

Abstract
In the S and G2 phases of the cell cycle, DNA double-strand breaks (DSBs) are processed into single-stranded DNA, triggering ATR-dependent checkpoint signaling and DSB repair by homologous recombination (HR). Previous work has implicated the MRE11 complex in such DSB processing events. Here, we show that the human CtIP protein confers resistance to DSB-inducing agents and is recruited to DSBs exclusively in S/G2. Moreover, we reveal that CtIP is required for DSB resection, and thereby for recruitment of RPA and ATR to DSBs and ensuing ATR activation. Furthermore, we establish that CtIP physically and functionally interacts with the MRE11 complex, and that both CtIP and MRE11 are required for efficient HR. Finally, we reveal that CtIP displays sequence homology with Sae2, which is involved in MRE11-dependent DSB processing in yeast. These findings establish evolutionarily conserved roles for CtIP-like proteins in controlling DSB resection, checkpoint signaling and HR.
DSBs are highly cytotoxic lesions induced by ionizing radiation and certain anti-cancer drugs. They also arise when replication forks encounter other lesions and are generated as intermediates during meiotic recombination1. Cells possess two main DSB repair mechanisms: non-homologous end-joining (NHEJ) and homologous recombination (HR). While NHEJ predominates in G0/G1 and is error-prone, HR is restricted to S and G2, when sister chromatids allow faithful repair2-4. HR is initiated by resection of DSBs to generate single-stranded DNA (ssDNA) that binds RPA. A ssDNA-RAD51 nucleoprotein filament then forms to initiate strand invasion. ssDNA-RPA also recruits the protein kinase ATR, triggering ATR-dependent checkpoint signaling by the protein kinase Chk15. As DSB resection is largely restricted to S and G2, both HR and checkpoint signaling are subject to cell-cycle control6-8.
A factor implicated in DSB resection is the MRE11-RAD50-NBS1 (MRN) complex, which binds DNA ends, possesses exo- and endo-nuclease activities and functions in triggering DNA-damage checkpoints9,10. Here, we show that human CtIP (RBBP8) physically and functionally interacts with MRN. CtIP – initially identified as an interactor for CtBP11 and the tumour suppressor proteins RB12 and BRCA113,14 – is recruited to DNA damage and complexes with BRCA1 to control the G2/M DNA-damage checkpoint15-17. We reveal that CtIP also promotes ATR activation and HR through it cooperating with MRN to mediate DSB resection. Based on these findings and our identification of sequence homologies between CtIP and Sae2 – a protein implicated in mitotic and meiotic DSB processing in Saccharomyces cerevisiae18-22 – we conclude that these proteins are functional homologues.
CtIP affects cellular responses to DSBs
To investigate CtIP function, we examined how its depletion affected clonogenic survival of human U2OS cells following their treatment with DNA-damaging agents. To circumvent possible effects arising from CtIP’s involvement in controlling cell-cycle progression, we employed acute (1 hour) drug treatments rather than continuous drug exposure. siRNA-mediated depletion of CtIP caused hypersensitivity towards the topoisomerase I inhibitor camptothecin and the topoisomerase II inhibitor etoposide (Fig. 1a,b) but did not alter cell-cycle distribution profiles and only modestly decreased the proportion of replicating cells (Supplementary Fig. 1a). Furthermore – and consistent with the major cytotoxic lesions caused by camptothecin and etoposide being DSBs arising during S-phase23 – the effects of CtIP depletion were largely abrogated by treating cells with the DNA-replication inhibitor aphidicolin (Fig. 1a,b). These data therefore suggested that CtIP depletion impairs cell survival when DSBs are generated during S-phase. Also, as DSBs generated in S-phase by camptothecin are repaired by HR24, these results suggested that CtIP promotes HR. Congruent with CtIP being unnecessary for NHEJ, its depletion caused only weak hypersensitivity towards bleocin, which generates DSBs at all cell-cycle stages (Fig. 1c and Supplementary Fig. 1b).
FIGURE 1. CtIP depletion causes hypersensitivity to DSB-inducing agents.
