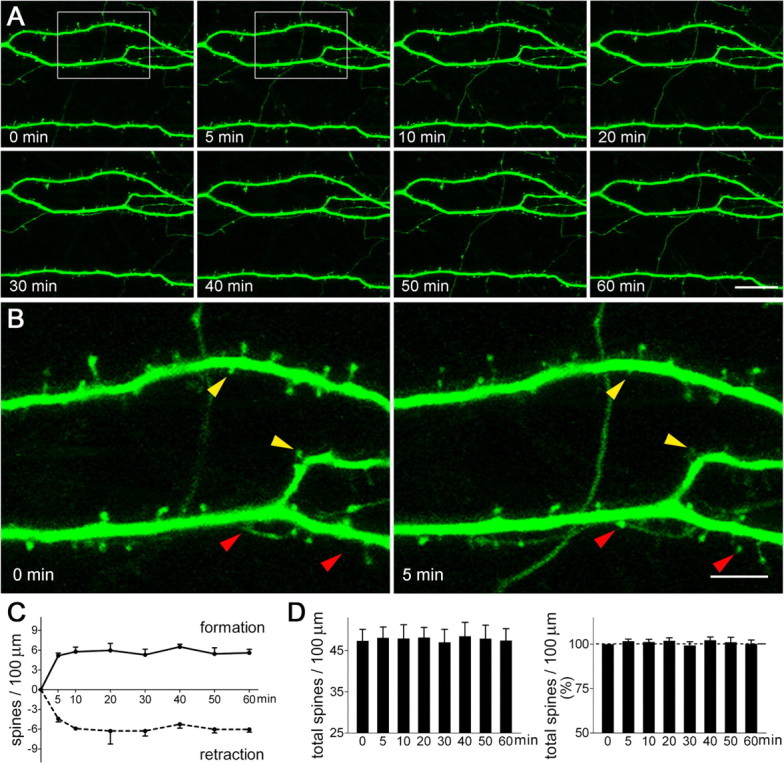
Dendritic spine dynamics in hippocampal pyramidal neurons, visualized using live, time-lapse two-photon microscopy. A, A series of images shows that the structure of the dendrites remained constant over the 60 min of imaging. B, During this period, new dendritic spines appeared (red arrowheads) and existing ones disappeared (retracted; yellow arrowheads) within minutes. C, Under the imaging conditions, spine formation and disappearance occurred at similar rates (F(1,48) = 0.02; p = 0.90). D, Spine density (left panel), expressed as numbers of total spines per unit dendritic length, was stable (p = 1), with little percentage change of spine density over the imaging period (right panel). A total of 2006 μm of dendritic branches from 12 neurons were imaged and analyzed blindly. Hippocampi were cultured on P1 and imaged on in vitro days 5–6. Error bars indicate SEM. Scale bars: A, 21.5 μm; B, 6.6 μm.
In vivo stress paradigms.
Three-month-old Thy1-YFP mice (three to six mice per group) experienced acute variable stress (Chen et al., 2004b, 2006). Briefly, mice were placed in a restrainer fashioned from a 50 ml plastic cylindrical tube for 4–5 h, and also subjected to concurrent jostling and noise arising from placing the restrainers on a laboratory rotator. To investigate the role of CRH in stress-induced spine loss, the CRFR1 antagonist NBI 30775 (15 μg in 1 μl) was infused into the lateral ventricle (intracerebroventricularly) of a group of mice 30 min before the stress. This was accomplished via cannulas implanted 6–7 d earlier as described previously (Chen et al., 2004b, 2006). Stressed animals were killed immediately after the end of the stress, and controls were killed under relatively “stress-free” conditions (Chen et al., 2001).
Experimental groups included the following: (1) stress-free (nonstressed mice with or without cannula insertion; these two subgroups were combined because the cannula did not alter spine density); (2) stress (with or without vehicle administration; the vehicle infusion did not alter spine density so that these two subgroups were combined); and (3) stressed animals that received NBI 30775. The stress paradigm increased plasma corticosterone levels to a similar degree regardless of the central administration of NBI 30775 (Chen et al., 2006), indicating that the intracerebroventricularly applied CRFR1 antagonist did not interact with pituitary CRH receptors and did not interfere with the systemic response to stress.
Immunocytochemistry.
Brains or cultures were sectioned transversely (20 μm) using a cryostat, and single or double immunocytochemistry was performed on free-floating sections (Chen et al., 2004a,b). Antibodies included mouse anti-GFP (1:8000; Sigma, St. Louis, MO) and rabbit anti-phospho-Cofilin 2 (Ser3; 1:1000; Upstate Biotechnology, Lake Placid, NY). Antibody binding was visualized with anti-mouse IgG conjugated to Alexa Fluor 488 or Alexa Fluor 568 anti-rabbit IgG (1:200; Invitrogen).
Image analysis of dendritic branches and spines.
