

Abstract
Immune cells invading the central nervous system (CNS) in response to Borna disease virus (BDV) antigens are central to the pathogenesis of Borna disease (BD). We speculate that the response of the resident cells of the brain to infection may be involved in the sensitization and recruitment of these inflammatory cells. To separate the responses of resident cells from those of cells infiltrating from the periphery, we used dexamethasone to inhibit inflammatory reactions in BD. Treatment with dexamethasone prevented the development of clinical signs of BD, and the brains of treated animals showed no neuropathological lesions and a virtual absence of markers of inflammation, cell infiltration, or activation normally seen in the CNS of BDV-infected rats. In contrast, treatment with dexamethasone exacerbated the expression of BDV RNA, which was paralleled by a similarly elevated expression of mRNAs for egr-1, c-fos, and c-jun. Furthermore, dexamethasone failed to inhibit the increase in expression of mRNAs for tumor necrosis factor α, macrophage inflammatory protein 1β, interleukin 6, and mob-1, which occurs in the CNS of animals infected with BDV. Our findings suggest that these genes, encoding transcription factors, chemokines, and proinflammatory cytokines, might be directly activated in CNS resident cells by BDV. This result supports the hypothesis that the initial phase of the inflammatory response to BDV infection in the brain may be dependent upon virus-induced activation of CNS resident cells.
Viral infections of the central nervous system (CNS) are responsible for many acute and chronic encephalopathies in humans (1–3). The pathogenic mechanisms underlying virus-induced neurological disease are extremely complex. Certain viruses are cytopathic for target cells in the CNS as a direct consequence of viral replication (4). Direct effects on neurons are also possible through noncytolytic effects, as is the case for rabies virus (5). Nevertheless, most virus-induced encephalopathies are not directly mediated by virus replication in the CNS but rather are the result of the host inflammatory response to the infection (6, 7). One example of this mechanism is Borna disease (BD). Although the natural hosts appear to be horses and sheep, Borna disease virus (BDV) can infect a wide range of animals, from birds to nonhuman primates (8). Much of the experimental data on BDV has been obtained in rat models, in which infection of adult immunocompetent rats with BDV causes either acute disease characterized by hyperactivity and aggressiveness followed by tremors, paralysis, and often death or a chronic disease in which the animals become listless and/or obese (9).
The neuropathological changes in BD are dependent on an inflammatory response resulting from sensitization against BDV antigens (10). The BDV-induced inflammatory process in the CNS has been likened to a delayed-type hypersensitivity reaction (11). Like a conventional delayed-type hypersensitivity, the inflammatory response in BD probably consists of a sequence of events including: (i) an immediate or early proinflammatory reaction, which must be mediated by locally resident cells, that ultimately results in both the sensitization and the trafficking of antigen-specific primed T cells into the area; (ii) restimulation and recruitment of antigen-specific T cells to produce locally the cytokines necessary for the induction and amplification of the reaction; and (iii) recruitment and activation of the nonspecific cells of the monocyte lineage responsible for the effector phase of the delayed-type hypersensitivity reaction (12, 13).
In BD and other neurotropic virus infections, CNS resident cells, such as neurons or glial cells, must furnish the factors required for the initiation of the inflammatory reaction. They must respond to injury or infection by affording immune cells access to any invading antigen and to the CNS, either by secreting vasoactive factors, which can cause microvascular permeability, or by activating the mast cells found in perivascular areas of the CNS (14). There is evidence of the direct activation of CNS resident cells by virus infection. For example, Newcastle disease virus directly induces immediate early response gene (IEG) cgr-2 in astrocytes and microglia (15). A member of the chemokine family, cgr-2 is the murine homologue of human inflammatory protein 10 (IP-10) and acts as a chemotactic factor for lymphocytes and monocytes (16). In addition to chemokines, it is conceivable that proinflammatory cytokines such as tumor necrosis factor α (TNF-α), which can be produced by neurons in the brain (17), are directly induced by virus infection of the CNS. We recently described evidence that the expression of mRNAs for several proinflammatory cytokines, including TNF-α, interleukin (IL)-1α, and IL-6, is up-regulated during BD (10). The purpose of this investigation is to determine whether this activity is the direct result of the stimulation of CNS resident cells by virus infection or whether it is mediated by invading immune cells. To distinguish between these alternatives, we used dexamethasone to separate the elements of the disease process and to enable us to examine the responses of resident cells in the absence of inflammation. Dexamethasone is one of the most potent and best characterized anti-inflammatory agents known. It exerts its major effects on macrophages by inhibiting the expression of numerous cytokines and chemokines through activation of IκB (thereby inhibiting NF-κB) (18) and by inducing the glucocorticoid response element (which interferes with the function of transcription factors such as AP-1) (19). We show here that treatment of BDV-infected rats with dexamethasone prevents both disease and any evidence of inflammation yet fails to inhibit virus replication and the activation of several immediate early and late host response genes that may be relevant to the induction of inflammation.
MATERIALS AND METHODS
Virus Infection and Treatment of Rats.
Six- to eight-week-old female Lewis rats were infected intranasally under anesthesia with 30 μl of PBS containing 105 focus-forming units of BDV. Following infection, the animals were examined daily for signs of disease. At the indicated times after infection, animals were anesthetized and perfused with PBS. Then their brains were removed, snap frozen, and stored at −80°C. To study the effect of dexamethasone, groups of BDV-infected rats were treated i.p. at days 15 and 20 postinfection with three doses of dexamethasone (5 mg/kg) in saline or with saline alone at 12-h intervals.
Immunohistochemical Analysis.
Rats were perfused transcardially with PBS containing procaine-HCl (5 g/liter) followed by Bouin–Hollande fixation solution (21). Brains were removed and dissected into anterior, middle, and posterior parts and postfixed for 24 h in the same fixative. After dehydration in a graded series of 2-propanol, tissues were embedded in Paraplast Plus (Merck) and cut into 7-μm-thick sections. Immunohistochemical analysis was performed on coronal sections, which were incubated with mouse monoclonal antibody ED1 (Camon, Wiesbaden, Germany), which recognizes rat macrophages, monocytes, and dentritic cells (20), or with mouse monoclonal antibody BO 18 (a gift from J. Richt, University of Giessen, Germany), which recognizes the BDV p38 antigen. Reactions were visualized with biotinylated sheep anti-mouse IgG and streptavidin–peroxidase complex (Amersham), using the nickel-enhanced diaminobenzadine reaction described previously (21).
RNA Extraction.
Total RNA was isolated from BDV-infected rat brains according to the RNAzol B method described in the manufacturer’s manual (Biotecx Laboratories, Houston). The RNA was subjected to reverse transcriptase–PCR (RT-PCR) followed by Southern blot or subjected to electrophoresis in a 1% agarose formaldehyde gel followed by Northern blot.
RT-PCR and Southern Blot Analysis.
Analyses for the mRNAs of the rat T-lymphocyte marker CD8, the rat major histocompatibility complex class II antigen RT1-B, the rat cytokines interferon (IFN)-γ, TNF-α, IL-6, the rat chemokines macrophage inflammatory protein 1β (MIP-1β), mob-1, and glyceraldehyde-3-phosphate dehydrogenase (G3PDH; as an internal control) were performed by RT-PCR followed by Southern blot. Gene-specific sense and antisense oligonucleotide primers used for RT-PCR, and the internal oligonucleotide probes used for Southern blot hybridization were designed from the published sequence data. The nucleotide sequences of the oligonucleotide primers used for RT-PCR are as follows: RT1-B antisense 27-mer, 5-TGTGACTCAAAGGGGCCCTGGGTGTCT-3; RT1-B sense 29-mer, 5-CAGCCCAACACCCTCATCTGCTTTGTAGA-3; segment of 446 bp was amplified (GenBank accession no. K02815); IL-6 antisense 33-mer, 5-AGGTAGAAACGGAACTCCAGAAGACCAGAGCAG-3; IL-6 sense 34-mer, 5-ATGAAGTTTCTCTCCGCAAGAGACTTCCAGCCAG-3; segment of 371 bp was amplified (GenBank accession no. M26744); MIP-1β antisense 26-mer, 5-TCAGTTCAACTCCAAGTCATTCACAT-3; MIP-1β sense, 5-ATGAAGCTCTGCGTGTCTGCCTTCT-3; segment of 279 bp was amplified (GenBank accession no. U06434); mob-1 antisense 25-mer, 5-GTTACGGAGCTCTTTTTGACCTTCT-3; mob-1 sense 28-mer, 5-ATGAACCCAAGTGCTGCTGTCGTCGTTC-3; segment of 298 bp was amplified (GenBank accession no. U17035); G3PDH antisense 20-mer, 5-AAGCAGTTGGTGGTGCAGGA-3; G3PDH sense 20-mer, 5-AAGGTGAAGGTCGGAGTCAA-3; segment of 464 bp was amplified (GenBank accession no. M33197).
The nucleotide sequence of CD8 antisense 28-mer, CD8 sense 31-mer, TNF-α antisense 32-mer, TNF-α sense 31-mer, IFN-γ antisense 32-mer, and IFN-γ sense 30-mer were described previously (22). Reverse transcriptase reactions were performed at 42°C for 1 h using avian myeloblastosis virus RT (Promega) as described previously (22). Total rat brain RNA (6 μg) and 1 μM antisense primer were used in the RT reaction. A portion of the RT product was subjected to PCR amplification using the antisense and the sense primers. Amplification was carried out for 35 cycles of denaturation at 94°C for 1 min, and annealing was done at 50°C for 1 min. Polymerization was done at 72°C for 1 min with a Taq DNA polymerase (Fisher Scientific), as described previously (22). The PCR products were subjected to electrophoresis on 1 or 1.5% agarose gel (Sigma) and then blotted onto GeneScreen Plus membrane (DuPont), hybridized with 32P-labeled internal oligonucleotide probes, and exposed to autoradiography film. Oligonucleotide probes were labeled with [γ-32P]ATP (ICN) (specific activity, 4500 Ci/mmol; 1 Ci = 37 GBq) using T4 polynucleotide kinase (Promega).
The nucleotide sequence of the probes are as follows: RT1-B 30-mer, 5-TTATGACTGCAAGGTGGAGCACTGGGGCCT-3; MIP-1β 33-mer, 5-CATCGGAACTTTGTGATGGATTACTATGAGACC-3; mob-1 36-mer, 5-TCCTATGGCCCTGGGTCTCAGCGTCTGTTCATGGAA-3; G3PDH 20-mer, 5-TTCTCCATGGTGGTGAAGAC-3.
The nucleotide sequence of the probes CD8 47-mer, IFN-γ 48-mer, TNF-α 40-mer, and IL-6 49-mer were described previously (22).
Northern Blot Analysis.
Total RNA (6 μg) was fractionated on a 1% agarose gel containing 2.2 M formaldehyde and MOPS buffer (pH 7.0) and blotted onto GeneScreen Plus membrane. Each cDNA probe was labeled with [α-32P]dCTP (ICN) (specific activity, 3000 Ci/mmol) using a nick translation kit (Boehringer Mannheim).
The following cDNA clones were used for probe: G3PDH (23), the p38 protein gene of BDV (24), egr-1 (5), c-jun (25), c-fos (26), inducible nitric oxide synthase (iNOS, ref. 27), rat T kininogen (28), rat cyclooxygenase-2 (COX-2, ref. 29).
RESULTS
Effects of Dexamethasone on BDV-Induced Neurological Signs and CNS Inflammation.
Because glucocorticoids influence a variety of important inflammatory actions, we studied the effect of dexamethasone on BD. Groups of 10 BDV-infected rats were treated with dexamethasone in saline or with saline alone as described in Materials and Methods. Although all of the animals that received saline alone developed severe signs of neurological disease at 20 days postinfection, none of the dexamethasone-treated rats showed any clinical signs up to 30 days postinfection. Furthermore, the brains of dexamethasone-treated, BDV-infected rats showed no neuropathological lesions and a virtual absence of the activated macrophages normally seen in the CNS of BDV-infected rats despite the presence of high levels of BDV antigen (Fig. 1).
Figure 1.