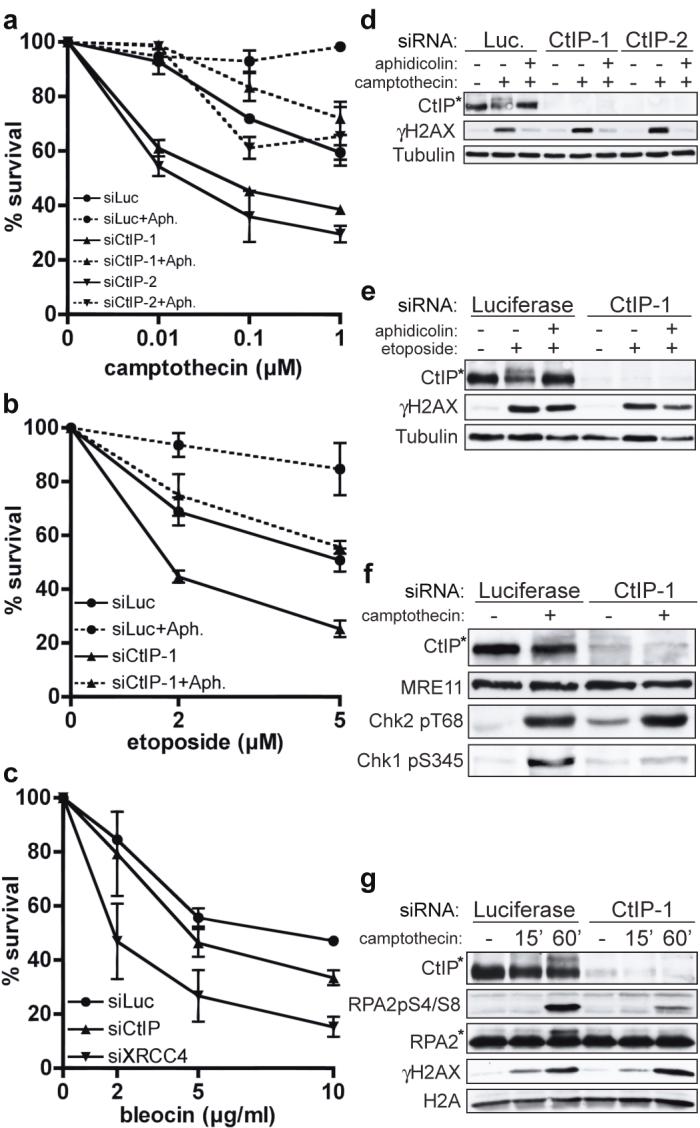
a-c, CtIP down-regulation causes replication-dependent camptothecin (CPT) and etoposide hypersensitivity, and weak bleocin hypersensitivity. U2OS cells were pre-incubated with aphidicolin (Aph) where indicated, then treated for 1 h with CPT, etoposide or bleocin. CtIP-1 and CtIP-2 are two independent siRNAs. Survival data represent mean +/- standard error of mean (±SEM) from ≥3 independent experiments.
d, e, Aphidicolin suppresses CPT- and etoposide-induced CtIP phosphorylation. Extracts from cells down-regulated for Luciferase or CtIP and treated with CPT or etoposide in the presence or absence of aphidicolin were immunoblotted as indicated.
f, g, CtIP depletion impairs Chk1 and RPA phosphorylation but not Chk2 phosphorylation after CPT treatment. Asterisks in d-g: hyper-phosphorylated CtIP and RPA2.
We next assessed the impact of CtIP on phosphorylations mediated by the protein kinases ATM and ATR. In response to DSBs, ATM phosphorylates the checkpoint kinase Chk2 and histone H2AX (generating a protein species termed γH2AX)25,26. By contrast, ATR activation by ssDNA elicits Chk1 phosphorylation5. As shown in Figure 1d, camptothecin triggered γH2AX formation and the generation of a slower-migrating, hyper-phosphorylated species of CtIP (CtIP hyper-phosphorylation was demonstrated by phosphatase treatments and a phospho-specific antibody; Supplementary Fig. 1c,d). Furthermore, both CtIP and H2AX phosphorylation were prevented when aphidicolin was co-administered with camptothecin. Etoposide also triggered modification of H2AX and CtIP; however, in this case, CtIP phosphorylation but not H2AX phosphorylation was abrogated by aphidicolin (Fig. 1e). These data are in accord with etoposide being able to generate DSBs throughout the cell cycle, and suggest that CtIP phosphorylation occurs most effectively in S-phase.
CtIP depletion did not affect H2AX phosphorylation triggered by either camptothecin or etoposide, revealing that CtIP is not required for DNA-lesion generation by these drugs (Fig. 1d,e). Similarly, camptothecin-induced phosphorylations of Chk2 and SMC1 were unaffected by CtIP depletion, indicating that ATM activation still occurs when CtIP is absent (Fig. 1f and data not shown). By contrast, CtIP depletion diminished camptothecin-induced Chk1 phosphorylation on Ser-345 and Ser-317 (Fig. 1f and data not shown). Notably, CtIP depletion markedly impaired Chk1 phosphorylation at late time-points but not at early time-points after continuous camptothecin exposure (Supplementary Fig. 1e). This suggested that CtIP is not required for initial Chk1 phosphorylation triggered by replicational stress induced by trapped DNA topoisomerase I molecules but is needed for signaling of camptothecin-induced replication-dependent DSBs, which – as for IR-induced DSBs7,27 – might require resection to elicit ATR activation. We reasoned that if this was the case, CtIP might influence hyper-phosphorylation of the amino-terminal region of RPA2 by ATM, ATR and DNA-PK in the context of replication-associated DSBs28,29. Indeed, camptothecin-induced RPA2 phosphorylation – as measured by a shift in RPA2 electrophoretic mobility and by phospho-specific antibodies against modified RPA2 serines 4 and 8 – was markedly impaired by CtIP depletion, while DSB formation as measured by γH2AX formation was unaffected (Fig. 1g). Similarly, CtIP depletion impaired RPA hyper-phosphorylation but not Chk2 or SMC1 phosphorylation in response to etoposide (data not shown). Together, these data indicated that CtIP is not required for DSB generation by topoisomerase inhibitors nor for DSB detection and signaling by ATM. Instead, CtIP is specifically needed for efficient activation and/or propagation of ATR-mediated signaling.
CtIP promotes ATR recruitment in S/G2
When we generated DSB-containing tracks in human U2OS cell nuclei by laser micro-irradiation30, CtIP was recruited to the damage with kinetics slower than that of γH2AX formation (Fig. 2a). Notably, while γH2AX tracks formed in all irradiated cells, only a subset displayed strong CtIP recruitment, suggesting that CtIP recruitment might be affected by cell-cycle status. Indeed, significant CtIP redistribution to laser tracks was only observed in cells staining positive for Cyclin A, indicating that CtIP responds predominantly to DSBs in S and G2 (Fig. 2b).
FIGURE 2. CtIP associates with sites of DNA damage in S/G2 and promotes ATR recruitment to DSBs.
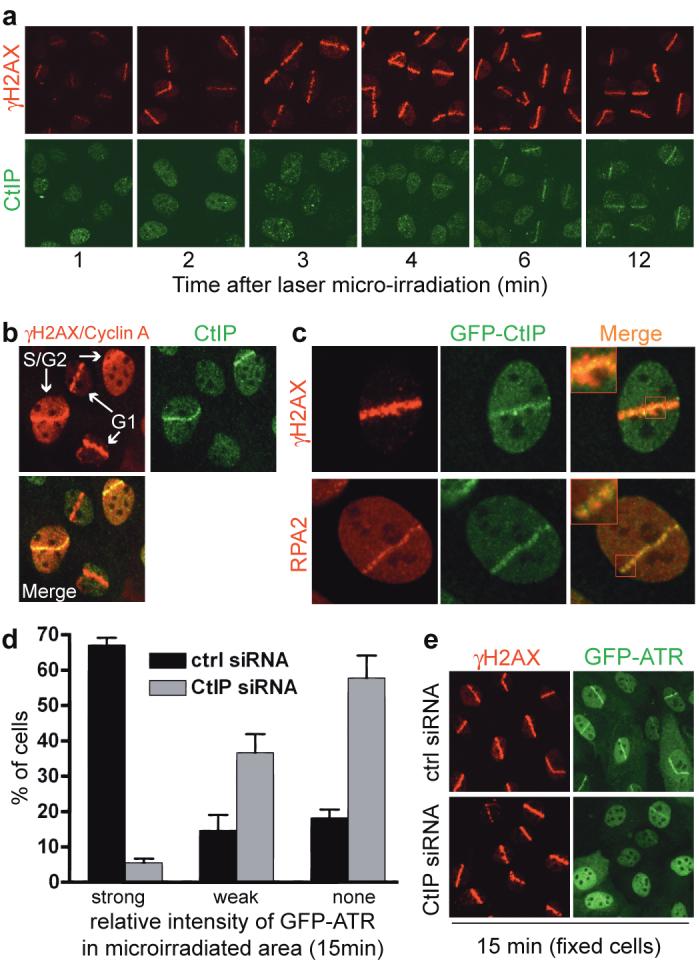
a, b, CtIP recruitment to laser-induced DSBs occurs in S/G2. Cells were stained for endogenous γH2AX, CtIP or Cyclin A. All cells display local γH2AX signals but only S/G2 cells have pan-nuclear Cyclin A staining.
c, GFP-CtIP co-localizes with RPA-ssDNA. Insets: higher magnifications.
d, CtIP down-regulation impairs ATR recruitment to DNA damage. GFP-ATR expressing cells were treated, micro-irradiated and monitored (Supplementary Fig. 2c). GFP-ATR DSB tracks were manually scored 15 min after micro-irradiation for control and CtIP-depleted cells (296 and 264 cells, respectively). Data represent mean (±SEM) from two experiments.
e, CtIP depletion does not affect γH2AX formation. Cells from d were fixed 15 min after micro-irradiation and immunostained.
In the above studies, we noted that – similar to what has been reported for RPA and ATR31 – CtIP stripes were narrower than those of γH2AX. Indeed, when we irradiated a U2OS cell line stably expressing GFP-tagged CtIP (GFP-CtIP; Supplementary Fig. 2a), GFP-CtIP did not cover the entire γH2AX-modified chromatin region but localized to smaller compartments that resembled those occupied by RPA (Fig. 2c). Consistent with this, endogenous CtIP co-localized with GFP-ATR (Supplementary Fig. 2b). Because ATR localizes to RPA foci, and since we had found that CtIP promotes ATR activation, we used a cell line expressing functional GFP-ATR7 to examine whether CtIP is needed for ATR recruitment. Indeed, while control cells exhibited strong GFP-ATR recruitment to laser tracks, this response was markedly attenuated in CtIP-depleted cells (Fig. 2d and Supplementary Fig. 2c; note that the siRNA CtIP-1 is used here and thereafter). Importantly, parallel immunostaining studies on fixed cells revealed that CtIP depletion impaired ATR recruitment but not γH2AX formation, implying that CtIP does not affect the extent of laser-induced DNA damage (Fig. 2e). Nevertheless, some residual ATR recruitment was observed in CtIP-depleted cells, indicating either that CtIP stimulates but is not absolutely required for ATR recruitment, or that it is essential for ATR recruitment but that CtIP siRNA-depletion was not complete.
CtIP facilitates the generation of ssDNA
Recruitment of the ATR-ATRIP complex to DNA damage is mediated by interactions with RPA bound to ssDNA5,32. To see whether CtIP might affect ssDNA generation, we analyzed DSB-induced RPA focus formation. Strikingly, CtIP depletion dramatically impaired RPA recruitment to laser-induced DNA damage (Fig. 3a; as shown in Supplementary Fig. 2d, this defect was ameliorated when cells stably expressed siRNA-resistant GFP-CtIP, indicating that the GFP-CtIP fusion is functional and that the phenotypes caused by the siRNA are CtIP-dependent). Similarly, camptothecin exposure generated large numbers of RPA foci in control cells (∼40 % displayed this pattern, reflecting the proportion of cells in S phase), while CtIP depleted cells generally lacked detectable RPA foci despite displaying normal γH2AX induction (Fig. 3b). Moreover, by using an anti-BrdU antibody staining technique that only detects DNA in single-stranded form, we found that camptothecin triggered substantial ssDNA formation in control cells but not in CtIP-depleted cells (Fig. 3c and Supplementary Fig. 3a,b). These data therefore established that CtIP promotes ssDNA formation.
FIGURE 3. CtIP depletion impairs DSB resection.

a, b, CtIP is required for RPA recruitment to laser- and CPT-induced DSBs. Cells treated with control or CtIP siRNA were either micro-irradiated and 30 min later co-immunostained for γH2AX and RPA2, or were treated with CPT for 1 h and immunostained.
c, CtIP depletion impairs ssDNA formation. After siRNA treatment, cells were tested for CPT-induced ssDNA formation by a non-denaturing BrdU staining procedure (see Methods and text for details). For each CPT-treated sample >100 cells were counted and the percentage exhibiting γH2AX foci, or both γH2AX and ssDNA foci, was determined (Supplementary Fig. 3a,b). Data represent the mean (±SEM) from two independent experiments.
CtIP interacts with MRN and promotes HR
MRN promotes the processing of DSBs to generate RPA-coated ssDNA that is needed for ATR recruitment and ensuing Chk1 phosphorylation7,33,34. Having found that CtIP was also required for these events, and since BRCA1 and CtIP co-purify with RAD50 in gel-filtration analyses15, we examined whether MRN and CtIP interact. Thus, we carried out immunoprecipitations from human HeLa cell nuclear extracts with antisera raised against the C- or N-terminal regions of CtIP. As shown in Figure 4a, MRN and BRCA1 were detected in CtIP immunoprecipitates. While most CtIP was recovered by such immunoprecipitations, BRCA1 was only inefficiently retrieved, possibly reflecting the G2-specific nature of the CtIP-BRCA1 interaction16. Furthermore, only a proportion of MRN was recovered in CtIP immunoprecipitates, suggesting that there might be different populations of MRN, with only a subset CtIP-associated. The interaction between CtIP and MRN was quite stable – surviving washing in 0.5 M NaCl – and persisted in the presence of ethidium bromide, indicating that it was not DNA-mediated (data not shown). Conversely, CtIP was retrieved in RAD50 and MRE11 immunoprecipitates (Fig. 4b and data not shown). The CtIP-MRN interaction was unaffected by camptothecin treatment and took place when BRCA1 was depleted (Fig. 4b). Furthermore, when we used extracts from human HCC1937 cells, CtIP was found in RAD50 immunoprecipitates and vice versa (Supplementary Fig. 4a). As HCC1937 cells express low levels of functionally impaired BRCA1 bearing a mutation in its C-terminal tandem BRCT domain that precludes its interaction with CtIP16, these data implied that BRCA1 does not bridge the CtIP-MRN interaction. Indeed, CtIP co-immunoprecipitated with RAD50 from mixtures only containing purified, recombinant CtIP and MRN, demonstrating a direct interaction between the two factors (Supplementary Fig. 4b).
FIGURE 4. CtIP interacts with MRN and promotes HR.

a, MRN co-immunoprecipitates with CtIP. HeLa nuclear extract (HNE) was immunoprecipitated (IP) with pre-immune or anti-CtIP antibodies and analyzed by immunoblotting (IB).
b, CtIP-MRN interaction after DNA damage and after BRCA1-depletion. Where indicated, cells were treated with 1 μM CPT for 1 h. Whole cell extracts (WCE) were immunoblotted directly or after immunoprecipitation.
c, The CtIP C-terminus binds MRN. Bacterially-expressed fusions (arrows) were tested for binding MRN in HNE by immunoblotting.
d, CtIP stimulates MR-dependent nuclease activity. PhiX174 substrate was incubated with MR (40 ng), BSA (200 ng) or CtIP (200 ng) in 5 mM MgCl2 or MnCl2, run on a agarose gel and stained with SYBR Gold. ssc: circular ssDNA.
e, CtIP or MRE11 down-regulation impairs HR (see Methods and text for details). Data represent the mean (±SEM) from four independent experiments.
To further characterize the CtIP-MRN interaction, we expressed in bacteria various regions of CtIP fused to GST and assessed their ability to retrieve MRN complexes from HeLa nuclear extracts (Fig. 4c). Whereas MRN did not bind to N-terminal regions of CtIP containing a putative coiled-coil or the BRCA1 interaction motif16 (lanes 4 and 5) it bound efficiently when GST was fused to full-length CtIP or to a region comprising the C-terminal 108 residues of CtIP (lanes 3 and 7; see Supplementary Fig. 6a for a schematic view of the CtIP fragments). Furthermore, when we used purified GST-fusions of CtIP (expressed in bacteria or insect cells), they bound directly to purified, baculovirus-expressed human MRE11-RAD50 (MR) (Supplementary Fig. 4c,d). However, while GST-fusions containing the CtIP C-terminal region interacted with MR (Supplementary Fig. 4c), a GST-CtIP fusion lacking the CtIP C-terminal region still interacted with MR (Supplementary Fig. 4d). These data therefore indicate that CtIP and MRN interact directly and that there is more than one point-of-contact between the two factors.
Since both CtIP and MRE11 are needed for effective DSB resection, we speculated that CtIP might affect nuclease activities associated with the MRE11 complex10,35-37. Thus, we expressed CtIP in insect cells, purified it to homogeneity (Supplementary Fig. 4b) and used it together with purified MR in an endonuclease assay with closed-circular single-stranded PhiX174 DNA (Fig. 4d). As shown previously38, MR exhibited nuclease activity in the presence of manganese (lane 12) but not in the presence of magnesium (lane 5). By contrast, CtIP did not display detectable nuclease activity with either metal cofactor (lanes 3 and 10). Strikingly, when CtIP was combined with MR, this produced endonuclease activity in the presence of magnesium (lane 6) and produced more endonuclease activity in the presence of manganese than was exhibited by MR alone (compare lanes 12 and 13). Heat denaturation of CtIP abolished its stimulatory effects and ATP was not required for MR activity nor for the stimulatory effect of CtIP (data not shown). Notably, when we carried out MR-dependent exonuclease assays with short (50 bp) radiolabeled oligonucleotide substrates, however, we did not detect CtIP-dependent effects (data not shown). We therefore speculate that CtIP mainly promotes endo- but not exo-nucleolytic activities in conjunction with the MRE11 complex.
Given that both CtIP and MRN regulate DSB resection, we speculated that they might facilitate HR. To address this, we used a U2OS cell line bearing one or two copies of an integrated HR reporter composed of two differentially mutated GFP genes oriented as direct repeats. Transient expression of I-SceI endonuclease in such cells generates a DSB that, when repaired by gene conversion, results in a functional GFP gene whose expression can be assessed by flow cytometry (Supplementary Fig. 5a)39. Significantly, depletion of CtIP or MRE11 decreased HR frequencies to levels similar to those achieved by depleting the key HR protein RAD51 (Fig. 4e and Supplementary Fig. 5b). Furthermore, depleting CtIP and MRE11 together did not decrease HR efficiency further than was achieved by CtIP or MRE11 depletion alone. These findings therefore revealed that both CtIP and MRE11 promote HR, and furthermore suggested that they do so through a common mechanism.
CtIP has homology to S. cerevisiae Sae2
Prompted by the above findings, we used regions of CtIP in PSI-Blast based database searches40. While most regions of CtIP only identified related proteins in higher eukaryotes, we identified proteins in a diverse range of eukaryotes that display homology with the C-terminal 108 amino acid region of CtIP (Supplementary Fig. 6a). Most strikingly, the only S. cerevisiae protein identified in this search was Sae2, a protein that genetically interacts with yeast Mre11 to govern camptothecin sensitivity and DSB processing (Fig. 5a and Supplementary Fig. 6a)18-22,41,42. To address the potential functional importance of the CtIP/Sae2 homology region, we generated a U2OS cell line stably expressing an siRNA-resistant GFP-tagged CtIP derivative (residues 1-789) lacking the C-terminal region. Analysis of these cells under conditions where the endogenous CtIP protein was down-regulated established that, unlike the wild-type protein, the CtIP truncation mutant did not promote efficient Chk1 phosphorylation, RPA2 hyper-phosphorylation or RPA focus formation upon camptothecin treatment (Fig. 5b-d). Similarly, wild-type CtIP promoted the formation of RPA-coated ssDNA in laser micro-irradiation studies, whereas the CtIP C-terminal truncation mutant did not (Fig. 5e; a similar proportion of wild-type and mutant cells stained positive for Cyclin A, ruling out cell-cycle effects – data not shown). As anticipated from previous experiments (Supplementary Fig. 4d), both wild-type CtIP and the truncation mutant could be co-immunoprecipitated with RAD50 (Supplementary Fig. 6b). Taken together, these data therefore indicated that the conserved C-terminal region of CtIP is functionally important.
FIGURE 5. Function and evolutionary conservation of the CtIP C-terminus.

a. Alignment of CtIP and Sae2 arising from BLAST-searches with the CtIP C-terminus (790-897).
b-e, Deletion of the CtIP C-terminus impairs CtIP function. Three days after siRNA transfection, U2OS cells stably expressing GFP- tagged siRNA-resistant wild-type CtIP (WT) or a deletion mutant (1-789) were either treated with 1 μM CPT for 1 h and analyzed by immunoblotting or co-immunostaining, or were micro-irradiated and 30 min later co-immunostained as indicated. Asterisks in b and c: hyper-phosphorylated CtIP and RPA2.
Discussion
We have shown that CtIP facilitates DSB resection and ssDNA formation, thus activating the ATR/Chk1 axis of the DNA-damage response and DNA repair by HR. Consequently, cells depleted of CtIP display hypersensitivity towards DSB-inducing agents, particularly those that generate DSBs specifically in S-phase. Furthermore, we have established that CtIP functionally interacts with the MRN complex. Taken together with the fact that MRN also promotes DSB resection, ATR signaling and HR, these findings identify CtIP as a critical regulator of the checkpoint signaling and DNA repair functions of the MRN complex.
We have also found that the CtIP C-terminus is highly conserved in CtIP orthologues in other organisms and is required for CtIP function in human cells. Strikingly, this domain displays homology with the C-terminus of S. cerevisiae Sae2, which genetically cooperates with the yeast Mre11 complex to promote DSB resection. We therefore conclude that CtIP and Sae2 are likely functional counterparts and that proteins related to these factors will turn out to control DSB resection, ssDNA-mediated checkpoint signaling and HR in diverse eukaryotes. Efficient DSB processing is restricted to S and G2, and requires CDK and ATM kinase activity7. It is therefore noteworthy that CtIP and Sae2 are phosphorylated by ATM and Mec143-45, respectively, and that the CtIP/Sae2 homology region contains conserved potential CDK and ATM target sites. This raises the exciting possibility that phosphorylation of these sites regulates DSB resection. In addition, CtIP interacts with and is ubiquitylated by BRCA113,14,16,17, and has been identified as an RB interaction-partner12, suggesting that these interactions might control the fate of DSBs during the cell cycle. Finally, it is significant that CtIP governs responses to commonly-used anti-cancer agents and that CtIP alterations have been reported in some cancer cells, thus highlighting CtIP and its interaction with MRN as potential targets for drug discovery13,46,47.
METHODS SUMMARY
HeLa, HCC1937, U2OS and U2OS-derived cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS) and standard antibiotics. Data for survival curves were generated by colony formation assays. In brief, U2OS cells were transfected with siRNA (see Supplementary Methods) and treated with DNA-damage-inducing drugs. After 1 h, the drug was removed and cells were left for 10-14 days at 37°C to allow colonies to form. Colonies were stained with 0.5% crystal violet/20% ethanol and counted. Where indicated, cells were pre-incubated with aphidicolin (10 μM) for 90 min, then treated with the specified drug and aphidicolin for 1 h. A U2OS-derived cell line stably expressing GFP-ATR was described previously7. The siRNA-resistant silent wild-type GFP-CtIP construct was generated by sub-cloning the CtIP cDNA into the pEGFP-C1 expression plasmid (BD Biosciences Clontech) and changing three nucleotides in the CtIP-1 siRNA targeting region by using a QuikChange site-directed mutagenesis kit (Stratagene, Inc.) as previously described16. The plasmid expressing the CtIP mutant lacking the C-terminus (1-789) was generated by changing residues 790 and 791 in the wild-type GFP-CtIP construct to two stop-codons. For generation of cell lines stably expressing siRNA-resistant GFP-tagged wild-type and mutant CtIP, U2OS cells were transfected with the appropriate constructs and, following antibiotic selection, resistant clones were tested for expression and nuclear localization of the transgene-product by immunofluorescence microscopy. To detect ssDNA by microscopy, cells were cultivated for 24 h in medium supplemented with 10 μM BrdU prior to camptothecin treatment and, after fixation, immunostained with an anti-BrdU antibody (see Methods) without any preceding DNA denaturation or nuclease treatment48. Laser micro-irradiation was performed as described previously30,31. Recombinant FLAG-GST-CtIP-6H was isolated from baculovirus-infected Sf9 cells as described previously49. Recombinant MRE11-RAD50 (MR) and MRE11-RAD50-NBS1 (MRN) complex were kind gifts from T. Paull.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Material
Acknowledgements
We thank T. Paull and J. Falck for providing reagents and P. Huertas, A. Meier, K. Dry, K. Miller and Y. Pommier for valuable advice and critical reading of the manuscript. This study was supported by Cancer Research UK and a Swiss National Science Foundation fellowship for advanced researcher (to A.A.S). Research in the S.P.J. laboratory is made possible by core infrastructure funding from Cancer Research UK and the Wellcome Trust. C.L., J.L., M.M and J.B were supported by grants from the Danish Cancer Society, Danish National Research Foundation, EU (DNA Repair), and John and Birthe Meyer Foundation. M.M and J.B are supported by grant-number MSM 6198959216. Research in the laboratory of R.B is supported by National Institute of Health grant CA97403 and S.F is supported by a fellowship from the New York State Breast Cancer Research Fund (C021330).
APPENDIX
METHODS
Immunofluorescence microscopy
U2OS cells were transfected with siRNA and after 3 days treated with 1 μM CPT for 1 h. After pre-extraction for 5 min on ice (25 mM Hepes 7.4, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose and 0.5% TritonX-100), cells were fixed with 4% formaldehyde (w/v) in PBS for 12 min. Coverslips were washed with PBS and then co-immunostained with primary antibodies against γH2AX (rabbit polyclonal, Cell Signaling Technology) either in combination with an antibody against RPA2 (mouse monoclonal, Lab Vision) or BrdU (mouse monoclonal, GE Healthcare). Appropriate Alexa Fluor-488 (green) and -594 (red) conjugated secondary antibodies (1:1000) were purchased from Molecular Probes (Paisley, UK). Slides were viewed with a Bio-Rad confocal laser microscope by sequential scanning of the two emission channels used.
Immunoblotting
Cell extracts were prepared in Laemmli buffer (4% SDS, 20% glycerol, 120 mM Tris-HCl pH 6.8), proteins were resolved by SDS-PAGE and transferred to nitrocellulose. Immunoblots were performed by using the appropriate antibodies (see Supplementary Methods).
Immunoprecipitation
If not specified otherwise, cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1mM EDTA and 0.1% SDS), supplemented with phosphatase inhibitor cocktails 1 and 2 (Sigma) and with complete protease inhibitor cocktail (Roche). Clarified extracts were pre-cleared with protein A or G beads (Sigma) for 1 h at 4 °C. Immunoprecipiting antibodies were added to the pre-cleared supernatant and incubated for 2 h at 4 °C. After a 1 h incubation with protein A or G beads, precipitated immunocomplexes were washed four times in lysis buffer (containing 0.5% NP-40, without SDS), boiled in SDS sample buffer and loaded on an SDS-polyacrylamide gel. Proteins were analysed by immunoblotting as described above.
GST pull-downs
GST-CtIP fragments were constructed by cloning polymerase chain reaction products into pGEX-4T1 (Amersham). GST fusion plasmids were grown in BL21 RIL (Codon+) Escherichia coli (Stratagene) and protein was expressed by incubation for 4 h at 30 °C after the addition of 100 μM IPTG. Proteins were purified from soluble extracts with glutathione Sepharose 4 fast flow beads (Amersham). GST fusion proteins bound to glutathione beads were mixed with 1 mg of HeLa nuclear extract and incubated for 1 h at 4 °C in 1 ml of TEN100 buffer (20 mM Tris-HCl, pH 7.4, 0.1 mM EDTA and 100 mM NaCl). Beads were then washed four times with NTEN300 buffer (0.5% NP-40, 0.1 mM EDTA, 20 mM Tris-HCl, pH 7.4, 300 mM NaCl), complexes were boiled in SDS sample buffer and analysed by SDS-PAGE followed by either coomassie staining or immunoblotting.
In vitro nuclease assay
The PhiX174 circular single-stranded virion DNA substrate (5386 nt, New England Biolabs), was mixed with MR together with BSA or CtIP in reaction buffer (30 mM potassium-MOPS, pH 7.2, 1 mM dithiothreitol, 1 mM ATP, 25 mM KCl, and 5 mM of either MgCl2 or MnCl2). After incubation for 3 h at 37°C, nuclease reactions were terminated by adding 1/10 volume of stop solution (3% SDS, 50 mM EDTA) and samples were run in a 0.8% agarose gel (1×TAE) for 90 min at 100 mA. DNA species were stained with SYBR Gold Nucleic Acid Gel Stain (Molecular Probes) for 20 min and visualised with a fluoroimager (FLA-5000, Fujifilm).
Homologous recombination assay
A U2OS clone with the integrated HR reporter DR-GFP was generated as described previously39. Two days after transfection with siRNA, U2OS-DR-GFP cells were co-transfected with an I-SceI expression vector (pCBA-I-SceI) together with a vector expressing monomeric red fluorescent protein (pCS2-mRFP). The latter plasmid was added in 1:3 ratio to mark the I-SceI-positive cells. Cells were harvested two days after I-SceI transfection and subjected to flow cytrometric analysis to examine recombination induced by double-strand breakage. Only RFP positive sub-populations of cells were analysed for HR efficiency to circumvent possible differences in transfection efficiencies. Fluorescence-activated cell sorting data were analysed with CellQuest software to reveal the percentage of GFP-positive cells relative to the number of transfected cells (RFP positive). The data were then related to a control siRNA treatment in each individual experiment. The dividing line between GFP (HR) positive and negative cells was set to 1 % background level of GFP positive cells in every internal control (not transfected with I-SceI). This gate was then applied to the RFP/I-SceI positive counterparts to determine HR efficiency (see Supplementary Fig. 5b).
Footnotes
Author information
The authors declare no competing financial interest.
References
- 1.Wyman C, Kanaar R. DNA double-strand break repair: all’s well that ends well. Annu. Rev. Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
- 2.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nature Rev. Mol. Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 3.West SC. Molecular views of recombination proteins and their control. Nature Rev. Mol. Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 4.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nature Rev. Mol. Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 5.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 6.Ira G, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jazayeri A, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nature Cell Biol. 2006;8:37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 8.Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lavin MF. The Mre11 complex and ATM: a two-way functional interaction in recognising and signaling DNA double strand breaks. DNA Repair (Amst) 2004;3:1515–1520. doi: 10.1016/j.dnarep.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 10.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nature Rev. Mol. Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 11.Schaeper U, Subramanian T, Lim L, Boyd JM, Chinnadurai G. Interaction between a cellular protein that binds to the C-terminal region of adenovirus E1A (CtBP) and a novel cellular protein is disrupted by E1A through a conserved PLDLS motif. J. Biol. Chem. 1998;273:8549–8552. doi: 10.1074/jbc.273.15.8549. [DOI] [PubMed] [Google Scholar]
- 12.Fusco C, Reymond A, Zervos AS. Molecular cloning and characterization of a novel retinoblastoma-binding protein. Genomics. 1998;51:351–358. doi: 10.1006/geno.1998.5368. [DOI] [PubMed] [Google Scholar]
- 13.Wong AKC, et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene. 1998;17:2279–2285. doi: 10.1038/sj.onc.1202150. [DOI] [PubMed] [Google Scholar]
- 14.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg RA, et al. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell Biol. 2004;24:9478–9486. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKee AH, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rattray AJ, McGill CB, Shafer BK, Strathern JN. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics. 2001;158:109–122. doi: 10.1093/genetics/158.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 22.Clerici M, Mantiero D, Lucchini G, Longhese MP. The S. cerevisiae Sae2 protein promotes resection and bridging of double-strand break ends. J. Biol. Chem. 2005;280:38631–38638. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- 23.D’Arpa P, Beardmore C, Liu LF. Involvement of nucleic acid synthesis in cell killing mechanisms of topoisomerase poisons. Cancer Res. 1990;50:6919–6924. [PubMed] [Google Scholar]
- 24.Saleh-Gohari N, et al. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol. Cell Biol. 2005;25:7158–7169. doi: 10.1128/MCB.25.16.7158-7169.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nature Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 26.Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118:9–17. doi: 10.1016/j.cell.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 27.Cuadrado M, et al. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med. 2006;203:297–303. doi: 10.1084/jem.20051923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao R-G, et al. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 1999;18:1397–1406. doi: 10.1093/emboj/18.5.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakasai R, et al. Differential involvement of phosphatidylinositol 3-kinase-related protein kinases in hyperphosphorylation of replication protein A2 in response to replication-mediated DNA double-strand breaks. Genes Cells. 2006;11:237–246. doi: 10.1111/j.1365-2443.2006.00942.x. [DOI] [PubMed] [Google Scholar]
- 30.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nature Cell Biol. 2003;5:255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 31.Bekker-Jensen S, et al. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ball HL, Myers JS, Cortez D. ATRIP Binding to RPA-ssDNA Promotes ATR-ATRIP Localization but Is Dispensable for Chk1 Phosphorylation. Mol. Biol. Cell. 2005;16:2372–2381. doi: 10.1091/mbc.E04-11-1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex. Oncogene. 2006;25:3894–3904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J. Biol. Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 36.Assenmacher N, Hopfner KP. MRE11/RAD50/NBS1: complex activities. Chromosoma. 2004;113:157–166. doi: 10.1007/s00412-004-0306-4. [DOI] [PubMed] [Google Scholar]
- 37.Larson ED, Cummings WJ, Bednarski DW, Maizels N. MRE11/RAD50 Cleaves DNA in the AID/UNG-Dependent Pathway of Immunoglobulin Gene Diversification. Mol. Cell. 2005;20:367–375. doi: 10.1016/j.molcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 38.Trujillo KM, Yuan S-SF, Lee E-YP, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 1998;273:21447–21450. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- 39.Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaffer AA, et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng C, Brown JA, You D, Brown JM. Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae. Genetics. 2005;170:591–600. doi: 10.1534/genetics.104.028795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 43.Li, et al. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature. 2000;13:210–215. doi: 10.1038/35018134. [DOI] [PubMed] [Google Scholar]
- 44.Baroni E, Viscardi V, Cartagena-Lirola H, Lucchini G, Longhese MP. The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol. Cell Biol. 2004;24:4151–4165. doi: 10.1128/MCB.24.10.4151-4165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cartagena-Lirola H, Guerini I, Viscardi V, Lucchini G, Longhese MP. Budding Yeast Sae2 is an In Vivo Target of the Mec1 and Tel1 Checkpoint Kinases During Meiosis. Cell Cycle. 2006;5:1549–1559. doi: 10.4161/cc.5.14.2916. [DOI] [PubMed] [Google Scholar]
- 46.Vilkki S, et al. Screening for microsatellite instability target genes in colorectal cancers. J. Med. Genet. 2002;39:785–789. doi: 10.1136/jmg.39.11.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu G, Lee WH. CtIP, a multivalent adaptor connecting transcriptional regulation, checkpoint control and tumor suppression. Cell Cycle. 2006;5:1592–1596. doi: 10.4161/cc.5.15.3127. [DOI] [PubMed] [Google Scholar]
- 48.Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl Acad. Sci. USA. 1999;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu-Baer F, Lagrazon K, Yuan W, Baer R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003;278:34743–34746. doi: 10.1074/jbc.C300249200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.