All analyses were performed without knowledge of treatment group. YFP-expressing individual CA3 pyramidal neurons were reconstructed and drawn using a Zeiss LSM Image Browser and Adobe Photoshop. For cultures, 10–14 neurons per group were analyzed, 50% each from CA3a and CA3b and including equal representation of long- and short-shaft populations. For in vivo studies, neurons were chosen using systematic unbiased sampling from the middle third of the anterior/posterior extent of the hippocampus (8–12 neurons per mouse) (Chen et al., 2001).
Dendritic branching was evaluated using Sholl analysis, measuring total dendritic length and number of intersections at concentric circles at increasing distance from the soma (data not shown) (Chen et al., 2004a). Spine density was quantified comparing dendritic branches of the same order. Spines were identified and characterized (Hering and Sheng, 2001), and then drawn and filopodia were excluded. Spine density was expressed as the number of spines per 10 or 100 μm of dendrite length. No correction factors were applied to the spine counts because high-magnification neuronal reconstruction permitted all spines of a given dendritic segment to be visualized.
Analysis of spine dynamics.
Newly formed spines were defined as those that were absent at one time point, emerged at a later time point, and remained for the duration of imaging. Eliminated (retracted) spines were defined as those that were present at one time point but not at a later one, and that did not reappear. Transient spines, present at a single time point only, were excluded from analysis. Changes in spine shape were not considered in the current studies.
Analysis of phospho-cofilin immunoreactive puncta.
Confocal images of phospho-cofilin (pCofilin) immunoreactive puncta and of YFP-labeled dendritic branches were obtained using identical settings for pinhole size, brightness, and contrast. At least 20 optical sections per group (1 μm; covering a 32 × 32 μm2 area) were collected from CA3a stratum radiatum. Images were converted to grayscale, and identified puncta (0.2–1 μm2) were quantified using the ImageTool software program (version 1.27; University of Texas Health Science Center, San Antonio, TX).
Statistical analysis.
Differences among groups were compared using one-way or two-way ANOVA (for genotype/treatment and distance from soma/branch order/time point) with Bonferroni's post hoc analysis (Prism; Graph Pad, San Diego, CA). Dendritic length and corticosterone were compared using Student's t test. Significance levels were set at 0.05, and data are expressed as mean ± SEM.
Results
Stress-induced spine loss in adult hippocampus is rapid, evident already within hours
Stress leads to dendritic remodeling in the hippocampus, and stress-induced spine loss of pyramidal neurons may be a key mediator of this effect (Pawlak et al., 2005; Donohue et al., 2006). To examine whether stress-induced hippocampal spine loss might be an early step in the process by which stress promotes dendritic remodeling, adult mice possessing hippocampal neurons expressing YFP controlled by the Thy-1 promoter (Fig. 1), were exposed to a 5 h restraint stress. At the end of this stress experience, spine densities on apical dendrites of CA3 pyramidal cells were lower than those of nonstressed controls (Fig. 1C,D). This finding suggested that stress-evoked spine loss, typically found after chronic stress (McEwen, 1999; Donohue et al., 2006), may be a rapid and dynamic process in adult hippocampus.
Figure 1.
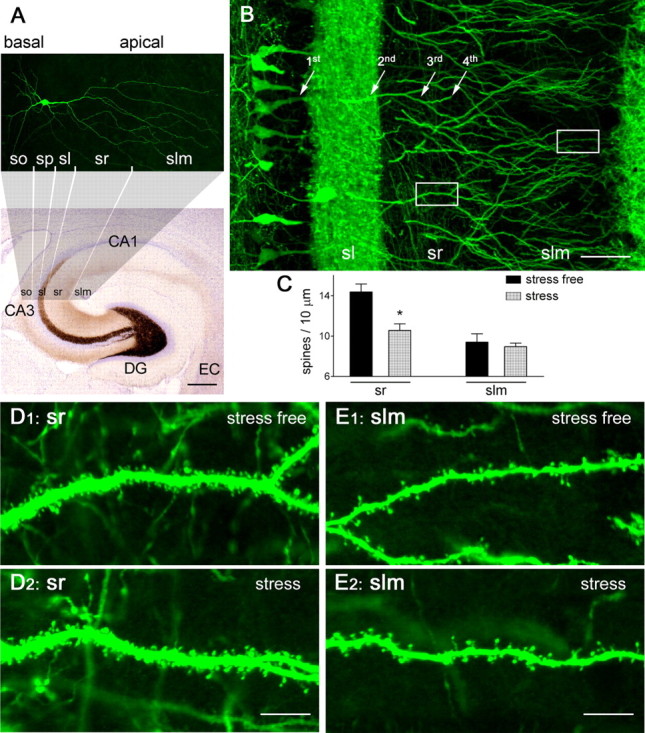
Reduced hippocampal dendritic spine density after a 5 h stress. A, Structure and organization of the hippocampus, clarifying the location and subcellular domains of a YFP-expressing CA3 pyramidal neuron. The location of the apical dendrites, emerging from stratum pyramidale (sp) and spanning stratum lucidum (sl), radiatum (sr), and lacunosum-moleculare (slm), is shown. Basal dendrites are in stratum oriens (so). EC, Enthorinal cortex. Scale bar, 260 μm. B–D2, Stress leads to spine loss in vulnerable domains of apical dendrites (third- and fourth-order branches in sr). In a 3-month-old, Thy1-YFP-expressing mouse, density of spines on the third/fourth dendritic branches was high under stress-free conditions (D1), and was significantly reduced immediately after a 5 h combined restraint/noise stress (D2), as quantified in C (*p < 0.05). Density of spines on the distal dendritic branches located in slm was not influenced by the acute stress (C, E1, E2). The frames in B denote areas magnified in D1 and E1. Error bars indicate SEM. Scale bars: B, 50 μm; D1–E2, 7 μm.
Stress-induced spine loss in selective dendritic domains requires activation of the CRH receptor CRFR1
The rapid, stress-induced reduction of spine density in apical dendrites of CA3 neurons was significant primarily on the third- and fourth-order dendritic branches (stress-free vs stress: 14.39 ± 0.80 vs 10.56 ± 0.65 spines/10 μm; p < 0.05) (Fig. 1C,D), the main postsynaptic target of excitatory commissural/associational fibers. In contrast, spine density in the distal dendrites was not influenced by this acute stress (stress-free vs stress: 9.41 ± 0.82 vs 8.96 ± 0.35 spines/10 μm; p = 0.56 in the fifth and sixth dendritic branches) (Fig. 1C,E). Interestingly, stress reduced spine density in the same dendritic domains where spine density was significantly increased in mutant mice lacking CRFR1, the receptor for the stress-activated neuropeptide CRH (YFP/CRFR1−/−; F(1,68) = 19.89, p < 0.0001) (Fig. 2). CRH is robustly expressed in interneurons within the hippocampal pyramidal cell layers (Chen et al., 2001) and is released during stress into the hippocampal intercellular space (Chen et al., 2004b; 2006). In addition, the principal hippocampal receptor for CRH, CRFR1, is located on dendritic spines (Chen et al., 2004b). These facts suggested that CRH–CRFR1 signaling might be involved in the effects of stress on dendritic spine integrity. To examine this possibility directly, we infused the selective CRFR1 antagonist NBI 30775 (15 μg in 1 μl) into the lateral cerebral ventricles of adult mice 30 min before the onset of stress, and then examined apical dendritic spines densities of CA3 pyramidal neurons. The CRFR1 antagonist abolished the stress-induced decline of spine density (stress vs stress plus antagonist: 10.56 ± 0.65 vs 13.83 ± 1.19 spines/10 μm; p < 0.05), indicating that activation of CRFR1, most likely by its endogenous ligand, CRH, is a necessary component of this rapid, stress-induced reduction of spine density.
Figure 2.

Deletion of CRH receptor CRFR1 enhances spine density. A, Apical dendrites of a CA3 pyramidal neuron expressing YFP (see Materials and Methods) from a 14-d-old mouse with a normal complement of the CRH receptor CRFR1 (wild type). B, The genetic absence of CRFR1 (CRFR1−/−) promoted both complex dendritic branching and increased spine density (F(1,68) = 19.89; p < 0.0001). C, The overall increased spine density in CRFR1−/− dendrites resulted from significant differences of spine densities in the third/fourth-order dendritic branches (*p < 0.05; n = 5 mice per group; Bonferroni's post hoc test). The framed areas in the top images are magnified in the bottom images, to show dendritic spines (filled arrowheads), including those with abnormal shapes (arrows and open arrowhead presented filopodia and big-head spine, respectively). Error bars indicate SEM. Scale bars: 50 μm (low magnification); 5.5 μm (high magnification).
CRH reduces spine density in apical dendrites of stress-vulnerable CA3 pyramidal cells
To study how CRH–CRFR1 signaling might contribute to a rapid stress-induced spine loss, we used hippocampal organotypic slice cultures. We first exposed the cultures to CRH (100 nm) for 7 or 13 d, and found that this treatment reduced the density of spines on apical dendrites of CA3 pyramidal neurons (F(1,60) = 10.74; p = 0.001). The distribution of CRH-provoked spine loss mirrored the effects of stress, being maximal in the third- and fourth-order dendritic branches (Fig. 3A,B) (n = 10 neurons per group; p < 0.05). This finding indicated that, chronically, CRH suppresses spine numbers in a pattern virtually identical with that induced by stress. As expected, growing hippocampal organotypic slice cultures in the chronic presence of the selective CRFR1 antagonist NBI 30775 led to increased density of spines on apical dendrites of CA3 pyramidal cells. In addition, this increase was selective to dendritic domains influenced by both CRH and stress (Fig. 3C,D).
Figure 3.

CRH application reduces spine density in hippocampal organotypic slice cultures, whereas neurons grown in the presence of a CRH receptor antagonist have increased spine density. A, Apical dendrites of a CA3 pyramidal neuron in control condition. B, Treatment of hippocampal slices with CRH (100 nm; 13 d) decreased spine density (F(1,60) = 10.74; p = 0.001), an effect resulting primarily from lower spine density on the third- and fourth-order dendritic branches (*p < 0.05; n = 10 neurons per group; Bonferroni's post hoc test) (D). C, Growing hippocampal slices in the presence of the CRH receptor antagonist NBI 30775 (1 μm; 13 d) led to higher spine density (F(1,58) = 4.17; p = 0.046), also stemming from increased density of spines on the third- and fourth-order dendritic branches (*p < 0.05; n = 10 neurons per group; post hoc test) (D). The framed areas are magnified to show individual spines (arrowheads) and filopodia (C, arrow) on fourth-order branches. Hippocampi were cultured on P1 and grown for 14 d. Error bars indicate SEM. Scale bars: 55 μm (low magnification); 6 μm (high magnification).
CRH disrupts balanced spine dynamics by selective acceleration of spine elimination
We then examined whether the rapid (within hours) reduction of spine density, induced by stress, could be reproduced by exposure to CRH. Spine density is governed by the dynamic formation of new spines and the disappearance (presumably retraction) of existing ones (Yuste and Bonhoeffer, 2004; Holtmaat et al., 2005; Segal, 2005). CRH could influence spine density by blunting spine formation, accelerating spine retraction, or a combination of these processes. To examine the dynamics of dendritic spines, we used time-lapse imaging of organotypic hippocampal slice cultures, using two-photon microscopy. In Thy1-YFP-expressing pyramidal neurons, dendrites were stable for the duration of the imaging period (up to 120 min), whereas spine distribution was dynamic (Fig. 4). The formation and the retraction of spines occurred at similar rates under the imaging conditions (Fig. 4C), resulting in a stable spine density (Fig. 4D). Superfusion of 100 nm CRH for 30 min resulted in a significant acceleration of spine retraction (Fig. 5A–C) with little effect on the rate of spine formation. This effect deranged spine dynamics, resulting in a net loss of dendritic spines (Fig. 5D). Notably, these actions of CRH were reversible: a 30 min washout period led to reduced rate of spine elimination and a gradual recovery of spine density (Fig. 5C,D). As shown in Figure 6, the presence of the selective antagonist of CRFR1 NBI 30775 abolished the actions of the peptide on spine elimination, whereas acute exposure to the antagonist alone had no effect on spine dynamics (Fig. 6A). In addition, CRH administration did not reduce spine density in hippocampal slices from mice lacking the CRH receptor CRFR1 (at 0 min vs at 30 min from CRH application: 47.04 ± 3.76 vs 46.74 ± 4.04 spines/100 μm; p = 0.85; n = 7 neurons from 3 YFP/CRFR1−/− mice). Together, these two observations support the specific role of CRFR1 occupancy in the effects of CRH on spine disappearance.
Figure 5.
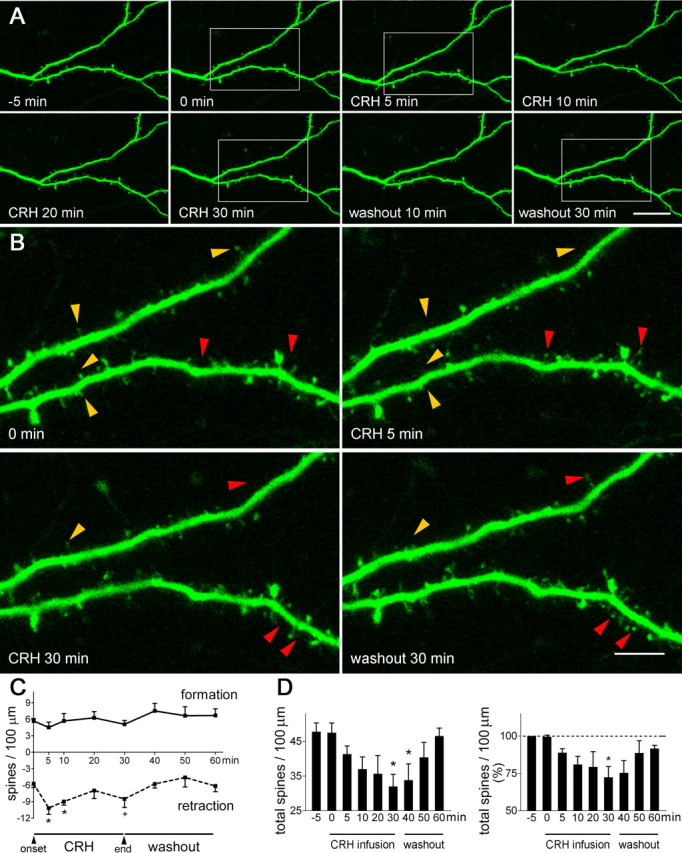
CRH alters spine dynamics rapidly and reversibly. A, Infusion of CRH (100 nm) for 30 min did not affect dendritic structure appreciably. B, High-magnification imaging revealed a rapid effect of CRH on the rate of spine retraction (yellow arrowheads): accelerated spine disappearance was apparent already by 5 min after CRH exposure, with little change in the rate of spine formation (red arrowheads; 0 min vs CRH 5 min). CRH-induced spine elimination was partially reversed by a 30 min washout. C, The effect of CRH (F(1,60) = 15.79; p = 0.0002, compared with vehicle infusion) was attributable primarily to increased retraction rates at the 5 min and 10 min time points (*p < 0.05; +p = 0.07). D, Accelerated spine disappearance with stable spine formation resulted in a significant net reduction of spine density (*p < 0.05, compared with the 0 min time point); this is presented as actual spine density (left panel) and percentage change of spine density (right panel). A total 2650 μm of dendrites was imaged and analyzed in six experiments, two to four neurons each. Hippocampi were cultured on P1 and imaged on in vitro days 5–6. Error bars indicate SEM. Scale bars: A, 21.5 μm; B, 6.6 μm.
Figure 6.
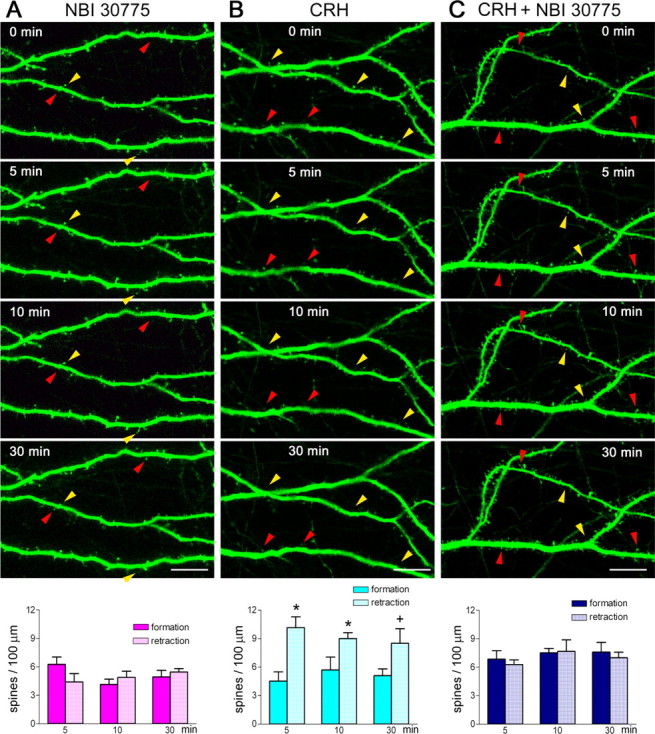
Spine disintegration induced by CRH requires activation of the receptor CRFR1. A, Application of the CRFR1 antagonist NBI 30775 (1 μm) for 30 min did not influence spine formation or elimination (F(1,36) = 0.1298; p = 0.7208, compared with untreated cultures). The red arrowheads denote newly formed spines, and the yellow ones show eliminated spines. B, Infusion of CRH (100 nm) accelerated spine disappearance (F(1,36) = 20.81; p < 0.0001). *p < 0.05, +p = 0.07, compared with formation at the same time point. C, This effect was blocked by infusion of the CRFR1 antagonist commencing 5–10 min before (and continuing during) CRH application (F(1,36) = 0.2529; p = 0.618, CRH+ antagonist compared with antagonist alone). Data are derived from five experiments, and 1367–2650 μm of dendrites were studied in each group. Organotypic cultures were initiated on P1 and grown for 5–6 d, followed by two-photon microscopy live imaging. Error bars indicate SEM. Scale bar, 11 μm.
CRH-induced acceleration of dendritic spine elimination involves regulation of F-actin
Spine retraction is mediated by a host of molecular cascades (Tada and Sheng, 2006; Fu et al., 2007). However, the rapidity of CRH-induced spine elimination suggested that the underlying mechanisms should operate within minutes. Spine integrity and morphology are modified within this timeframe during synaptic potentiation and depression (Zhou et al., 2004; Chen et al., 2007; Fu et al., 2007) associated with learning and memory processes (Bliss and Collingridge, 1993, Chen et al., 2007). Formation of polymerized, filamentous actin (F-actin) and the destabilization of this actin species by selective regulatory proteins provide a final common pathway for spine remodeling (Penzes et al., 2003; Tada and Sheng, 2006). Therefore, we examined whether the regulation of F-actin within dendritic spines changed after CRH application. We focused on activation (dephosphorylation) of the F-actin destabilizing protein cofilin, because the phosphorylation state of this protein changes within minutes by signals impacting spine stability (Chen et al., 2007).
In control organotypic hippocampal cultures, pCofilin was amply detectable within YFP-expressing dendritic spines (Fig. 7A). Within 5–10 min of exposure to 100 nm CRH, a significant reduction in pCofilin puncta within spines was apparent (Fig. 7B,C), denoting dephosphorylation of pCofilin and generation of the active form that contributes to disassociation of monomers from F-actin. The decline in pCofilin-immunoreactive puncta progressed with time (Fig. 7D), and the reduced number of pCofilin puncta per spine was significant even after accounting for the CRH-induced decrease in total spine density (Fig. 7D, right panel).
Figure 7.

CRH leads to rapid dephosphorylation (activation) of the F-actin-regulating protein, cofilin, within dendritic spines. A, Confocal images (1 μm) of YFP-expressing dendrites labeled also for phosphorylated (inactive) cofilin (pCofilin) demonstrate the presence of pCofilin immunoreactive puncta (red) within spine heads (arrows). B–D, CRH exposure (100 nm) for 5–30 min resulted in a significant reduction of pCofilin puncta (*p < 0.05). The CRH-induced reduction of pCofilin immunoreactive puncta per spine was still significant when the reduction of spine density by the peptide was taken into consideration (D, right panel). Note the similar time course of CRH-induced dephosphorylation of cofilin and of CRH-induced spine loss. Hippocampi were cultured on P1 and grown for 6 d. Data are derived from 20–40 sections per group, obtained in five experiments (n = 5). Error bars indicate SEM. Scale bar, 5 μm.
Discussion
The principal findings presented here are as follows: Stress reduces spine density within hours in vulnerable dendritic domains of hippocampal pyramidal cells. Blocking the principal receptor for the stress-activated neuropeptide, CRH, abrogates this rapid, stress-evoked spine loss; conversely, mice lacking CRFR1 have increased spine density in the same, stress-vulnerable dendritic domains. CRH activates CRFR1 to reduce spine density by accelerating spine retraction, an effect associated with activation of F-actin-destabilizing molecular cascades within the spine. Together, these findings indicate that CRH-mediated spine retraction contributes critically to stress-induced spine loss and associated dendritic atrophy.
Dendritic spines and synaptic function
Dendritic spines form, retract, and change their morphology in both developing and mature brain (Engert and Bonhoeffer, 1999; Segal, 2005; Park et al., 2006), and these dynamic changes constitute a structural component of synaptic plasticity (Calabrese and Halpain, 2005; Lin et al., 2005; Chen et al., 2007). Reduced spine number, such as resulting from stress-induced spine retraction, reduces the total postsynaptic area of excitatory synapses, influencing receptor density, synaptic signaling (Kennedy et al., 2005), and the number of functional excitatory synapses (Lin et al., 2005; Fu et al., 2007). These changes, in turn, may influence synaptic plasticity (Chen et al., 2007), as supported by the association of new spines and activated synapses (Engert and Bonhoeffer, 1999) and of spine loss with long-term depression (Nägerl et al., 2004; Okamoto et al., 2004; Zhou et al., 2004). Synaptic plasticity is generally believed to underlie hippocampus-mediated learning and memory functions (Bliss and Collingridge, 1993; Roman et al., 1987; Lin et al., 2005). Therefore, the increase in spine retraction provoked by stress and involving CRH may provide a neurobiological foundation for the established adverse actions of chronic stress (Kim and Diamond, 2002; Alfarez et al., 2003; Brunson et al., 2005) and of CRH (Brunson et al., 2001) on long-term potentiation and on hippocampus-mediated learning and memory.
Selective effects of stress and CRH on spine density
In this respect, it is interesting to note that both the stress- and the CRH-induced spine and dendritic losses were most apparent in a spatially circumscribed region within the apical dendrites of hippocampal pyramidal cells: The third- and fourth-order dendritic branches reside in stratum radiatum and provide postsynaptic anchoring sites for axons of the commissural/associational fibers. Whereas the basis of the specific vulnerability of these regions is unclear, reduced spine density in these regions predicts selective derangement of synaptic function in these pathways after stress. Indeed, profound deficits in commissural/associational synapses within CA3, and preservation of the more proximal mossy fiber synapses, have been recently demonstrated after chronic early-life stress (Brunson et al., 2005).
Mechanisms of CRH-induced spine disintegration
How might CRH, released during stress, cause spine loss and consequent dendritic atrophy? Of the two major CRH receptors, the current studies point to CRFR1 as the mediator of this effect. Time-lapse imaging demonstrated that CRH application led to rapid, selective elimination of dendritic spines, with little influence on the formation of new ones. This was associated with an equally rapid activation (dephosphorylation) of cofilin, an actin-regulating protein that permits removal of actin monomers from the filamentous, polymerized form of actin (F-actin). The rapid time course (5–10 min) of both these processes supports the possibility that activation of CRFR1 triggers selective structural destabilization of mature spines. In line with this speculation, electron microscopy has demonstrated that CRFR1 resides within the postsynaptic density (PSD) of spines, suggesting that the expression of this receptor is confined to mature, PSD-possessing spines (Chen et al., 2004b).
The signal cascades downstream of the G-protein-coupled CRFR1, which eventually result in spine disintegration, are unknown. Stabilization, degradation, and remodeling of filamentous actin, a molecule that provides the bulk of the cytoskeleton of the spine, are regulated by a family of Rho GTPases, including RhoA, Rac1, and cdc42 (Hall, 1998; Bamburg, 1999; Hering and Sheng 2001; Ethell and Pasquale, 2005). A PKA (protein kinase A)-dependent, Rac1-mediated mechanism has been found for the effects of CRFR1 activation on actin cytoskeleton in locus ceruleus neurons (Swinny and Valentino, 2006), and might operate in the hippocampus as well. Alternatively, RhoA, when constitutively expressed, promotes spine elimination (Govek et al., 2005; Sfakianos et al., 2007), although this might be a result of its redistribution away from the spine (Schubert et al., 2006). Because CRH provokes spine disintegration, it is reasonable to consider RhoA as a plausible key signaling molecule between CRFR1 activation and the actin cytoskeleton of the spine.
The rapidity of these actions of CRH on spine integrity also raises questions about the time course of detectable loss of spines after exposure to acute stress. Remarkably, in adult mice subjected to restraint stress, significant reduction of spine density was apparent within 5 h. This indicates that substantial spine plasticity persists in the adult hippocampus, at least in selective, stress-vulnerable dendritic domains.
Significance and relevance of CRH-induced spine derangements
Abrogation of stress-induced spine loss by an antagonist of CRH receptor activation provides direct evidence for a physiologically relevant role of the CRH–CRFR1 interaction in the effects of stress on spine and dendritic integrity. In addition to stress, CRH-mediated spine loss and the associated dendritic regression may contribute to the dendritic atrophy and cognitive decline associated with a number of human conditions. These include aging (Schierhorn, 1981; Arancio and Chao, 2007) and chronic depression, a disorder associated with high brain CRH levels and hippocampal dendritic atrophy (Nemeroff, 2004). Recently, CRH has been implicated in the molecular pathology of Alzheimer's disease (Holtzman, 2007). In addition, dendritic atrophy and spine loss characterize several developmental neurological disorders, including autism and Rett syndrome (Armstrong, 2001), and recent studies in a mouse model of the latter disorder demonstrated increased expression of the CRH gene (McGill et al., 2006). Together, this body of evidence supports a high relevance of CRH-mediated spine loss and related dendritic atrophy to both normal and pathological processes in the human brain.
In summary, the data presented here demonstrate a rapid effect of stress on dendritic spine integrity in the in vivo mature hippocampus. This effect involves acceleration of spine elimination via CRH–CRFR1 signaling and a net reduction of spine density with subsequent dendritic regression (Chen et al., 2004b; Pawlak et al., 2005) and impaired synaptic plasticity (Brunson et al., 2001, 2005; Segal, 2005). Therefore, future therapeutic methods targeting this novel molecular mechanism might allow prevention of cognitive decline associated with stress and a number of human disorders (McEwen, 1999; Brunson et al., 2005; De Kloet et al., 2005; Holtzman, 2007).
Footnotes
This work was supported by National Alliance for Research on Schizophrenia and Depression Young Investigator Award (Y.C.) and National Institutes of Health Grants NS 28912 and MH73136 (T.Z.B.).
References
- Alfarez D, Joels M, Krugers H. Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci. 2003;17:1928–1934. doi: 10.1046/j.1460-9568.2003.02622.x. [DOI] [PubMed] [Google Scholar]
- Arancio O, Chao MV. Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Rett syndrome: neuropathology (2000) Brain Dev. 2001;23:S72–S76. doi: 10.1016/s0387-7604(01)00332-1. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Brunson KL, Eghbal-Ahmadi M, Bender R, Chen Y, Baram TZ. Long-term progressive hippocampal cell loss and dysfunction induced by early-life administration of corticotropin releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci USA. 2001;98:8856–8861. doi: 10.1073/pnas.151224898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson KL, Kramar E, Lin B, Chen Y, Colgin LL, Yanagihara TK, Lynch G, Baram TZ. Mechanisms of late-onset cognitive decline after early-life stress. J Neurosci. 2005;25:9328–9338. doi: 10.1523/JNEUROSCI.2281-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Halpain S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron. 2005;48:77–90. doi: 10.1016/j.neuron.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Casale MS, Gall CM, Lynch G. Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci. 2007;27:5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Muller MB, Cariaga W, Baram TZ. Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF1)-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J Comp Neurol. 2000;420:305–323. doi: 10.1002/(sici)1096-9861(20000508)420:3<305::aid-cne3>3.0.co;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W, Baram TZ. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci USA. 2004a;101:15782–15787. doi: 10.1073/pnas.0403975101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Brunson KL, Adelmann G, Bender RA, Frotscher M, Baram TZ. Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience. 2004b;126:533–540. doi: 10.1016/j.neuroscience.2004.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fenoglio KA, Dube CM, Grigoriadis DE, Baram TZ. Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol Psychiatry. 2006;11:992–1002. doi: 10.1038/sj.mp.4001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Donohue HS, Gabbott PL, Davies HA, Rodríguez JJ, Cordero MI, Sandi C, Medvedev NI, Popov VI, Colyer FM, Peddie CJ, Stewart MG. Chronic restraint stress induces changes in synapse morphology in stratum lacunosum-moleculare CA1 rat hippocampus: a stereological and three-dimensional ultrastructural study. Neuroscience. 2006;140:597–606. doi: 10.1016/j.neuroscience.2006.02.072. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Ethell IM, Pasquale EB. Molecular mechanisms of dendritic spine development and remodeling. Prog Neurobiol. 2005;75:161–205. doi: 10.1016/j.pneurobio.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Fenoglio KA, Chen Y, Baram TZ. Neuroplasticity of the hypothalamic-pituitary-adrenal axis early in life requires recurrent recruitment of stress-regulating brain regions. J Neurosci. 2006;26:2434–2442. doi: 10.1523/JNEUROSCI.4080-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu WY, Chen Y, Sahin M, Zhao XS, Shi L, Bikoff JB, Lai KO, Yung WH, Fu AK, Greenberg ME, Ip NY. Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat Neurosci. 2007;10:67–76. doi: 10.1038/nn1811. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Holtzman J. Disparities in adverse birth outcomes may reflect influence of stress. Am J Public Health. 2007;97:1541. doi: 10.2105/AJPH.2007.114843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Khan S, Milot M, Lecompte-Collin J, Plamondon H. Time-dependent changes in CRH concentrations and release in discrete brain regions following global ischemia: effects of MK-801 pretreatment. Brain Res. 2004;1016:48–57. doi: 10.1016/j.brainres.2004.04.062. [DOI] [PubMed] [Google Scholar]
- Kim J, Diamond D. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci USA. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Kramar EA, Bi X, Brucher FA, Gall CM, Lynch G. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci. 2005;25:2062–2069. doi: 10.1523/JNEUROSCI.4283-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magarinos A, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995;69:89–98. doi: 10.1016/0306-4522(95)00259-l. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C, Zoghbi HY. Enhanced anxiety and stress-induced corticosterone release are associated with increased Crh expression in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2006;103:18267–18272. doi: 10.1073/pnas.0608702103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z, McIntosh J, Kent P, Michaud D, Anisman H. Aversive and appetitive events evoke the release of corticotropin-releasing hormone and bombesin-like peptides at the central nucleus of the amygdala. J Neurosci. 1998;18:4758–4766. doi: 10.1523/JNEUROSCI.18-12-04758.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. Early-life adversity, CRF dysregulation, and vulnerability to mood and anxiety disorders. Psychopharmacol Bull. 2004;38:14–20. [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, Ehlers MD. Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron. 2006;52:817–830. doi: 10.1016/j.neuron.2006.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci USA. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Preil J, Muller MB, Gesing A, Reul JM, Sillaber I, van Gaalen MM, Landgrebe J, Holsboer F, Stenzel-Poore M, Wurst W. Regulation of the hypothalamic-pituitary-adrenocortical system in mice deficient for CRH receptors 1 and 2. Endocrinology. 2001;142:4946–4955. doi: 10.1210/endo.142.11.8507. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- Roelandse M, Matus A. Hypothermia-associated loss of dendritic spines. J Neurosci. 2004;24:7843–7847. doi: 10.1523/JNEUROSCI.2872-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman F, Staubli U, Lynch G. Evidence for synaptic potentiation in a cortical network during learning. Brain Res. 1987;418:221–226. doi: 10.1016/0006-8993(87)90089-8. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57:925–935. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- Schierhorn H. Structural change in neocortical pyramidal neurons in man in the 5th to 9th decade. Psychiatr Neurol Med Psychol. 1981;33:664–673. [PubMed] [Google Scholar]
- Schubert V, Da Silva JS, Dotti CG. Localized recruitment and activation of RhoA underlies dendritic spine morphology in a glutamate receptor-dependent manner. J Cell Biol. 2006;172:453–467. doi: 10.1083/jcb.200506136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Sfakianos MK, Eisman A, Gourley SL, Bradley WD, Scheetz AJ, Settleman J, Taylor JR, Greer CA, Williamson A, Koleske AJ. Inhibition of Rho via Arg and p190RhoGAP in the postnatal mouse hippocampus regulates dendritic spine maturation, synapse and dendrite stability, and behavior. J Neurosci. 2007;27:10982–10992. doi: 10.1523/JNEUROSCI.0793-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Swinny JD, Valentino RJ. Corticotropin-releasing factor promotes growth of brain norepinephrine neuronal processes through Rho GTPase regulators of the actin cytoskeleton in rat. Eur J Neurosci. 2006;24:2481–2490. doi: 10.1111/j.1460-9568.2006.05129.x. [DOI] [PubMed] [Google Scholar]
- Tada T, Sheng M. Molecular mechanisms of dendritic spine morphogenesis. Curr Opin Neurobiol. 2006;16:95–101. doi: 10.1016/j.conb.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat Rev Neurosci. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]