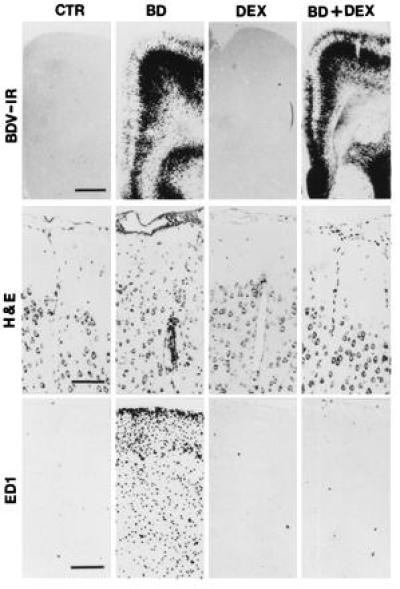
Immunohistochemical analysis showing the distribution of BDV p38-positive cells (BDV-IR) and ED1-positive cells (ED1) in normal rat brain (CTR), BDV-infected brain at 22 days postinfection (BD), brain from an uninfected dexamethasone-treated rat (DEX), and brain from a dexamethasone-treated infected rat (BD+DEX). Brain sections from each experimental group were also stained with hematoxylin–eosin (H&E). Immunohistochemical analysis was performed on sections from paraffin-embedded brains as described in Materials and Methods. (Bars: BDV-IR = 250 μm; H & E = 100 μm; ED1 = 200 μm.)
To further study the effects of dexamethasone on the BDV-induced recruitment and/or activation of effector CD8 T cells and inflammatory cells in the CNS, we analyzed the expression of mRNAs for the CD8 T cell antigen and the major histocompatibility complex class II antigen, RT-1B. For these experiments, we used Southern blot analysis following RT-PCR amplification as described in Materials and Methods. Infection with BDV results in the appearance of mRNA for CD8 and RT-1B in the CNS (Fig. 2). The virus-induced expression of these mRNAs is strongly inhibited in brains of dexamethasone-treated, BDV-infected rats (Fig. 2).
Figure 2.
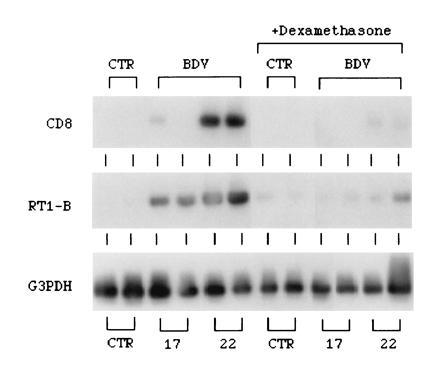
Effect of dexamethasone on the expression of mRNAs for CD8, RT1-B, and G3PDH in BDV-infected rat brain. On different days after infection, RNA was isolated from the brain, and 6 μg of total RNA was subjected to each RT-PCR. The PCR products were subjected to Southern blotting and identified by using gene-specific, 32P-labeled oligonucleotide probe as described in Materials and Methods. CTR, brain RNA from normal uninfected rats; BDV, brain RNA from BDV-infected rats at days 17 and 22 postinfection; +Dexamethasone, brain RNA from dexamethasone-treated rats. The RT-PCR products obtained from two rats per time point are displayed.
Expression of effector molecules of inflammation, including iNOS, the enzyme responsible for NO production, the bradykinin precursor T kininogen, and COX-2, the rate-limiting enzyme in prostaglandin E synthesis, was also examined in the CNS of BDV-infected rats either left untreated or treated with dexamethasone. Northern blot analysis showed that BDV infection triggers the up-regulated expression of all three of these mRNAs and that treatment with dexamethasone either totally blocks (T kininogen) or strongly inhibits (iNOS and COX-2) expression of these mRNAs (Fig. 3).
Figure 3.
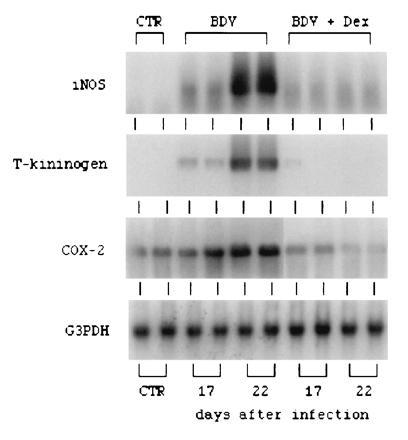
Northern blot analysis of mRNAs for iNOS, T kininogen, and COX-2. RNAs were isolated from normal rat brains (CTR), BDV-infected rat brains at days 17 and 22 postinfection (BDV), and dexamethasone-treated, BDV-infected rat brains at days 17 and 22 postinfection (BDV+Dex). RNA was electrophoresed in a 1% agarose gel containing formaldehyde, transferred onto nylon membrane, and hybridized with specific 32P-labeled probes as described in Materials and Methods.
Identification of CNS Host Gene Responses to BDV Infection That Occur in the Absence of Inflammation.
Based on the hypothesis that virus-induced events must occur before the invasion of inflammatory cells and on the level at which dexamethasone acts, we examined the expression of genes that we considered likely to be up-regulated directly by virus infection. Foremost among these are IEGs. We have shown previously that infection with BDV induces the up-regulation of several IEGs, including the transcription factor egr-1 (5). The influence of dexamethasone on BDV transcription and on the BDV-induced expression of egr-1, c-fos, and c-jun, transcription factors that may play a role in inflammation, was examined using Northern blot analysis. (Fig. 4). The expression of mRNAs for egr-1, c-fos, and c-jun, which was, surprisingly, somewhat up-regulated in normal rats by treatment with dexamethasone, substantially increased in the CNS of BDV-infected, dexamethasone-treated rats (Fig. 4).
Figure 4.
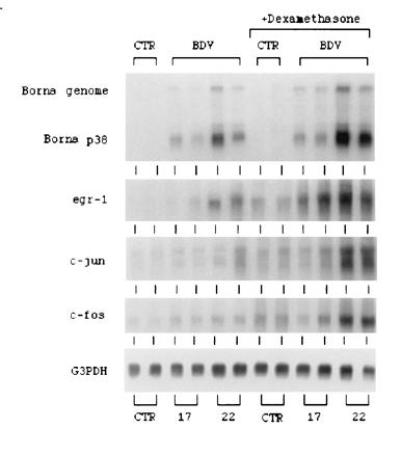
Northern blot analysis of BDV genomic RNA and mRNAs of BDV p38, egr-1, c-jun, and c-fos. RNAs were isolated from normal rat brains (CTR), BDV-infected rat brains at days 17 and 22 postinfection (BDV), brain RNA from dexamethasone-treated rats (+Dexamethasone). RNA was electrophoresed in a 1% agarose gel containing formaldehyde, transferred onto nylon membrane, and hybridized with specific 32P-labeled probes as described in Materials and Methods.
The fact that the expression of a number of transcription factors is enhanced in the CNS of BDV-infected rats treated with dexamethasone suggested that late host response genes, such as those responsible for the production of inflammatory mediators, may also be activated as a direct consequence of virus replication. We showed previously that the expression of mRNAs for the proinflammatory cytokines TNF-α, IL-1α, IL-6 as well as the lymphokine IFN-γ, is strongly increased in the brain during the acute phase of BD (22). We therefore assessed, using RT-PCR (Fig. 5), whether or not dexamethasone can alter the expression levels of these factors. Treatment with dexamethasone blocks the BDV-induced stimulation of IFN-γ mRNA expression in the brains of BDV-infected rats. In contrast, BDV-induced expression of mRNAs for TNF-α and IL-6 still occurs in animals receiving dexamethasone.
Figure 5.
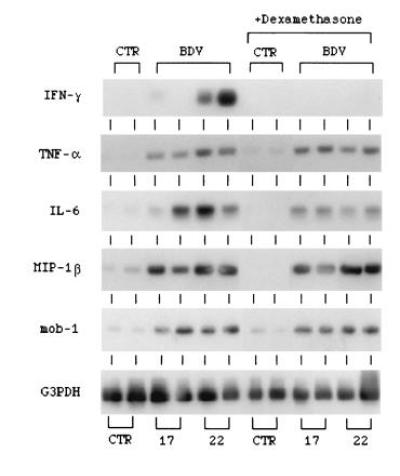
Effect of dexamethasone on the expression of mRNAs for the cytokines IFN-γ, TNF-α, and IL-6 and chemokines MIP-1β and mob-1 in BDV-infected rat brains. Total RNA was subjected to RT-PCR by using gene-specific primers, and the amplified DNA was analyzed by Southern blotting as described in Materials and Methods. CTR, brain RNA from normal uninfected rats; BDV, brain RNA from BDV-infected rats at days 17 and 22 postinfection; +Dexamethasone, brain RNA from dexamethasone-treated rats.
We have also studied the effects of dexamethasone on the expression of mRNAs for chemokines that are potentially involved in the development of neuropathologic lesions in BD. These include members of both the CC chemokine family (MIP-1β) (30) and the CXC chemokine family (mob-1) (31). The results show that levels of mRNAs for MIP-1β and mob-1 (Fig. 5) are increased in BDV-infected rat brains at 17 and 22 days postinfection and that treatment with dexamethasone has no notable effect on their expression.
DISCUSSION
These data confirm the hypothesis that neither the neuropathological lesions nor the clinical signs of BD are directly related to the level of BDV replication in the CNS. Treatment with dexamethasone, although protecting the animal from disease, enhanced the levels of viral antigen and RNA detected in the CNS. The clinical benefit of treatment with dexamethasone correlated with the ablation of signs of CNS inflammation including cell infiltration, the presence of monocytic cells expressing the activation marker ED-1, as well as the expression of mRNAs specific for CD8, IFN-γ, RT-1B, iNOS, T kininogen, and COX-2. Although the action of glucocorticoids is multifaceted, their predominant effect is anti-inflammatory, with both early and late phase inflammatory reactions being susceptible to the inhibitory action of these compounds (32, 33).
With respect to affording antigen and immune cells access through the blood–brain barrier, vasoactive substances including bradykinin, the action of which is potentiated by prostaglandin E2 (34) and NO (35), are likely to play major roles. Our results indicate that BDV infection strongly induces mRNAs encoding precursor proteins or enzymes involved in the generation of these effector molecules and that dexamethasone blocks this response. Thus, in addition to its other anti-inflammatory activities, treatment with dexamethasone may help stabilize the blood–brain barrier in BD by interfering with the production of vasoactive substances.
Treatment with dexamethasone, which evidently prevents the influx of inflammatory cells from the periphery into the brain, has enabled us to separate more clearly the response of CNS-resident cells to BDV-infection from the influence of peripheral immune cells. We define responses of CNS-resident cells as those that occur in the presence of dexamethasone. Under these conditions, the expression of immediate early genes encoding transcription factors, certain proinflammatory cytokines, such as TNF-α, and chemokines including the IP-10 analogue mob-1, is up-regulated by BDV infection. We expect that the first host response to BDV infection is the activation of IEGs including egr-1, c-fos, and c-jun. This hypothesis is supported by our observation that the expression of these IEGs and that of BDV RNA is coordinated spatiotemporally in the CNS (5). Our assumption is that the next stage in the contribution of resident cells to the induction of the inflammatory response, dependent either directly or indirectly on IEG products such as AP-1, would be the activation of late response genes encoding proinflammatory factors. In BD, these include TNF-α, IL-6, mob-1, and MIP-1β, the production of which is not blocked by dexamethasone.
The observation that TNF-α is not inhibited by dexamethasone in this model requires some explanation because glucocorticoids are known to inhibit the expression of TNF-α and numerous other cytokines and chemokines (18, 19). Several possibilities exist. (i) The blood–brain barrier may limit the amount of dexamethasone that reaches the brain such that the level attained is insufficient to inhibit expression of certain cytokine and chemokine genes in the CNS. (ii) The CNS resident cells expressing these cytokine or chemokine mRNAs may lack glucocorticoid receptors or other components linking glucocorticoids to the inhibition of the induction of TNF-α and other cytokines. In any case, treatment with dexamethasone prevents clinical symptoms of BD, which suggests that the products of these cytokine and chemokine genes are not directly responsible for the disease. Based on their known immunologic functions, we would expect that the main role of these factors in the induction of BD is to activate and attract immune cells from the periphery. BD is known to be triggered by factors produced by activated BDV-specific T cells (10). At the outset of BD, these must be sensitized and attracted to the CNS such that they may produce, locally, the factors stimulatory for macrophages and other non-antigen-specific effector cells, the most important of which is IFN-γ. This latter activity causes the massive inflammation that is characteristic of BD.
The implication of our findings is that CNS-resident cells, directly in response to infection, are capable of elaborating factors that may forge the link between peripheral immune cells and the CNS. The ability to produce factors that attract both T cells and monocytes, such as CXC chemokines (16), suggests that resident cells may be at the front line of the inflammatory response in the CNS, a hypothesis that will be further tested in experiments targeting the expression of genes implicated in the response of CNS resident cells.
Acknowledgments
We thank H. Preibsch, S. Roscher, P. Sack, E. Rodenberg, P. Lattermann, and H. Schneider for excellent technical help. We also thank Dr. L. E. Eiden, National Institute of Mental Health, for his helpful comments. This work was supported by grants from Deutsche Forschungsgemeinschaft, Volkswagenstitung, and Kempkes-Stitung (to E.W.).
Footnotes
Abbreviations: CNS, central nervous system; BD, Borna disease; BDV, BD virus; TNF, tumor necrosis factor; MIP, macrophage inflammatory protein; IL, interleukin; IEG, immediate early response gene; G3PDH, glyceraldehyde-3-phosphate dehydrogenase; RT, reverse transcriptase; iNOS, inducible nitric oxide synthase; COX, cyclooxygenase; IFN, interferon.
References
- 1.Grant I, Atkinson J H. In: Immunogenic Mechanisms in Neurologic and Psychiatric Disease. Waksman B H, editor. New York: Raven; 1990. pp. 291–304. [Google Scholar]
- 2.Price R W, Brew B J, Rosenblum M. In: Immunogenic Mechanisms in Neurologic and Psychiatric Disease. Waksman B H, editor. New York: Raven; 1990. pp. 269–290. [Google Scholar]
- 3.Wolinski J S. In: Immunogenic Mechanisms in Neurologic and Psychiatric Disease. Waksman B H, editor. New York: Raven; 1990. pp. 259–268. [Google Scholar]
- 4.Schneider R J, Shenk T. Annu Rev Biochem. 1987;56:317–332. doi: 10.1146/annurev.bi.56.070187.001533. [DOI] [PubMed] [Google Scholar]
- 5.Fu Z F, Weihe E, Zheng Y M, Schäfer M K-H, Sheng H, Corisdeo S, Rauscher F J, III, Koprowski H, Dietzschold B. J Virol. 1993;67:6674–6681. doi: 10.1128/jvi.67.11.6674-6681.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrne A, Oldstone M B A. J Virol. 1984;51:682–686. doi: 10.1128/jvi.51.3.682-686.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty P C, Dunlop M B C, Parish C R, Zinkernagel R M. J Immunol. 1976;117:187–190. [PubMed] [Google Scholar]
- 8.Ludwig H, Becht H. In: Slow Virus Infections of the Central Nervous System. ter Meulen V, Katz M, editors. New York: Springer; 1977. pp. 75–83. [Google Scholar]
- 9.Narayan O, Herzog S, Frese K, Scheffers H, Rott R. J Infect Dis. 1983;148:305–315. doi: 10.1093/infdis/148.2.305. [DOI] [PubMed] [Google Scholar]
- 10.Stitz L, Dietzschold B, Carbone K M. Curr Top Microbiol Immunol. 1995;190:75–92. doi: 10.1007/978-3-642-78618-1_5. [DOI] [PubMed] [Google Scholar]
- 11.Richt J, Stitz L, Deschl U, Frese K, Rott R. J Gen Virol. 1990;71:2565–2573. doi: 10.1099/0022-1317-71-11-2565. [DOI] [PubMed] [Google Scholar]
- 12.Meltzer M S, Nacy C A. In: Fundamental Immunology. 2nd Ed. Paul W E, editor. New York: Raven; 1989. pp. 765–777. [Google Scholar]
- 13.Ptak P J, Herzog W R, Askenase P W. J Immunol. 1993;146:469–475. [PubMed] [Google Scholar]
- 14.Theoharides T C, Dimitriadou V, Letourneau R, Rozniecki J J, Vliagoftis H, