

SUMMARY
Escherichia coli possesses a unique RNase activity that cleaves stop codons in the ribosomal aminoacyl-tRNA binding site (A-site) during inefficient translation termination. This A-site mRNA cleavage allows recycling of arrested ribosomes by facilitating recruitment of the tmRNA•SmpB ribosome rescue system. To test whether A-site nuclease activity also cleaves sense codons, we induced ribosome pausing at each of the six arginine codons using three strategies – rare codon usage, arginine starvation, and inactivation of arginine tRNAs with colicin D. In each instance, ribosome pausing induced mRNA cleavage within the target arginine codons, and resulted in tmRNA-mediated SsrA-peptide tagging of the nascent polypeptide. A-site mRNA cleavage did not require the stringent factor (ppGpp), or bacterial toxins such as RelE, which mediates a similar nuclease activity. However, the efficiency of A-site cleavage was modulated by the identity of the two codons immediately upstream (5′ side) of the A-site codon. Starvation for histidine and tryptophan also induced A-site cleavage at histidine and tryptophan codons, respectively. Thus, A-site mRNA cleavage is a general response to ribosome pausing, capable of cleaving a variety of sense and stop codons. The induction of A-site cleavage during amino acid starvation suggests this nuclease activity may help to regulate protein synthesis during nutritional stress.
Keywords: amino acid starvation, A-site mRNA cleavage, colicin D, ribosome pausing, tmRNA
INTRODUCTION
A-site mRNA cleavage is a novel RNase activity specific for codons within the aminoacyl-tRNA binding site of the ribosome. A-site cleavage is mediated by at least two A-site nucleases in Escherichia coli, of which only one has been identified. The E. coli RelE protein is a sequence-specific A-site nuclease that preferentially cleaves CAG and UAG codons 1. The other unidentified A-site nuclease activity has been shown to cleave UAA and UGA stop codons during inefficient translation termination 2; 3; 4. This latter activity is dramatically influenced by the identity of the last two amino acid residues of the nascent polypeptide, with Asp-Pro and Pro-Pro sequences favoring cleavage of A-site stop codons 2; 3. The physiological function of A-site mRNA cleavage is unclear, but Gerdes and colleagues have suggested that the RelE-mediated activity helps to regulate global protein synthesis during acute nutritional stress 5; 6. The role of RelE-independent A-site mRNA cleavage remains obscure, but may represent a mechanism for translational quality control 3. Both A-site nuclease activities produce truncated mRNAs that lack in-frame stop codons. Ribosomes accumulate at the 3′ ends of such nonstop messages and all eubacteria use the tmRNA•SmpB quality control system to “rescue” these stalled ribosomes 7; 8. tmRNA (transfer-messenger RNA) is a specialized RNA that functions first as a tRNA to bind the A-site of paused ribosomes, and then as an mRNA to direct the addition of the SsrA peptide tag to the C-terminus of the nascent polypeptide chain 7; 8. SmpB is a tmRNA-binding protein that is required for the delivery of tmRNA to arrested ribosomes, and for translation of the tmRNA-encoded SsrA peptide tag 9; 10. As a result of tmRNA•SmpB activity, the nascent polypeptide is SsrA-tagged for rapid proteolysis and the stalled ribosome undergoes normal translation termination and recycling.
In principle, ribosome pausing during translation elongation could induce the same A-site mRNA cleavage activity observed during inefficient translation termination. Several groups have reported that ribosome pausing during translation elongation leads to mRNA cleavage. However, these mRNA cleavages all occur at either the 5′- or 3′-boundaries of paused ribosomes, not within the A-site codon 11; 12; 13; 14; 15; 16. In the case of SecM-mediated translational arrest in E. coli, prolyl-tRNAPro is bound to the A-site and may inhibit efficient A-site cleavage 13; 17. In contrast, it is unclear why A-site cleavage has not been observed during translational pausing at non-preferred codons (also termed rare codons). Ribosomes tend to idle at rare codons with empty A-sites because the cognate tRNAs are typically present at low levels in the cell 18; 19; 20; 21, and it is well established that tmRNA acts readily on ribosomes paused at rare Arg codons 14; 22; 23; 24. However, a recent study showed that rare Arg codons in the A-site were not cleaved during ribosome pausing. Instead, the mRNA was cleaved at sites 10–12 nucleotides downstream of the paused ribosome A-site 14. Cleavage at these positions could be due to 3′Π5′ exonuclease activity degrading downstream mRNA up to the 3′-boundary of the paused ribosome 13. Taken together, these findings indicate that an unoccupied A-site is not sufficient to induce A-site mRNA cleavage during ribosome pausing.
The apparent lack of A-site cleavage at rare Arg codons (and sense codons in general) could indicate that A-site nuclease activity is specific for stop codons. Alternatively, there may be other requirements for efficient A-site mRNA cleavage such as specific nascent peptide sequences, which play an important role in A-site cleavage of stop codons 2; 3; 4. We developed strategies to pause ribosomes at specific sense codons such that the nascent peptide contained two consecutive C-terminal Pro residues and tested for A-site mRNA cleavage. In this model system, A-site mRNA cleavage occurred readily in response to a variety of experimentally induced translational arrests. Replacing the nascent peptide Pro-Pro residues with other dipeptides had a significant effect on mRNA cleavage, demonstrating that A-site cleavage activity can be modulated by sequence context. Our results show that several sense codons, including those encoding arginine (AGA, AGG, CGA, CGC, CGG and CGU), histidine (CAC and CAU), and tryptophan (UGG) are subject to A-site cleavage that is independent of RelE and its homologues. These data demonstrate that A-site mRNA cleavage is not limited to translation termination and suggest a role for A-site nuclease activity in regulating protein synthesis during acute amino acid starvation.
RESULTS
A-site mRNA cleavage of sense codons
Efficient A-site cleavage of stop codons requires an unoccupied A-site and specific C-terminal nascent dipeptide sequences, such as Pro-Pro or Asp-Pro 2; 3. We hypothesized that sense codons can be cleaved by A-site nuclease activity provided the ribosome carries a permissive nascent peptide during translational pausing. We used inducible, plasmid-borne constructs to express Flag-TrxA-PPRR, an N-terminal FLAG-tagged derivative of the E. coli thioredoxin (TrxA) protein in which the C-terminal Leu-Ala residues were replaced by Pro-Pro-Arg-Arg (Fig. 1a). Additionally, the sole Arg codon of wild-type trxA was mutated to Lys so that Flag-TrxA-PPRR only contains two Arg residues at its C-terminus. Initially, we analyzed three constructs in which the C-terminal Arg residues were coded as tandem rare AGG, AGA or CGG codons (Fig. 1a). Ribosomes pause at clusters of rare Arg codons because the corresponding cognate tRNAArg species are expressed at low levels in E. coli 18; 19; 21. Therefore, paused ribosomes will carry C-terminal nascent peptide sequences (Pro-Pro at the first rare codon and Pro-Arg at the second), which support efficient A-site mRNA cleavage at stop codons 2; 3.
Figure 1. A-site mRNA cleavage of rare Arg codons.

(a) The model flag-trxA(PPRR) mRNA is depicted with FLAG-encoding region and oligonucleotide probe binding sites indicated. Arg-84 of all flag-trxA(PPRR) constructs was changed to Lys as indicated (R84K). Flag-TrxA-PPRR residues Ala-116 to Arg-121 are shown along with the encoding mRNA sequence and the complementary sequence of the S1 nuclease protection probe. Arg-120 and Arg-121 were coded as tandem AGG, AGA or CGG codons as indicated. The 3′ end of the truncated in vitro transcript used in Northern blot and S1 protection analysis is shown. Arrows indicate the positions of BsaJI, NlaIV and BamHI endonuclease cleavages in the S1 protection probe used to generate gel migration standards. (b) Northern blot analyses of RNA from ΔtmRNA cells. Samples from cells expressing mRNA containing tandem AGG, AGA or CGG codons (labeled accordingly) were probed with a radiolabeled oligonucleotide probe specific for the ribosome binding site (RBS) of mRNA as depicted in (a). Lanes labeled 5, 4 and 3 indicate the samples came from ΔtmRNA cells overproducing tRNA5Arg (decodes AGG), tRNA4Arg (decodes AGA and AGG) and tRNA3Arg (decodes CGG), respectively 18; 60. The position of truncated flag-trxA(PPRR)AGG mRNA is indicated by the arrow. The control in vitro transcript was truncated after the first AGG codon as shown in (a). Northern analysis of tRNAArg species was performed using labeled oligonucleotide probes specific for each tRNA. (c) S1 nuclease protection of truncated flag-trxA(PPRR)AGG transcripts from ΔtmRNA cells was similar to that of the truncated control in vitro transcript, consistent with cleavage in and around the AGG codons. These protections were not seen in samples taken from tmRNA+ cells or ΔtmRNA cells overproducing tRNA4Arg or tRNA5Arg. No S1 protection was observed if the flag-trxA(PPRR)AGG mRNA was not induced with IPTG. The relative positions of BamHI, BsaJI and NlaIV digestion sites in the S1 probe with respect to the tandem AGG codons are shown in (a).
Northern blot analysis revealed the accumulation of truncated mRNA from each rare codon construct in cells lacking tmRNA (ΔtmRNA) (Fig. 1b). The accumulation of truncated mRNA was dependent on ribosome pausing at the rare Arg codons as determined by four criteria. First, cells expressing flag-trxA, which lacks rare Arg codons, did not accumulate truncated mRNA (data not shown). Second, cleaved mRNA appeared to be the same size as a control in vitro transcript truncated between the tandem AGG codons of flag-trxA(PPRR)AGG mRNA (Figs. 1a & 1b). Third, truncated mRNA did not accumulate to high levels in tmRNA+ cells (Fig. 1c and data not shown), presumably because tmRNA facilitates rapid non-stop mRNA degradation by delivering RNase R to the stalled ribosome complex 25. Finally, overproduction of cognate tRNAArg suppressed the accumulation of truncated mRNA (Figs. 1b & 1c). Additionally, Flag-TrxA-PPRR protein from each construct was efficiently tagged with SsrA(His6), a proteolysis-resistant, hexahistidine-containing SsrA peptide encoded by tmRNA(His6) 23; 26, and this tagging was inhibited by cognate tRNAArg overproduction (data not shown). Mass spectrometry of tagged Flag-TrxA-PPRR showed SsrA peptide tagging only at positions corresponding to the Arg residues (data not shown) consistent with previous studies 14; 22; 23; 24. Taken together, these data indicate that the observed mRNA cleavage activity is associated with translational pausing at rare Arg codons.
The mRNA cleavage sites were mapped by S1 nuclease protection analysis of flag-trxA(PPRR)AGG mRNA, using a 3′-radiolabeled oligonucleotide probe that hybridized to a region spanning the tandem AGG codons (Figs. 1a & 1c). S1 protection products corresponding to cleavage in the AGG codons and the stop codon were detected in samples from ΔtmRNA cells (Fig. 1c). This protection pattern was similar to that obtained with the control truncated in vitro transcript (Fig. 1c), consistent with A-site nuclease activity. S1 products corresponding to A-site cleaved mRNA were not detected in tmRNA+ cells and were significantly reduced by the overproduction of tRNA4Arg or tRNA5Arg (but not tRNA3Arg) in ΔtmRNA cells (Fig. 1c). Additional cleavages were detected 12 – 17 nucleotides downstream (3′ side) of the first AGG codon in samples taken from both tmRNA+ and ΔtmRNA cells (Fig. 1c). These downstream cleavages were not detected in cells overexpressing tRNA4Arg or tRNA5Arg suggesting that they are produced in response to ribosome pausing (Fig. 1c). Based on their position and dependence upon translational pausing, the downstream cleavages could be produced by 3′Π5′ exonucleases degrading mRNA to the 3′-boundary of the paused ribosome as described previously 13.
E. coli contains a family of small RNases that have been termed “toxins” because their expression inhibits protein synthesis and rapidly arrests cell growth 6. One member of this family, RelE, mediates ribosome-dependent A-site mRNA cleavage 1, whereas the other homologues (MazF, ChpBK, YoeB, and YafQ) appear to have RNase activity independent of the ribosome 27; 28; 29; 30. We analyzed flag-trxA(PPRR)AGG mRNA from cells in which the relE, mazF, chpBK, yoeB, and yafQ genes were deleted and found no significant difference in the extent or pattern of mRNA cleavage compared to wild-type cells (Fig. 1c). While we were performing these studies, a sixth member (YhaV) of the toxin family was identified and characterized 31. Further deletion of the yhaV gene in our five-toxin deletion strain resulted in no change in A-site cleavage at AGG codons (data not shown). We conclude that the six known E. coli toxin RNases are not required for the cleavage of rare Arg codons during ribosome pausing.
Nascent peptide and A-site mRNA cleavage
We next examined the effect of nascent peptide sequence on A-site mRNA cleavage by changing the tandem Pro residues of Flag-TrxA-PPRR to Asp-Thr, Lys-Lys, Ser-His and Leu-Ala. These sequences were chosen based on their frequency at the C-terminus of E. coli proteins. Pro-Pro (PP) and Asp-Thr (DT) are not found at the C-terminus of any E. coli protein, whereas Lys-Lys (KK) and Ser-His (SH) are statistically overrepresented as C-terminal residues 2. In contrast, Leu-Ala (LA) is the C-terminal sequence of wild-type thioredoxin and does not appear to be under any positive or negative selection in E. coli 2. We reasoned that the natural bias against certain dipeptide sequences at the C-terminus of E. coli proteins may reflect a selective pressure against A-site mRNA cleavage during translation termination, and that these dipeptide sequences could enhance or inhibit A-site mRNA cleavage at sense codons.
Northern blot analysis detected significant levels of truncated mRNA in ΔtmRNA cells expressing each nascent peptide variant construct (Fig. 2a). The accumulation of truncated mRNA was inhibited in cells overproducing tRNA5Arg, indicating that translational pausing at the tandem AGG codons was required for cleavage (Fig. 2a). The major species of truncated flag-trxA(SHRR)AGG and flag-trxA(DTRR)AGG mRNA were similar to that of flag-trxA(PPRR)AGG, consistent with cleavage in the AGG codons (Fig. 2a). In contrast, truncated flag-trxA(KKRR)AGG message migrated at a higher position in polyacrylamide gels than truncated flag-trxA(PPRR)AGG (Fig. 2a), suggesting cleavage at positions downstream of the AGG codons. Two discernable species of truncated flag-trxA(LARR)AGG mRNA were detected by Northern analysis (Fig. 2a), consistent with cleavage at both the AGG codons and downstream positions. S1 nuclease protection analysis of the flag-trxA(KKRR)AGG message detected cleavage primarily at positions 12 – 17 nucleotides downstream of the first AGG codon, with less S1 protection corresponding to cleavage within the second AGG codon (Fig. 2b). These data are not consistent with efficient A-site cleavage activity because most of the mRNA cleavage occurred downstream of the flag-trxA(KKRR)AGG stop codon (Fig. 2b). To determine whether these downstream cleavages led to tmRNA-mediated peptide tagging activity, we identified tagging sites in each of the protein variants (Fig. 2c and data not shown). As expected, SsrA(His6) peptide tags were observed at positions corresponding to the Arg residues of each protein (Fig. 2c and data not shown). However, there was no evidence of additional tagging corresponding to the downstream cleavage sites within flag-trxA(KKRR)AGG and flag-trxA(LARR)AGG messages (Fig. 2c and data not shown). These data are consistent with mRNA cleavage at the 3′-boundary of ribosomes paused at the AGG codons. SsrA peptide tagging of Flag-TrxA(KKRR) suggests that this 3′-boundary cleavage is sufficient for tmRNA recruitment to the A-site, consistent with in vitro work from Ehrenberg and colleagues 32. However, a low level of A-site cleavage could also account for the observed tmRNA activity.
Figure 2. Effect of nascent peptide on mRNA cleavage.
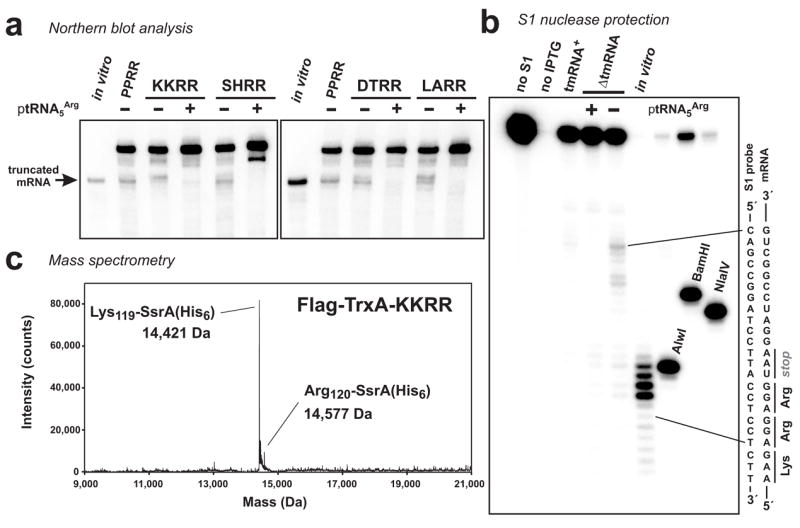
(a) Northern blot analyses of RNA isolated from ΔtmRNA cells. Translation of each mRNA leads to ribosome pausing at tandem rare AGG Arg codons in the context of Pro-Pro (PPRR), Leu-Ala (LARR), Ser-His (SHRR), Asp-Thr (DTRR) or Lys-Lys (KKRR) nascent peptides. Overproduction of tRNA5Arg suppressed the accumulation of truncated mRNA in each case. The position of truncated flag-trxA(PPRR)AGG mRNA (as described in Fig. 1a) is indicated by the arrow. Larger truncated transcripts were observed in cells expressing flag-trxA(LARR)AGG and flag-trxA(KKRR)AGG messages. (b) S1 nuclease protection of flag-trxA(KKRR)AGG message from ΔtmRNA cells showed prominent cleavages 12 – 17 nucleotides downstream of the first AGG codon. These protections were not seen ΔtmRNA cells overproducing tRNA5Arg. No S1 protection was observed if the flag-trxA(KKRR)AGG mRNA was not induced with IPTG. (c) Flag-TrxA-KKRR was expressed in tmRNA(His6) cells and purified by Ni2+ affinity chromatography. Mass spectrometry detected two species corresponding to the SsrA(His6) tag added after Lys-119 (calculated mass, 14,423.3 Da) and Arg-120 (calculated mass, 14,579.5). The same SsrA-peptide tagging sites were identified in each protein variant (data not shown).
In principle, downstream mRNA cleavage could also arise from a translational frameshift event that allows the ribosome to pause at a second site downstream of the stop codon. We examined each TrxA variant by Western blot using antibodies specific for the N-terminal FLAG epitope and detected additional products that migrated above the full-length proteins on SDS polyacrylamide gels (Fig. 3a). These alternative translation products accumulated to equal levels in ΔtmRNA and tmRNA+ cells, but were suppressed by tRNA5Arg overproduction (Fig. 3a and data not shown). To identify the alternative translation event, we subcloned each trxA gene into a series of three pET21-derived plasmids that encode a hexahistidine tag in each reading frame downstream of the stop codon. His6-tagged Flag-TrxA proteins were only produced when the His6 tag was in the +1 reading frame (data not shown). To determine the relationship between mRNA cleavage and +1 frameshifting, we quantified the percentage of frameshifting and compared it to the relative amounts of mRNA cleavage for each construct. Although +1 frameshifting occurred at a lower frequency during translation of Flag-TrxA-PPRR compared to the KKRR, DTRR, SHRR and LARR variants, there was no quantitative correlation between +1 frameshifting and downstream mRNA cleavage (Figs. 3b & 3c). Moreover, we did not observe tmRNA-mediated peptide tagging of any +1 frameshift products (Fig. 2c and data not shown). Taken together, these data suggest that the downstream cleavages were not a consequence of translational frameshifting. It appears that translational pausing does not elicit efficient A-site cleavage of certain messages, and instead allows degradation of downstream mRNA to the 3′-boundary of the paused ribosome.
Figure 3. Analysis of translational frameshifting at rare Arg codons.
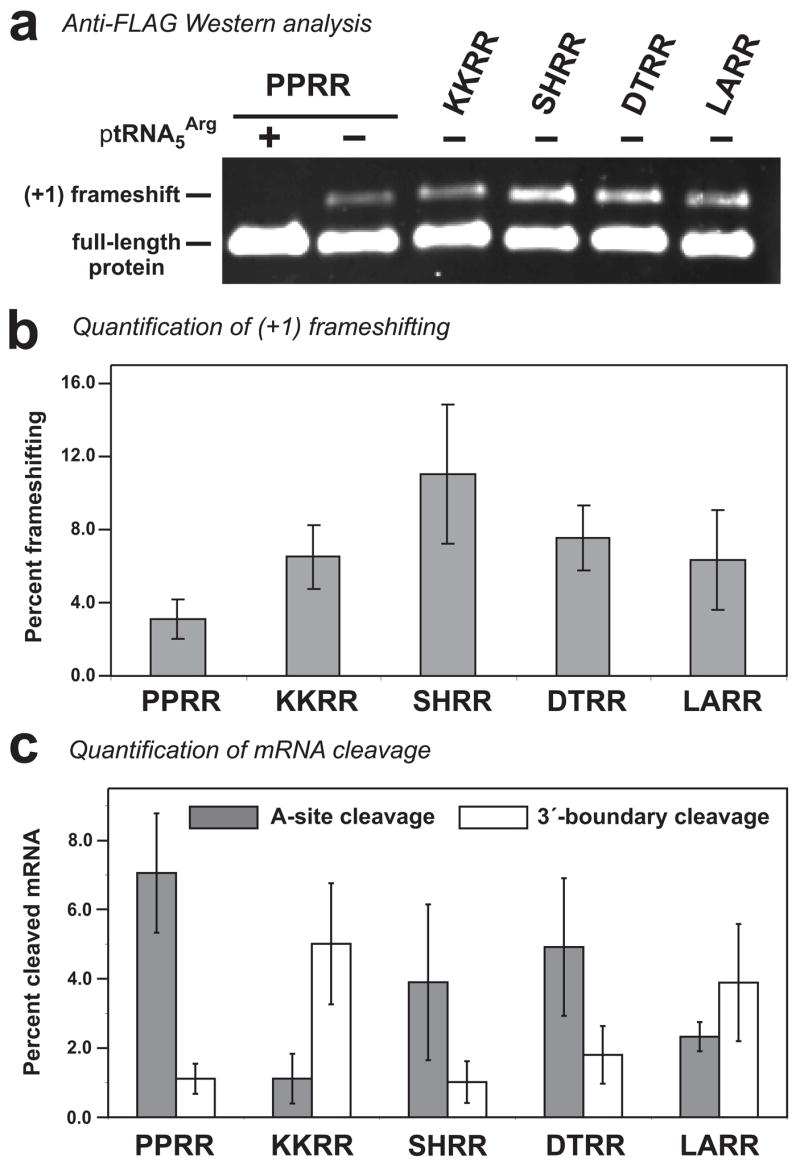
(a) Flag-TrxA protein variants were produced in ΔtmRNA cells and total protein analyzed by Western blot using a monoclonal antibody specific for the N-terminal FLAG epitope. The +1 frameshift products and full-length proteins are indicated. Overproduction of tRNA5Arg suppressed +1 frameshifting in each construct (+ptRNA5Arg, and data not shown). (b) The percentages of +1 frameshifting were determined from anti-FLAG fluorescent Western blot data using LI-COR® Odyssey software. The reported values are the average ± standard deviation determined from five independently prepared total protein samples. (c) The percentages of A-site cleaved and 3′-boundary cleaved products relative to full-length mRNA were determined from Northern blot phosphorimager data using the Quantity One (BioRad) software package. Reported values are the average ± standard deviation determined from five independently prepared total RNA samples.
Induction of A-site mRNA cleavage by amino acid starvation
Ribosome pausing should occur at any sense codon if the concentration of cognate aminoacylated tRNA is reduced sufficiently. We reasoned that starvation for an individual amino acid would lead to deacylation of specific tRNAs, allowing the corresponding codons to be tested as A-site cleavage substrates. Additional flag-trxA(PPRR) constructs were made in which the C-terminal Arg residues were encoded as tandem CGA, CGC, or CGU codons. These codons are decoded by the abundant tRNA2Arg isoacceptor in E. coli 18. Because these constructs only contain the two tandem Arg codons, ribosome pausing was specifically targeted to these codons during arginine starvation.
The three constructs were introduced into an arginine auxotrophic strain and the resulting cells grown in defined glucose minimal medium supplemented with arginine. After induction of mRNA expression, cultures were split in two, harvested by filtration, washed, and resuspended in defined media that either contained or lacked arginine. To confirm that the arginine starvation protocol decreased aminoacylation of tRNAArg, we resolved total RNA on acid-urea polyacrylamide gels and performed Northern blot analysis for tRNA2Arg (Fig. 4a). Under these electrophoresis conditions, arginyl-tRNA2Arg was resolved from deacylated tRNA2Arg, showing that virtually all tRNA2Arg was deacylated during arginine starvation (Fig. 4a). Northern blot analysis showed that each flag-trxA(PPRR) mRNA was truncated only in ΔtmRNA cells that had been starved for arginine (Fig. 4a). S1 nuclease protection analysis of the flag-trxA(PPRR)CGU mRNA demonstrated that cleavage occurred primarily within the two CGU codons during arginine starvation (Fig. 5b). Surprisingly, some S1 products corresponding to cleavage within the CGU codons were detected in the sample from arginine-fed ΔtmRNA cells, even though truncated mRNA was not readily apparent by Northern analysis (Figs. 4a & 5b).
Figure 4. Arginine starvation induces mRNA cleavage and tmRNA-mediated peptide tagging activities.
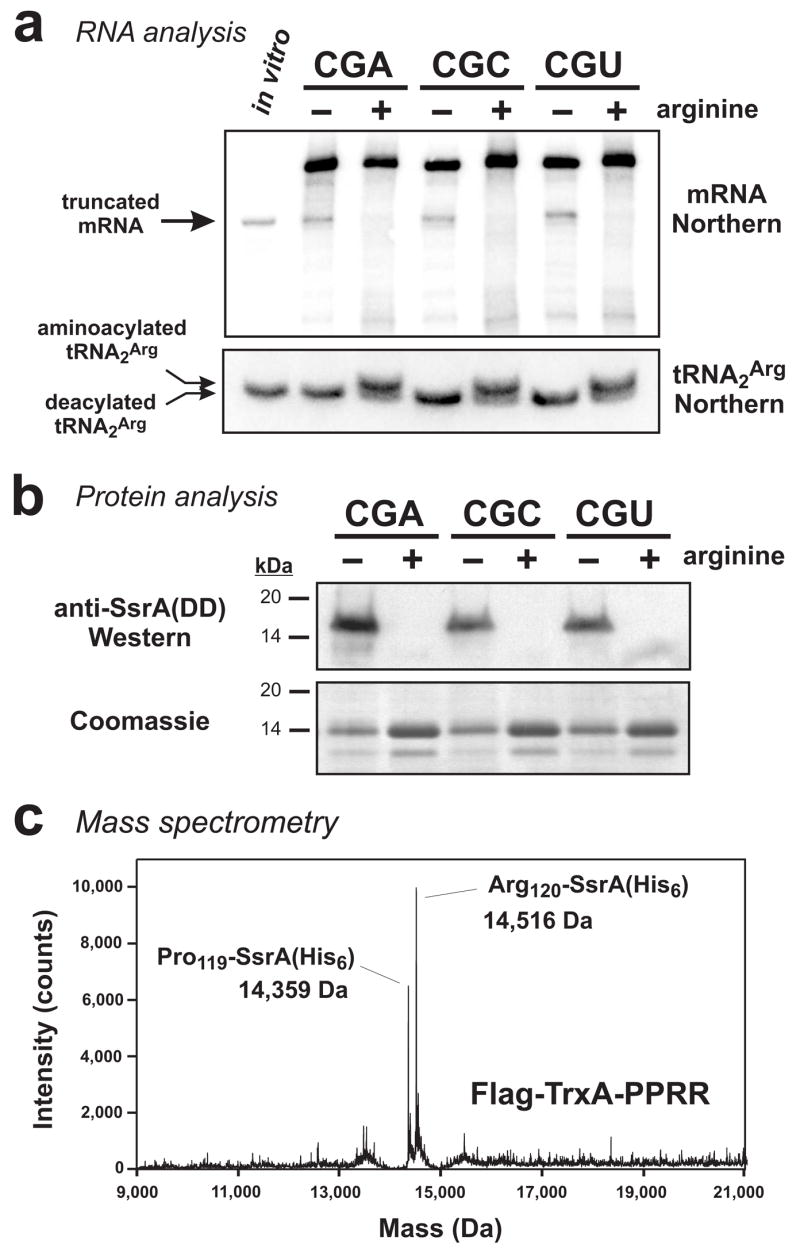
(a) Northern blot analyses of RNA isolated from ΔtmRNA cells that were arginine-fed (+) or starved for arginine (−). For each codon construct, truncated mRNA accumulated under arginine starvation conditions. The position of truncated mRNA indicated by the arrow corresponds to a control in vitro transcript of flag-trxA(PPRR)CGU that was truncated after the first CGU codon. RNA was resolved at low pH on acid-urea gels for Northern analysis of tRNA2Arg. Under these conditions, aminoacylated tRNA2Arg was resolved from deacylated tRNA2Arg. The sample loaded in the in vitro lane contained 0.2 pmole of truncated in vitro transcript mixed with 10 μg of total RNA that had been alkali-treated to deacylate tRNA. (b) Flag-TrxA-PPRR was produced in tmRNA(DD) cells under arginine-fed (+) or arginine-starved (−) conditions. Total protein was analyzed by SDS-PAGE followed by Coomassie blue staining, and by Western blot analysis using antibodies specific for the SsrA(DD) peptide tag. SsrA(DD) peptide tagging was only observed in response to arginine starvation. (c) Flag-TrxA-PPRR from the flag-trxA(PPRR)CGU construct was expressed in tmRNA(His6) cells under arginine starvation conditions and purified by Ni2+ affinity chromatography. Mass spectrometry detected two species corresponding to SsrA(His6) tag addition after Pro-119 (calculated mass, 14,361.2 Da) and Arg-120 (calculated mass, 14,517.4). Essentially identical results were obtained with Flag-TrxA-PPRR protein produced from the CGA and CGC coded constructs (data not shown).
Figure 5. S1 nuclease mapping of arginine starvation and colicin D-induced mRNA cleavages.
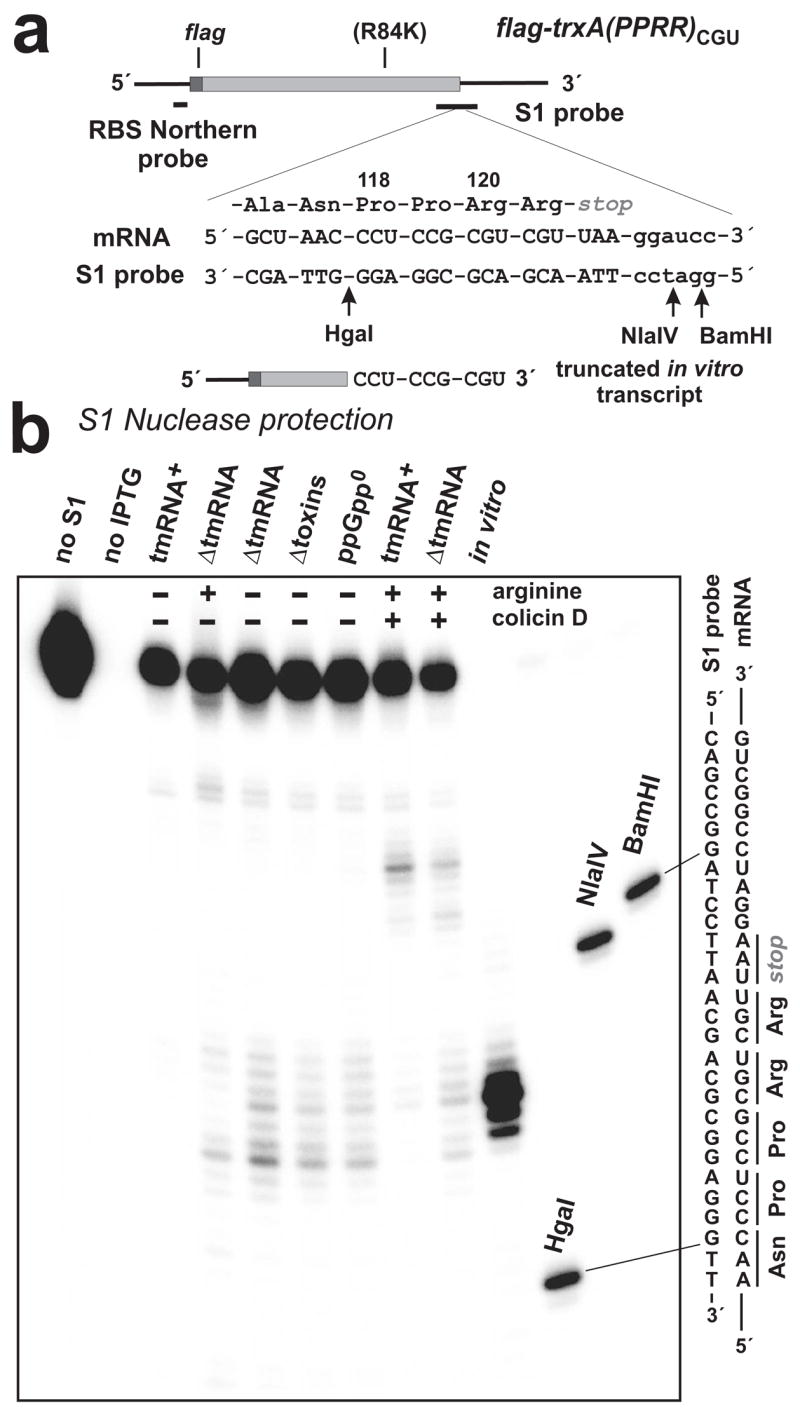
(a) The trxA(PPRR)CGU mRNA is depicted with FLAG encoding region and oligonucleotide probe binding sites indicated. Residues Ala-116 to Arg-121 of Flag-TrxA-PPRR are shown along with the encoding mRNA sequence and the complementary sequence of the S1 nuclease protection probe. Arg-120 and Arg-121 were coded as tandem CGU codons, and Arg-84 was changed to Lys as indicated (R84K). The truncated in vitro transcript used in Northern blot (Fig. 3a) and S1 protection analysis is shown, indicating the position of the 3′ terminus. Arrows indicate the positions of HgaI, NlaIV and BamHI endonuclease cleavages in the S1 probe used to generate gel migration standards. (b) The main S1 nuclease protections seen in ΔtmRNA cells subjected to arginine starvation mapped to the tandem CGU codons. Similar protections were seen in arginine-starved ΔtmRNA cells that were also deleted for RelE and related toxins (Δtoxins), and ΔtmRNA cells lacking ppGpp (ppGpp0). These protections were not seen in samples taken from tmRNA+ cells. In ΔtmRNA cells, colicin D treatment resulted in cleavages in the CGU codons and at sites 12 nucleotides downstream of second CGU codon, whereas only the downstream cleavages were detected in tmRNA+ cells. No S1 protection was observed in uninduced cells (no IPTG). The relative positions of HgaI, NlaIV and BamHI digestion sites in the S1 probe with respect to the tandem CGU codons are shown in (a).
A-site mRNA cleavage during arginine starvation should induce tmRNA-mediated SsrA tagging of Flag-TrxA-PPRR protein. We produced Flag-TrxA-PPRR protein in cells expressing tmRNA(DD), which encodes the proteolysis-resistant SsrA(DD) peptide tag 24, and analyzed protein produced under arginine starvation and arginine-fed conditions (Fig. 4b). SDS-PAGE analysis of cell extracts showed that less Flag-TrxA-PPRR was produced under arginine starvation conditions compared to the arginine-fed cells (Fig. 4b). Western blot analysis detected SsrA(DD)-tagged Flag-TrxA-PPRR only under arginine starvation conditions (Fig. 4b). To identify peptide tagging sites, we performed the same arginine starvation protocol in cells expressing tmRNA(His6), and analyzed purified SsrA(His6)-tagged proteins by mass spectrometry. Two protein species were identified with masses corresponding to tag addition after residues Pro-119 and Arg-120 of Flag-TrxA-PPRR produced from each construct (Figs. 4c and data not shown). These data, combined with results showing mRNA cleavage depends on translational pausing, strongly suggest that the Arg codons occupy the A-site when they are cleaved.
Arginine starvation has been shown to induce the stringent response in E. coli 33. The prokaryotic stringent response is mediated largely by guanosine tetraphosphate (ppGpp), which is synthesized by RelA in response to deacylated tRNA binding to the A-site 34; 35. Although ppGpp has been reported to inhibit transcription by T7 RNA polymerase 36, we saw no significant change in steady-state flag-trxA(PPRR) mRNA levels during arginine starvation (Figs. 4a & 5b). To uncover possible effects of RelA and ppGpp on A-site mRNA cleavage, we examined RNA samples prepared from arginine-starved ppGpp0 cells (which lack both ppGpp synthetases, RelA and SpoT), but found no significant difference in mRNA cleavage compared to relA+ spoT+ cells (Fig. 5b and data not shown). We conclude that the stringent response and ppGpp are not required for A-site mRNA cleavage induced by amino acid starvation. The RNase activities of RelE, MazF and other toxins are also induced by amino acid starvation 37; 38; 39. However, arginine starvation still induced cleavage of the CGU codons in cells lacking RelE, MazF, ChpBK, YoeB, and YafQ (Fig. 5b and data not shown). Deletion of the recently identified yhaV toxin gene 31, either individually or in combination with the other five toxin genes, had no effect on mRNA cleavage in response to arginine starvation (data not shown). Therefore, the six known toxin RNases are not required for mRNA cleavage at CGA, CGC and CGU codons during arginine starvation.
Colicin D treatment induces A-site cleavage of arginine codons
The results presented thus far suggest that A-site mRNA cleavage is a response to slow decoding. To test this hypothesis, we elicited translational pausing at Arg codons with colicin D. Colicin D is a plasmid-encoded tRNase that enters cells by the TonB pathway and specifically cleaves tRNAArg isoacceptors between residues 38 and 39 in the anticodon loop 40; 41; 42; 43. We reasoned that colicin D-cleaved tRNAArg would not be efficiently delivered to the ribosome, and elicit translational pausing specifically at Arg codons.
Northern blot analysis showed that colicin D treatment of cells resulted in the cleavage of a significant proportion of tRNA2Arg, as well as the accumulation of truncated flag-trxA(PPRR) mRNA (Fig. 6a). Two distinct truncated mRNA products were detected in ΔtmRNA cells, whereas only the higher molecular weight species accumulated in tmRNA+ cells (Fig. 6a). S1 protection mapping indicated that the smaller truncated messages were cleaved within the tandem Arg codons, whereas the larger products were cleaved at sites 12 – 17 nucleotides downstream of the first CGU codon (Fig. 5b). The colicin D-induced mRNA cleavage pattern differed from that observed with arginine starvation, where downstream cleavages were not readily apparent (Figs. 4a & 5b). The discrepancy in mRNA cleavage pattern could be explained if colicin D-cleaved tRNAArg is still delivered to the ribosomal A-site, but unable to add arginine to the nascent chain (see Discussion). If colicin D-cleaved tRNAArg is indeed delivered to the ribosome, its binding to the A-site must be transitory because colicin D treatment induced tmRNA-mediated peptide tagging of Flag-TrxA-PPRR (Fig. 6b), which requires an unoccupied A-site for tmRNA binding 13.
Figure 6. Colicin D treatment induces mRNA cleavage and tmRNA-mediated peptide tagging activities.
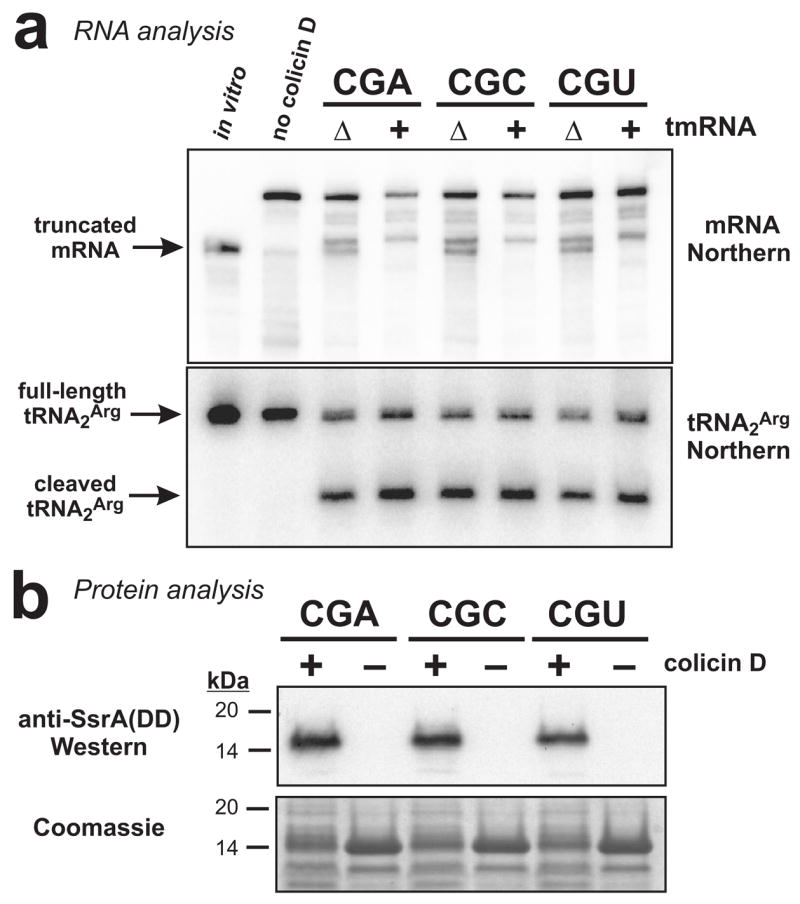
(a) Northern blot analyses of RNA isolated from cells treated with colicin D. For each codon construct, colicin D treatment produced truncated mRNA not observed in the untreated sample (no colicin D). Two distinct truncated messages were detected in ΔtmRNA cells, whereas only one accumulated in tmRNA+ cells. The position indicated by the arrow corresponds to a control in vitro transcript of trxA(PPRR)CGU truncated after the first CGU codon (in vitro). Northern analysis for tRNA2Arg showed a substantial fraction of the tRNA was cleaved in response to colicin D treatment. The sample loaded in the in vitro lane contained 0.2 pmole of truncated in vitro transcript mixed with 10 μg of total RNA isolated from cells that had not been incubated with IPTG or colicin D. (b) Flag-TrxA-PPRR was produced in tmRNA(DD) cells either treated with colicin D (+) or not (−). Total protein was analyzed by SDS-PAGE followed by Coomassie blue staining, and by Western blot analysis using antibodies specific for the SsrA(DD) peptide tag. SsrA(DD) peptide tag addition was only observed in samples taken from colicin D treated cells.
A-site mRNA cleavage at other sense codons
It is possible that Arg codons and stop codons are unique in their ability to be cleaved by A-site nuclease activity. Because amino acid starvation can be used to pause ribosomes at many codons, we tested whether histidine (His) and tryptophan (Trp) codons are also subject to A-site mRNA cleavage. Constructs analogous to flag-trxA(PPRR) were generated by mutating all internal His or Trp codons, followed by insertion of tandem His (CAC and CAU) or Trp (UGG) codons immediately downstream of the Pro-Pro coding sequence. Expression of the resulting flag-trxA(PPHH)CAC, flag-trxA(PPHH)CAU and flag-trxA(PPWW)UGG messages led to significant mRNA cleavage even in rich media (data not shown). This cleavage occurred within the stop codon of each mRNA (data not shown), which interfered with our attempts to induce ribosome pausing at the His and Trp codons by amino acid starvation. We suspected that the His-His and Trp-Trp nascent peptide sequences influenced stop codon cleavage, and sought to suppress this unwanted mRNA cleavage by inserting a Leu-Ala coding sequence in between the tandem His (or Trp) codons and the stop codon. The resulting flag-trxA(PPHHLA)CAC, flag-trxA(PPHHLA)CAU and flag-trxA(PPWWLA)UGG constructs no longer exhibited stop codon cleavage, and were introduced into the appropriate auxotrophic strains for amino acid starvation studies.
Northern blot analysis for tRNAs resolved on acid-urea gels confirmed that starvation for histidine and tryptophan resulted in essentially complete deacylation of tRNAHis and tRNATrp, respectively (Fig. 7a). Northern blot and S1 nuclease protection analyses revealed that each message was cleaved within the tandem His or Trp codons in response to amino acid starvation (Figs. 7a, 8b and data not shown). Additionally, the starvation protocols induced tmRNA-mediated peptide tagging of Flag-TrxA-PPHHLA and Flag-TrxA-PPWWLA proteins (Fig. 7b), with the SsrA(His6) peptide tag added at positions corresponding to the tandem Trp residues in the case of tryptophan starvation (Fig. 7c). As with the arginine starvation results, the convergence of translation arrest-dependent mRNA cleavage and tmRNA recruitment at the same sites strongly suggests that the His and Trp codons were cleaved by A-site nuclease activity.
Figure 7. Histidine and tryptophan starvation induces mRNA cleavage and tmRNA-mediated peptide tagging activities.
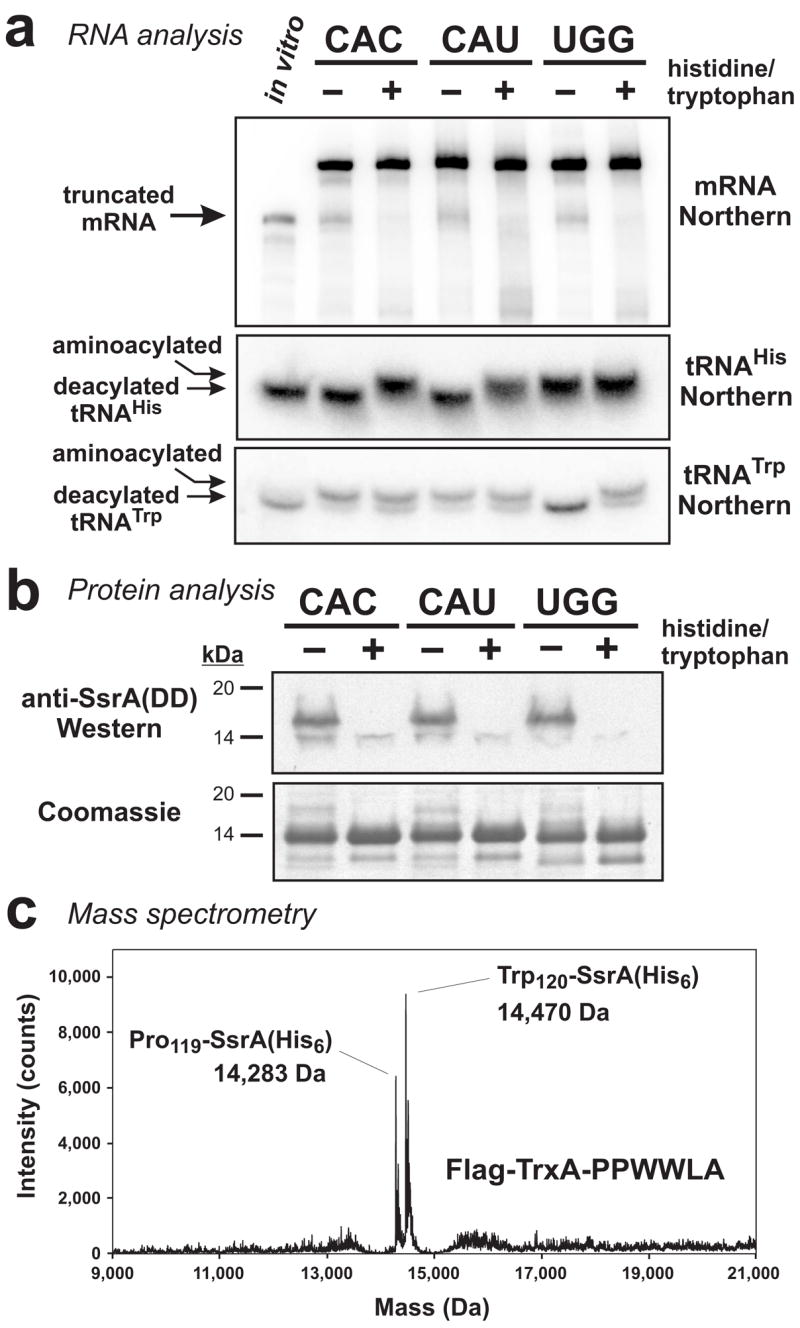
(a) Northern blot analyses of RNA isolated from ΔtmRNA cells that were fed (+) or starved (−) for histidine or tryptophan. Truncated mRNA from the CAC and CAU containing constructs was only produced during histidine starvation, and tryptophan starvation induced cleavage of the UGG containing mRNA. The position of truncated mRNA indicated by the arrow corresponds to a control in vitro transcript of flag-trxA(PPHHLA)CAU that was truncated after the first CAU codon. RNA was resolved at low pH on acid-urea gels for Northern analysis of tRNAHis and tRNATrp. Under these electrophoresis conditions, aminoacylated tRNAs were resolved from deacylated tRNAs. The sample loaded in the in vitro lane contained 0.2 pmole of truncated in vitro transcript mixed with 10 μg of total RNA that had been alkali-treated to deacylate tRNA. (b) Flag-TrxA-PPHHLA and Flag-TrxA-PPWWLA proteins were produced in tmRNA(DD) cells under amino acid-fed (+) or amino acid-starved (−) conditions. Total protein was analyzed by SDS-PAGE followed by Coomassie blue staining, and by Western blot analysis using antibodies specific for the SsrA(DD) peptide tag. SsrA(DD) peptide tag addition was only observed during amino acid starvation. (c) Flag-TrxA-PPWWLA was expressed in tmRNA(His6) cells under tryptophan starvation conditions and purified by Ni2+ affinity chromatography. Mass spectrometry detected two species corresponding to SsrA(His6) peptide tag addition after Pro-119 (calculated mass, 14,283.1 Da) and Trp-120 (calculated mass, 14,469.3 Da).
Figure 8. S1 nuclease mapping of tryptophan starvation-induced mRNA cleavages.
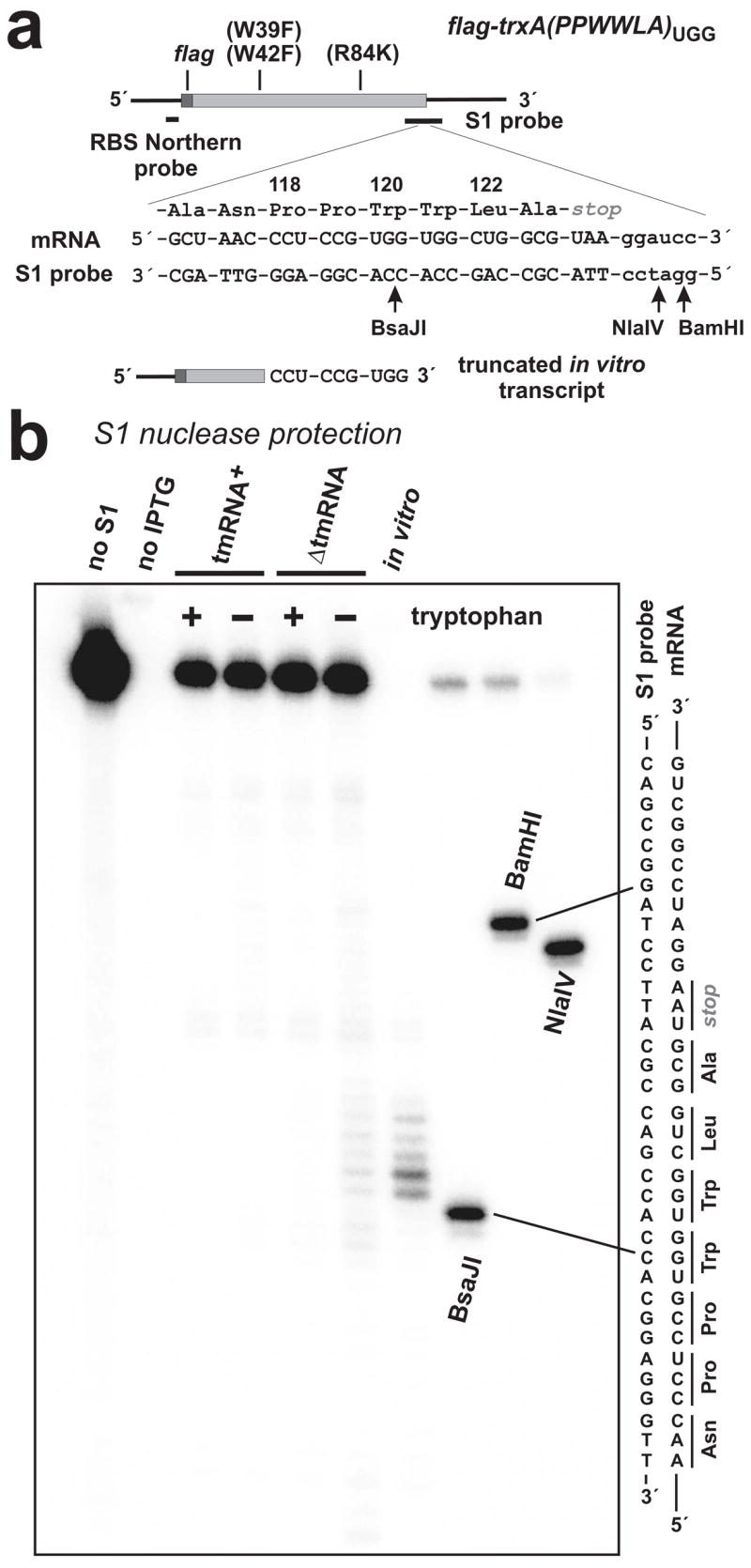
(a) The trxA(PPWWLA) mRNA is depicted with FLAG coding region and oligonucleotide probe binding sites indicated. Trp-39 and Trp-42 were changed to Phe residues as indicated. Residues Ala-116 to Ala-123 of Flag-TrxA-PPWWLA are shown along with the encoding mRNA sequence and the complementary sequence of the S1 nuclease probe. The truncated in vitro transcript used in Northern blot and S1 protection analysis is shown, indicating the position of the 3′-terminus. Arrows indicate the positions of HgaI, NlaIV and BamHI endonuclease cleavages in the S1 probe used to generate gel migration standards. (b) S1 nuclease protections detected in ΔtmRNA cells subjected to tryptophan starvation mapped to the tandem UGG codons. These protections were not seen in tmRNA+ cells or ΔtmRNA cells that were fed tryptophan. No S1 protection was observed in uninduced cells (no IPTG). The relative positions of HgaI, NlaIV and BamHI digestion sites in the S1 probe with respect to the tandem UGG codons are shown in (a).
DISCUSSION
The experiments presented here show that mRNA is cleaved at a variety of sense codons in response to ribosome pausing. Several observations argue that this mRNA cleavage occurs within the A-site codon during translational arrest. Perhaps most importantly, codons were only cleaved under conditions that interfere with rapid decoding of the A-site. Accordingly, mRNA cleavage was remarkably specific for the A-site codon during ribosome arrest: His codons were cleaved only in response to histidine starvation, Trp codons only during tryptophan deprivation, and Arg codons only during arginine starvation or colicin D treatment. Moreover, three independent translational arrest strategies – rare codon usage, starvation for a single amino acid, and specific tRNA inactivation – all led to cleavage of target codons, strongly suggesting that mRNA cleavage requires an unoccupied ribosome A-site. Additionally, ribosome pausing at target codons was confirmed by SsrA peptide tag addition at corresponding positions within the thioredoxin protein variants. Although it is formally possible that mRNA cleavage occurred prior to ribosome arrest, it is unclear how a nuclease could differentiate between rapidly and slowly decoded codons without using the A-site as a framework. The simplest model accounting for these data is that prolonged ribosome pausing with an empty A-site induces cleavage of the A-site codon.
The importance of an empty A-site suggests the existence of a trans-acting A-site nuclease that competes with translation factors for access to the A-site codon. Indeed, the E. coli RelE protein appears to be such a factor 1. However, the RNase activity described in this work was not mediated by RelE or any of the known toxin proteins. It is possible that E. coli contains an additional uncharacterized, trans-acting A-site nuclease(s). Alternatively, A-site mRNA cleavage may be a latent activity of the ribosome itself 3. The ribosome is required for A-site mRNA cleavage and several highly conserved 16S rRNA residues are near enough to the A-site codon to mediate cleavage 44; 45. Although an empty A-site is presumably required for a trans-acting nuclease to gain access to the A-site codon, this requirement does not necessarily exclude a catalytic role for the ribosome. A-site occupancy may explain the difference in mRNA cleavage pattern between arginine-starved and colicin D-treated cells. The prominent 3′-boundary cleavage seen in colicin D-treated cells could be indicative of cleaved tRNAArg binding the A-site transiently. Colicin D cuts tRNAArg at the 3′ end of the anticodon loop, leaving the anticodon triplet intact 43. Furthermore, acid-urea gel analysis showed that the majority of colicin D-cleaved tRNA2Arg is charged with arginine (data not shown), which would be required to form a ternary complex with EF-Tu and GTP for delivery to the A-site. We have been unable to directly demonstrate binding of colicin D-cleaved tRNAArg to the ribosome, presumably because the interaction is weak. However, similar cleavages occur at the 3′-boundary of SecM-arrested ribosomes, which have prolyl-tRNAPro bound stably to the A-site 13; 17.
A-site cleavage activity has now been demonstrated at several sense (AGA, AGG, CAC, CAU, CGA, CGC, CGG and UGG) and stop codons (UAA and UGA) 3. Although the tested codons all contain a purine at the second position, this A-site nuclease activity appears to display little sequence specificity. This contrasts sharply with RelE activity, which cleaves CAG ~3,600 times more rapidly than CGA 1. In fact, the A-site codon appears to exert less of an effect on cleavage activity than the codons immediately upstream (5′ side) of the A-site. The two upstream codons could modulate A-site cleavage directly by virtue of their position in the E- and P-sites of the paused ribosome, or indirectly via their cognate tRNAs. Alternatively, the encoded nascent peptide may modulate A-site cleavage, consistent with work showing that C-terminal residues of the nascent peptide are critical for A-site cleavage of stop codons 3; 4; 23; 46. The role of the nascent peptide is probably indirect because the C-terminal residues of the nascent chain are near the peptidyltransferase center on the 50S subunit, whereas the A-site codon is bound by the 30S subunit some 75 – 80 Å away. We envision at least two models to account for the nascent peptide effect on A-site cleavage. The nascent peptide may directly bind to a trans-acting nuclease, or perhaps induce conformational changes in the ribosome that allow the nuclease to bind the A-site. Alternatively, the nascent peptide may induce a latent A-site nuclease activity present within the ribosome itself. Regulation of ribosome activity by the nascent peptide has been reported in many organisms including bacteria, fungi, vascular plants, and mammals 47; 48; 49. Perhaps A-site mRNA cleavage is yet another example of this widespread phenomenon. Although A-site cleavage has not yet been observed in purified translation systems 32, to our knowledge, no one has studied nascent peptides that support efficient A-site mRNA cleavage.
Because translational pausing at rare codons is a function of cognate tRNA availability, ribosomes should pause at a given codon to the same extent independent of genetic context. However, A-site cleavage of rare AGG codons varies depending on upstream codon sequence. These findings raise the possibility that rare codons are embedded in sequence contexts designed to either facilitate or inhibit A-site mRNA cleavage. In general, Pro:ArgRARE codon pairs are not particularly underrepresented in the E. coli genome, and several codon pairs with potential for efficient A-site cleavage (CCG:AGG, CCC:AGG, CCA:AGA and CCC:AGA) are statistically overrepresented in E. coli 50. Other potential hotspots for A-site cleavage are messages that undergo regulated ribosome pausing. In general, regulatory ribosome pauses in prokaryotes serve to modify mRNA secondary structure, and thereby influence either transcription termination (transcriptional attenuation), or translation initiation at a downstream cistron (translational attenuation) 47; 51; 52. A-site cleavage activity physically separates paused ribosomes from downstream mRNA and could conceivably interfere with both transcriptional and translational attenuation. We have previously shown that the E. coli SecM-mediated ribosome pause does not elicit A-site cleavage, presumably because the A-site is occupied by prolyl-tRNA during ribosome arrest 13. Similarly, E. coli TnaC-arrested ribosomes probably contain release factor-2 bound in the A-site 53, preventing A-site cleavage of the stop codon. In contrast, many operons are regulated by transcriptional attenuation in which reduced levels of aminoacylated-tRNA induce site-specific ribosome pausing during the synthesis of small regulatory leader peptides. The best characterized of these systems is the E. coli trp operon, in which ribosome pausing at two tandem Trp codons in the trp leader sequence facilitates transcription antitermination and expression of the entire trp operon. The E. coli his operon and several others involved in amino acid biosynthesis are regulated in a similar fashion. Although Trp and His codons are subject to A-site cleavage, this activity could interfere with attenuation. Therefore we suspect that A-site cleavage does not occur during ribosome pausing on the trp and his leaders. On the other hand, A-site nuclease activity may not influence the switch between terminator and antiterminator structures, and consequently have little effect on the attenuation mechanism. Because we can induce efficient A-site cleavage at Trp and His codons, we are in a position to test whether A-site mRNA cleavage affects transcriptional attenuation.
The prokaryotic stringent response controls many changes in gene expression during amino acid starvation, such as increased transcription of genes involved in amino acid biosynthesis and decreased synthesis of rRNA, tRNA and translation factors 34; 35. These transcriptional effects serve to streamline protein synthesis capacity and allow adaptation to new environments where amino acids are scarce. Our results show that acute amino acid starvation also leads to A-site mRNA cleavage. Gerdes and colleagues have previously reported RelE-catalyzed A-site mRNA cleavage occurs in response to amino acid starvation. Moreover, they have proposed that RelE acts as a stress response regulator that transiently inhibits protein synthesis in response to amino acid starvation, and facilitates amino acid recovery via tmRNA-mediated peptide tagging and subsequent proteolysis 5; 6; 38. The RelE-independent A-site nuclease activity described here may play a similar role, and perhaps the two RNase activities are complementary to ensure cleavage at a wide variety of codons. In addition, A-site cleavage could play a role in the attenuation of ppGpp synthesis during amino acid starvation. The concentration of ppGpp rises dramatically upon amino acid starvation and then decreases within several minutes to lower levels 34; 54. A-site cleavage may help to shut down RelA-dependent ppGpp synthesis by preventing the binding of deacylated tRNA to the A-site. Moreover, A-site cleavage removes mRNA protruding from the 3′-boundary of paused ribosomes, which has recently been shown to be critical for RelA binding 55. It will be important to determine the extent to which A-site mRNA cleavage and tmRNA•SmpB-mediated ribosome rescue work with the stringent response to control bacterial gene expression during amino acid starvation.
MATERIALS AND METHODS
Bacterial strains and plasmids
Table I lists the bacterial strains and plasmids used in this study. All bacterial strains were derivatives of E. coli strain X90 2; 13. The argH (encoding argininosuccinate lyase) and hisD (encoding histidinol dehydrogenase) genes were deleted using the phage λ Red recombination method with minor modifications as described 2; 56. The trpE (encoding anthranilate synthase component I) deletion was obtained from the Keio collection of E. coli single-gene knockout mutants 57. All gene disruptions and deletions were introduced into strains CH12 and CH113 2; 13 by phage P1-mediated transduction. Strain CH2385 containing the ssrA(DD) allele of tmRNA was constructed as described 13. Strain CH1256 containing deletions of the relBE, chpBIK, yefM-yoeB, mazEF and dinJ-yafQ antitoxin-toxin modules was constructed by sequential deletion followed by removal of kanamycin resistance cassettes using the FLP recombinase as described 3; 56. Strain CH3525 was generated by removal of the kanamycin cassette in strain CH1256, followed by transduction of the ΔyhaV::kan allele from the Keio collection 57. The details of all strain constructions are available upon request.
TABLE I.
Bacterial strains and plasmids
| Strain or plasmid | Genotype or descriptiona | Reference |
|---|---|---|
| Strains | ||
| X90 | F′ lacIq lac′ pro′/ara Δ[lac-pro] nalA argE[am] rifr thi-1 | (2) |
| CH12 | X90 (DE3) | (23) |
| CH113 | X90 ssrA::cat (DE3), Cmr | (23) |
| CH1256 | CH113 ΔrelBE ΔchpBIK ΔyefM-yoeB ΔmazEF ΔdinJ-yafQ ΔargH::kan, Cmr, Kanr | This study |
| CH2046 | CH113 ΔargH::kan, Cmr, Kanr | This study |
| CH2073 | CH12 ΔargH::kan, Kanr | This study |
| CH2092 | CH113 ΔhisD::kan, Cmr, Kanr | This study |
| CH2198 | CH12 ssrA(his6) | (13) |
| CH2385 | CH12 ssrA(DD) | This study |
| CH2618 | CH12 ΔssrA ΔargH ΔrelA::kan spoT::cat | (3) and this study |
| CH2819 | CH12 ssrA(his6) ΔtrpE::kan, Kanr | This study |
| CH2824 | CH12 ssrA(DD) ΔhisD::kan, Kanr | This study |
| CH2825 | CH12 ssrA(DD) ΔtrpE::kan, Kanr | This study |
| CH3038 | CH113 ΔtrpE::kan, Cmr, Kanr | (57) and this study |
| CH3039 | CH12 ΔtrpE::kan, Kanr | (57) and this study |
| CH3040 | CH12 ΔhisD::kan, Kanr | This study |
| CH3278 | CH12 ssrA(DD) ΔargH::kan, Kanr | This study |
| CH3525 | CH1256 ΔargH ΔyhaV:kan, Cmr, Kanr | (57) and this study |
| Plasmids | ||
| pET11d | T7 RNA polymerase expression plasmid, Ampr | Novagen (EMD) |
| pFG11d | Derivative of pET11d encoding the FLAG peptide epitope, Ampr | This study |
| pET21b | T7 RNA polymerase expression plasmid encoding His6 peptide epitope, Ampr | Novagen (EMD) |
| pFG21a | Derivative of pET21b with His6 epitope coded in the (0) reading frame, Ampr | This study |
| pFG21b | Derivative of pET21b with His6 epitope coded in the (+1) reading frame, Ampr | This study |
| pFG21c | Derivative of pET21b with His6 epitope coded in the (−1) reading frame, Ampr | This study |
| pET21b::colD-immD | Plasmid expressing colicin D and immunity protein, Ampr | This study |
| pCH201 | Derivative of pACYC184 expressing tmRNA(His6), Tetr | (23) |
| pCH405Δ | Derivative of pACYC184 containing multi-cloning site, Tetr | (2) |
| ptRNA5Arg | pCH405Δ::argW, overproduces tRNA5Arg, Tetr | (2) |
| ptRNA4Arg | pCH405Δ::argU, overproduces tRNA4Arg, Tetr | This study |
| ptRNA3Arg | pCH405Δ::argX-hisR, overproduces tRNA3Arg and tRNAHis, Tetr | This study |
| pFlag-TrxA | pFG11d::trxA(R73K), Ampr | This study |
| pFlag-TrxA(PPRR)AGA | Pro-Pro-Arg-Arg in place of Leu118Ala119, Arg coded as AGA, Ampr | This study |
| pFlag-TrxA(PPRR)AGG | Pro-Pro-Arg-Arg in place of Leu118Ala119, Arg coded as AGG, Ampr | This study |
| pFlag-TrxA(PPRR)CGG | Pro-Pro-Arg-Arg in place of Leu118Ala119, Arg coded as CGG, Ampr | This study |
| pFlag-TrxA(PPRR)CGA | Pro-Pro-Arg-Arg in place of Leu118Ala119, Arg coded as CGA, Ampr | This study |
| pFlag-TrxA(PPRR)CGC | Pro-Pro-Arg-Arg in place of Leu118Ala119, Arg coded as CGC, Ampr | This study |
| pFlag-TrxA(PPRR)CGU | Pro-Pro-Arg-Arg in place of Leu118Ala119, Arg coded as CGT, Ampr | This study |
| pFlag-TrxA(LARR)AGG | tandem AGG codons introduced downstream of Leu118Ala119, Ampr | This study |
| pFlag-TrxA(DTRR)AGG | Asp-Thr-Arg-Arg in place of Leu118Ala119, Arg coded as AGG, Ampr | This study |
| pFlag-TrxA(SHRR)AGG | Ser-His-Arg-Arg in place of Leu118Ala119, Arg coded as AGG, Ampr | This study |
| pFlag-TrxA(KKRR)AGG | Lys-Lys-Arg-Arg in place of Leu118Ala119, Arg coded as AGG, Ampr | This study |
| pFlag-TrxA(PPHHLA)CAC | Pro-Pro-His-His placed between Asn117 and Leu118, His coded as CAC, Ampr | This study |
| pFlag-TrxA(PPHHLA)CAU | Pro-Pro-His-His placed between Asn117 and Leu118, His coded as CAT, Ampr | This study |
| pFlag-TrxA(PPWWLA)UGG | Pro-Pro-Trp-Trp placed between Asn117 and Leu118, Ampr | This study |
Ampr, ampicillin resistant; Cmr, chloramphenicol resistant; Kanr, kanamycin resistant; Tetr, tetracycline resistant
Plasmid pFG11d is a derivative of pET11d (Novagen) encoding the Met-Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys-His-Met peptide, where the underlined residues indicate the FLAG epitope tag. The His-Met peptide linker is encoded by an NdeI restriction site (CAT-ATG), which allows the initiating Met codon of any gene to be fused in-frame to the FLAG encoding sequence. All thioredoxin gene (trxA) variants were ligated to pFG11d via NdeI and BamHI restriction sites. The trxA gene was PCR amplified from E. coli genomic DNA using the following oligonucleotide primers containing restriction endonuclease sites (underlined residues): trxA-Nde, (5′-GTG GAG TTA CAT ATG AGC GAT AAA ATT ATT CAC C); and trxA-Bam, (5′-ATC GGA TCC TTA CGC CAG GTT AGC GTC GAG G). The sole Arg codon of wild-type trxA (Arg-73) was changed to lysine using primer trxA-R73K, (5′-CGA AAT ATG GCA TCA AAG GTA TCC CGA C) by the PCR megaprimer method 58, in conjunction with primers trxA-Nde and trxA-Bam. The resulting plasmid, pFlag-TrxA, expresses a FLAG-tagged version of thioredoxin containing eleven additional residues at the N-terminus. In this report, we use Flag-TrxA as the reference for amino acid residue number. Thus, Arg-73 of wild-type thioredoxin becomes Arg-84 of Flag-TrxA.
Plasmid pFlag-TrxA was used as a PCR template to generate a series of constructs encoding modified C-terminal tetrapeptide sequences. Plasmids pFlag-TrxA(PPRR)AGG, pFlag-TrxA(PPRR)AGA, pFlag-TrxA(PPRR)CGG, pFlag-TrxA(PPRR)CGU, pFlag-TrxA(PPRR)CGC, pFlag-TrxA(PPRR)CGA, pFlag-TrxA(LARR)AGG, pFlag-TrxA(SHRR)AGG, pFlag-TrxA(DTRR)AGG and pFlag-TrxA(KKRR)AGG were constructed using primer trxA-Nde in conjunction with primers: trxA(PPRR)AGG, (5′-ATC GGA TCC TTA CCT CCT CGG AGG GTT AGC GTC GAG); trxA(PPRR)AGA, (5′-GCC GGA TCC TTA TCT TCT CGG AGG GTT AGC G); trxA(PPRR)CGG, (5′-GCC GGA TCC TTA CCG CCG CGG AGG GTT AGC G); trxA(PPRR)CGU, (5′-GCC GGA TCC TTA ACG ACG CGG AGG GTT AGC G); trxA(PPRR)CGC, (5′-GCC GGA TCC TTA GCG GCG CGG AGG GTT AGC G); trxA(PPRR)CGA, (5′-GCC GGA TCC TTA TCG TCG CGG AGG GTT AGC G); trxA(LARR)AGG, (5′-ATC GGA TCC TTA CCT CCT CGC CAG GTT AGC GTC GAG G); trxA(SHRR)AGG, (5′-CCG GAT CCT TAC CTC CTA TGC GAG TTA GCG TCG AGG); trxA(DTRR)AGG (5′-CCG GAT CCT TAC CTC CTG GTG TCG TTA GCG TCG AGG); trxA(KKRR)AGG, (5′-CCG GAT CCT TAC CTC CTC TTC TTG TTA GCG TCG AGG), respectively.
The pFlag-TrxA(PPHHLA) expression plasmids were derived from plasmid pFlag-TrxA(PPRR)CGU using the PCR megaprimer method 58. Codons His-10 and His-17 were mutated to glutamine codons during the first PCR using oligonuleotides: trxA(His→Gln), (5′ - CAG TTG AAT AAT TTT ATC GCT CAT TTG CTT G) and pET-Sph, (5′ - CAA GGA ATG GTG CAT GCA AGG AGA TGG CGC CC). The second PCR used primer trxA(PPHHLA)CAC, (5′ - CGG ATC CTT ACG CCA GGT GGT GCG GAG GGT TAG CG); or trxA(PPHHLA)CAU, (5′ - CCG GAT CCT TAC GCC AGA TGA TGC GGA GGG TTA GCG) in conjunction with the megaprimer to add the His-His-Leu-Ala coding sequence. A similar megaprimer strategy was used to construct pFlag-TrxA(PPWWLA). Trp-39 and Trp-42 were changed to Phe codons using oligonucleotides: trxA(Trp→Phe), (5′ - CCG CAA AAC TCT GCA AAG AAA TCG ACG) and pET-Sph. The second PCR used oligonucleotide trxA(PPWWLA)UGG, (5′ - CCG GAT CCT TAC GCC AGC CAC CAC GGA GGG TTA GCG) to introduce the Trp-Trp-Leu-Ala sequence. Final PCR products were digested with SphI and BamHI and ligated to plasmid pFG11d.
The ptRNA4Arg overproduction plasmid was constructed by PCR amplification of the argU gene and its promoter with primers: argU-Sac, (5′-ATT GAG CTC TGAT ACA TGA AAA TAC GGG) and argU-Kpn, (5′-ATG GGT ACC TTG CGC CTA ATC ATT TGA CAG AGC), followed by ligation of the resulting PCR product into SacI/KpnI-digested plasmid pCH405Δ 2; 13. Similarly, the tRNA3Arg and tRNAHis overproduction plasmid was constructed from a PCR product using primers: argX-Sac, (5′-ATG GAG CTC TTT GTT GGC ATC ATC TTT ATG CTG G) and hisR-Kpn, (5′-TTG GTA CCA AAA AAG CCT GCT CGT TGA GCA GGC TTT TCT CGA GTT CTA ATA ATG GGG TGG CTA ATG G). The hisR-Kpn primer also contains a rho-independent transcription terminator.
Plasmids pFG21a, pFG21b and pFG21c are derivatives of pET21b (Novagen EMD) encoding the FLAG epitope between NcoI and NdeI restriction sites. Plasmids pFG21a and pFG21c contain an insertion of two and one thymidylate residues (respectively) between the NotI and XhoI sites of the polylinker. The genes encoding colicin D and its immunity protein were PCR amplified from plasmid ColD-CA23 (a kind gift from Haruhiko Masaki, University of Tokyo) using oligonucleotides; colD-Nde, (5′ - AGA GGT GTT CAT ATG AGT GAT TAC GAA GGT AGT GG), and immD-Xho, (5′ -TGG ACT CGA GTA ATT TAA ATT TTT CCA AG). The resulting product digested with NdeI and XhoI followed by ligation to NdeI/XhoI-digested plasmid pET21b. The sequences of all plasmid constructs were confirmed by DNA sequencing.
Colicin D purification
Colicin D (ColD) and its C-terminally His6-tagged immunity protein (ImmD-His6) were overexpressed from plasmid pET21b::colD-immD in E. coli strain CH12 ΔslyD::kan. Cells were grown at 37°C with aeration in 1.0 L of LB media (supplemented with 150 μg/mL of ampicillin) in a baffled Fernbach flask. At OD600 of 1.0, colicin expression was induced by isopropyl-β-D-thiogalactopyranoside (IPTG) at 1.5 mM followed by further culture for 6 hr. Cells were harvested by centrifugation and the cell pellet frozen at −80°C. The frozen cell pellet was resuspended in 25 mL of cold sodium phosphate lysis buffer [20 mM sodium phosphate (pH 7.0) – 150 mM NaCl – 1 mM phenylmethylsulfonyl fluoride], and cells disrupted by two passages through a French press at 20,000 psi. The cell lysate was clarified by centrifugation at 30,000 5g for 15 min at 4°C. 300 μL of Ni2+-NTA agarose resin (Qiagen) was added to the supernatant and incubated at 4°C for 1.5 hr. The Ni2+-NTA agarose resin was collected by centrifugation and batch washed twice with 5 mL of sodium phosphate wash buffer [20 mM sodium phosphate (pH 7.0) – 150 mM NaCl – 20 mM imidazole] at 4°C. The washed resin was poured into a gravity column and the resin washed further with 5 mL of sodium phosphate buffer. The ColD/ImmD-His6 complex was eluted from the resin with 500 μL of 20 mM sodium phosphate – 150 mM NaCl – 250 mM imidazole and dialyzed against 20 mM sodium phosphate – 150 mM NaCl. The His6-ColD/ImmD complex was approximately 90% pure by SDS-PAGE, and protein concentrations determined using an extinction coefficient at 280 nM of 76,445 M−1 cm−1.
mRNA expression and RNA analysis
For analysis of mRNA cleavage at rare Arg codons (AGG, AGA and CGG), E. coli cultures were grown and processed essentially as described previously 13. For analysis of mRNA cleavage in response to amino acid starvation, amino acid auxotrophic strains of E. coli were grown overnight in MOPS minimal defined medium 59 supplemented with 0.5% D-glucose, 1 μg/ml thiamine, 150 μg/mL ampicillin and appropriate L-amino acids (500 μM arginine, 500 μM histidine or 100 μM tryptophan). The following day, cells were resuspended at an optical density at 436 nm (OD436) of 0.05 in 35 ml of fresh glucose-MOPS medium and grown at 37°C with aeration. After growth to an OD436 of 0.8 – 0.9, mRNA expression was induced with IPTG at 1.5 mM. After incubation for 30 min, cultures were split and each 15 ml fraction was vacuum-filtered through Whatman 0.45 μm nitrocellulose filters. Filters containing control cells (not amino acid starved) were washed twice with 10 ml of glucose-MOPS media containing 1.5 mM IPTG and appropriate amino acids. Amino acid-starved cells were washed twice with 10 ml of glucose-MOPS containing 1.5 mM IPTG. Filters containing washed cells were then added to 15 ml of glucose-MOPS medium containing 1.5 mM IPTG (with or without auxotrophic amino acids) and incubated for 45 min at 37°C with aeration. Strain CH2618 (ΔrelA::kan spoT::cat) was grown in glucose-MOPS medium supplemented with 1 mM of each amino acid and arginine starvation was induced by filtration followed by washing and resuspension in glucose-MOPS containing all amino acids except arginine. Cultures were poured into an equal volume of ice-cold methanol, the cells collected by centrifugation, and cell pellets frozen at −80°C. Total RNA was isolated from frozen cell pellets as described 13.
Colicin D treated cells were grown at 37°C with aeration in LB media supplemented with 150 μg/mL ampicillin. Once at an OD600 of 0.3 – 0.4, IPTG was added to 1.5 mM to induce expression of flag-trxA(PPRR) mRNA. After 30 min, purified colicin D/immunity protein was added to a final concentration of 10 nM, followed by continued culture for 40 min at 37°C with aeration. Colicin D treated cultures were poured into an equal volume of ice-cold methanol to arrest growth and total RNA was isolated as described 13.
Northern blot and S1 nuclease protection analyses were performed essentially as described 3; 13. The following DNA oligonucleotides were 5′-radiolabeled with 32P and used for Northern blot hybridizations: RBS for pET-derived mRNA ribosome binding site, (5′-GTA TAT CTC CTT CTT AAA GTT AAA C); argQ for tRNA2Arg, (5′ - GCT GAG CTA CGG ATG C); argW for tRNA5Arg, (5′-CCT GCA ATT AGC CCT TAG G); argU for tRNA4Arg, (5′-CCT GCG GCC CAC GAC TTA G); argX for tRNA3Arg, (5′-CCT GAG ACC TCT GCC TCC GGA); hisR for tRNAHis, (5′ - CAC GAC AAC TGG AAT CAC); and trpT for tRNATrp, (5′ - CCC AAC ACC CGG TTT TGG). The following DNA oligonucleotides were 3′-radiolabeled using terminal deoxynucleotide transferase and α-[32P]-3′-deoxyadenosine triphosphate, and used as probes for S1 nuclease protection analyses: PPRRCGU S1 probe, (5′-CCT TTC GGG CTT TGT TAG CAG CCG GAT CCT TAA CGA CGC GGA GGG TTA GCG TCG AGG AAC TCT TTC AAC TGA CCT TTA G); KKRRAGG S1 probe, (5′-CCT TTC GGG CTT TGT TAG CAG CCG GAT CCT TAC CTC CTC TTC TTG TTA GCG TCG AGG AAC TCT TTC AAC TGA CCT TTA G); PPRRAGG S1 probe, (5′-CCT TTC GGG CTT TGT TAG CAG CCG GAT CCT TAC CTC CTC GGA GGG TTA GCG TCG AGG AAC TCT TTC AAC TGA CCT TTA G); and PPWWLA S1 probe, (5′ - CCT TTC GGG CTT TGT TAG CAG CCG GAT CCT TAC GCC AGC CAC CAC GGA GGG TTA GCG TCG AGG AAC TCT TTC AAC TGA CCT TTA G). In vitro transcripts were prepared using T7 RNA polymerase as described 3.
Protein expression and analysis
Strains were cultured as described above for RNA analysis. Protein extraction and Western blot analysis using anti-SsrA(DD) peptide antisera were conducted as described 23. Fluorescent anti-FLAG Western analysis was performed using the LI-COR® Odyssey infrared imaging system according to manufacturer’s instructions with minor modifications. Briefly, 0.2 – 0.4 μg of total urea-soluble protein was resolved by SDS-PAGE on 10% polyacrylamide gels followed by electrotransfer to nitrocellulose membranes. Blots were blocked with 2% dried milk in phosphate buffered saline (2.7 mM KCl – 1.8 mM KH2PO4 – 137 mM NaCl – 10.1 mM Na2HPO4, pH 7.4), followed by overnight incubation with anti-FLAG M2 monoclonal antibody (Sigma-Aldrich). IRDye™ 800-conjugated anti-mouse secondary antibodies (Rockland) were used for fluorescent detection. The +1 frameshift products from five independently prepared total protein samples were quantified using LI-COR® Odyssey software. All quantitative determinations were made within the linear response range of the instrument.
SsrA(His6) tagged proteins were purified by Ni2+-NTA agarose (Qiagen) affinity chromatography as described 2; 23. Ni2+-NTA purified protein was further purified by reverse phase-HPLC using a Vydac 15 5 300 mm C4 column. Purified samples were dried, dissolved in 0.5% formic acid/50% acetonitrile, and infused into a Waters Q-Tof II™ mass spectrometer for electrospray ionization.
Acknowledgments
*We thank Brian Janssen for helpful discussions, Jim Curran for suggesting an analysis of translational frameshifting, and Haruhiko Masaki for generously providing plasmid ColD-CA23. Mass spectrometry was performed in the Department of Chemistry & Biochemistry Mass Spectrometry Facility at UC Santa Barbara with the assistance of Dr. James Pavlovich. This work was supported by grant GM078634 from the National Institutes of Health. J. G. G. was supported in part by the Stipanich Fellowship for Undergraduate Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–140. doi: 10.1016/s0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- 2.Hayes CS, Bose B, Sauer RT. Proline residues at the C terminus of nascent chains induce SsrA tagging during translation termination. J Biol Chem. 2002;277:33825–33832. doi: 10.1074/jbc.M205405200. [DOI] [PubMed] [Google Scholar]
- 3.Hayes CS, Sauer RT. Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol Cell. 2003;12:903–11. doi: 10.1016/s1097-2765(03)00385-x. [DOI] [PubMed] [Google Scholar]
- 4.Sunohara T, Jojima K, Yamamoto Y, Inada T, Aiba H. Nascent-peptide-mediated ribosome stalling at a stop codon induces mRNA cleavage resulting in nonstop mRNA that is recognized by tmRNA. RNA. 2004;10:378–386. doi: 10.1261/rna.5169404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerdes K. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J Bacteriol. 2000;182:561–572. doi: 10.1128/jb.182.3.561-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 7.Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- 8.Moore SD, Sauer RT. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem. 2007;76:101–124. doi: 10.1146/annurev.biochem.75.103004.142733. [DOI] [PubMed] [Google Scholar]
- 9.Karzai AW, Susskind MM, Sauer RT. SmpB, a unique RNA-binding protein essential for the peptide-tagging activity of SsrA (tmRNA) EMBO J. 1999;18:3793–3799. doi: 10.1093/emboj/18.13.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sundermeier TR, Dulebohn DP, Cho HJ, Karzai AW. A previously uncharacterized role for small protein B (SmpB) in transfer messenger RNA-mediated trans-translation. Proc Natl Acad Sci U S A. 2005;102:2316–2321. doi: 10.1073/pnas.0409694102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjornsson A, Isaksson LA. Accumulation of a mRNA decay intermediate by ribosomal pausing at a stop codon. Nucleic Acids Res. 1996;24:1753–1757. doi: 10.1093/nar/24.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drider D, DiChiara JM, Wei J, Sharp JS, Bechhofer DH. Endonuclease cleavage of messenger RNA in Bacillus subtilis. Mol Microbiol. 2002;43:1319–1329. doi: 10.1046/j.1365-2958.2002.02830.x. [DOI] [PubMed] [Google Scholar]
- 13.Garza-Sánchez F, Janssen BD, Hayes CS. Prolyl-tRNAPro in the A-site of SecM-arrested ribosomes inhibits the recruitment of transfer-messenger RNA. J Biol Chem. 2006;281:34258–34268. doi: 10.1074/jbc.M608052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Hirano R, Tagami H, Aiba H. Protein tagging at rare codons is caused by tmRNA action at the 3′ end of nonstop mRNA generated in response to ribosome stalling. RNA. 2006;12:248–255. doi: 10.1261/rna.2212606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loomis WP, Koo JT, Cheung TP, Moseley SL. A tripeptide sequence within the nascent DaaP protein is required for mRNA processing of a fimbrial operon in Escherichia coli. Mol Microbiol. 2001;39:693–707. doi: 10.1046/j.1365-2958.2001.02241.x. [DOI] [PubMed] [Google Scholar]
- 16.Sunohara T, Jojima K, Tagami H, Inada T, Aiba H. Ribosome stalling during translation elongation induces cleavage of mRNA being translated in Escherichia coli. J Biol Chem. 2004;279:15368–15375. doi: 10.1074/jbc.M312805200. [DOI] [PubMed] [Google Scholar]
- 17.Muto H, Nakatogawa H, Ito K. Genetically encoded but nonpolypeptide prolyl-tRNA functions in the A site for SecM-mediated ribosomal stall. Mol Cell. 2006;22:545–552. doi: 10.1016/j.molcel.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 18.Dong H, Nilsson L, Kurland CG. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J Mol Biol. 1996;260:649–663. doi: 10.1006/jmbi.1996.0428. [DOI] [PubMed] [Google Scholar]
- 19.Andersson SG, Kurland CG. Codon preferences in free-living microorganisms. Microbiol Rev. 1990;54:198–210. doi: 10.1128/mr.54.2.198-210.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen KS, Peters TC, Walker JR. A minor arginine tRNA mutant limits translation preferentially of a protein dependent on the cognate codon. J Bacteriol. 1990;172:2504–2510. doi: 10.1128/jb.172.5.2504-2510.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorensen MA, Kurland CG, Pedersen S. Codon usage determines translation rate in Escherichia coli. J Mol Biol. 1989;207:365–377. doi: 10.1016/0022-2836(89)90260-x. [DOI] [PubMed] [Google Scholar]
- 22.Collier J, Binet E, Bouloc P. Competition between SsrA tagging and translational termination at weak stop codons in Escherichia coli. Mol Microbiol. 2002;45:745–754. doi: 10.1046/j.1365-2958.2002.03045.x. [DOI] [PubMed] [Google Scholar]
- 23.Hayes CS, Bose B, Sauer RT. Stop codons preceded by rare arginine codons are efficient determinants of SsrA tagging in Escherichia coli. Proc Natl Acad Sci U S A. 2002;99:3440–3445. doi: 10.1073/pnas.052707199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roche ED, Sauer RT. SsrA-mediated peptide tagging caused by rare codons and tRNA scarcity. EMBO J. 1999;18:4579–4589. doi: 10.1093/emboj/18.16.4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richards J, Mehta P, Karzai AW. RNase R degrades non-stop mRNAs selectively in an SmpB-tmRNA-dependent manner. Mol Microbiol. 2006;62:1700–1712. doi: 10.1111/j.1365-2958.2006.05472.x. [DOI] [PubMed] [Google Scholar]
- 26.Roche ED, Sauer RT. Identification of endogenous SsrA-tagged proteins reveals tagging at positions corresponding to stop codons. J Biol Chem. 2001;276:28509–28515. doi: 10.1074/jbc.M103864200. [DOI] [PubMed] [Google Scholar]
- 27.Kamada K, Hanaoka F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol Cell. 2005;19:497–509. doi: 10.1016/j.molcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Motiejunaite R, Armalyte J, Markuckas A, Suziedeliene E. Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol Lett. 2007;268:112–119. doi: 10.1111/j.1574-6968.2006.00563.x. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell. 2003;12:913–923. doi: 10.1016/s1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Zhu L, Zhang J, Inouye M. Characterization of ChpBK, an mRNA interferase from Escherichia coli. J Biol Chem. 2005;280:26080–26088. doi: 10.1074/jbc.M502050200. [DOI] [PubMed] [Google Scholar]
- 31.Schmidt O, Schuenemann VJ, Hand NJ, Silhavy TJ, Martin J, Lupas AN, Djuranovic S. prlF and yhaV encode a new toxin-antitoxin system in Escherichia coli. J Mol Biol. 2007;372:894–905. doi: 10.1016/j.jmb.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ivanova N, Pavlov MY, Felden B, Ehrenberg M. Ribosome rescue by tmRNA requires truncated mRNAs. J Mol Biol. 2004;338:33–41. doi: 10.1016/j.jmb.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 33.Cashel M. The control of ribonucleic acid synthesis in Escherichia coli. IV Relevance of unusual phosphorylated compounds from amino acid-starved stringent strains. J Biol Chem. 1969;244:3133–3141. [PubMed] [Google Scholar]
- 34.Cashel M, Gentry DR, Hernandez VJ, Vinella D. The Stringent Response. In: Neidhardt FC, editor. Escherichia coli and Salmonella: Cellular and Molecular Biology. I. American Society for Microbiology; Washington, D. C.: 1996. pp. 1458–1524. [Google Scholar]
- 35.Magnusson LU, Farewell A, Nystrom T. ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 2005;13:236–242. doi: 10.1016/j.tim.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Yamagishi M, Cole JR, Nomura M, Studier FW, Dunn JJ. Stringent control in Escherichia coli applies also to transcription by T7 RNA polymerase. J Biol Chem. 1987;262:3940–3943. [PubMed] [Google Scholar]
- 37.Aizenman E, Engelberg-Kulka H, Glaser G. An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci U S A. 1996;93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc Natl Acad Sci U S A. 2001;98:14328–14333. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christensen SK, Pedersen K, Hansen FG, Gerdes K. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J Mol Biol. 2003;332:809–819. doi: 10.1016/s0022-2836(03)00922-7. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science. 1999;283:2097–2100. doi: 10.1126/science.283.5410.2097. [DOI] [PubMed] [Google Scholar]
- 41.Masaki H, Ogawa T. The modes of action of colicins E5 and D, and related cytotoxic tRNases. Biochimie. 2002;84:433–438. doi: 10.1016/s0300-9084(02)01425-6. [DOI] [PubMed] [Google Scholar]
- 42.Mora L, Diaz N, Buckingham RH, de Zamaroczy M. Import of the transfer RNase colicin D requires site-specific interaction with the energy-transducing protein TonB. J Bacteriol. 2005;187:2693–2697. doi: 10.1128/JB.187.8.2693-2697.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomita K, Ogawa T, Uozumi T, Watanabe K, Masaki H. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc Natl Acad Sci U S A. 2000;97:8278–8283. doi: 10.1073/pnas.140213797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogle JM, Brodersen DE, Clemons WM, Jr, Tarry MJ, Carter AP, Ramakrishnan V. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 2001;292:897–902. doi: 10.1126/science.1060612. [DOI] [PubMed] [Google Scholar]
- 45.Ogle JM, Carter AP, Ramakrishnan V. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci. 2003;28:259–266. doi: 10.1016/S0968-0004(03)00066-5. [DOI] [PubMed] [Google Scholar]
- 46.Sunohara T, Abo T, Inada T, Aiba H. The C-terminal amino acid sequence of nascent peptide is a major determinant of SsrA tagging at all three stop codons. RNA. 2002;8:1416–1427. doi: 10.1017/s1355838202020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lovett PS, Rogers EJ. Ribosome regulation by the nascent peptide. Microbiol Rev. 1996;60:366–385. doi: 10.1128/mr.60.2.366-385.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mankin AS. Nascent peptide in the “birth canal” of the ribosome. Trends Biochem Sci. 2006;31:11–13. doi: 10.1016/j.tibs.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Tenson T, Ehrenberg M. Regulatory nascent peptides in the ribosomal tunnel. Cell. 2002;108:591–594. doi: 10.1016/s0092-8674(02)00669-4. [DOI] [PubMed] [Google Scholar]
- 50.Boycheva S, Chkodrov G, Ivanov I. Codon pairs in the genome of Escherichia coli. Bioinformatics. 2003;19:987–998. doi: 10.1093/bioinformatics/btg082. [DOI] [PubMed] [Google Scholar]
- 51.Yanofsky C. Attenuation in the control of expression of bacterial operons. Nature. 1981;289:751–758. doi: 10.1038/289751a0. [DOI] [PubMed] [Google Scholar]
- 52.Yanofsky C, Konan KV, Sarsero JP. Some novel transcription attenuation mechanisms used by bacteria. Biochimie. 1996;78:1017–1024. doi: 10.1016/s0300-9084(97)86725-9. [DOI] [PubMed] [Google Scholar]
- 53.Gong F, Ito K, Nakamura Y, Yanofsky C. The mechanism of tryptophan induction of tryptophanase operon expression: tryptophan inhibits release factor-mediated cleavage of TnaC-peptidyl-tRNAPro. Proc Natl Acad Sci U S A. 2001;98:8997–9001. doi: 10.1073/pnas.171299298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiil NP, von Meyenburg K, Friesen JD. Accumulation and turnover of guanosine tetraphosphate in Escherichia coli. J Mol Biol. 1972;71:769–783. doi: 10.1016/s0022-2836(72)80037-8. [DOI] [PubMed] [Google Scholar]
- 55.Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH. Dissection of the mechanism for the stringent factor RelA. Mol Cell. 2002;10:779–788. doi: 10.1016/s1097-2765(02)00656-1. [DOI] [PubMed] [Google Scholar]
- 56.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aiyar A, Leis J. Modification of the megaprimer method of PCR mutagenesis: improved amplification of the final product. Biotechniques. 1993;14:366–369. [PubMed] [Google Scholar]
- 59.Neidhardt FC, Bloch PL, Smith DF. Culture medium for enterobacteria. J Bacteriol. 1974;119:736–747. doi: 10.1128/jb.119.3.736-747.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakamoto K, Ishimaru S, Kobayashi T, Walker JR, Yokoyama S. The Escherichia coli argU10(Ts) phenotype is caused by a reduction in the cellular level of the argU tRNA for the rare codons AGA and AGG. J Bacteriol. 2004;186:5899–5905. doi: 10.1128/JB.186.17.5899-5905.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]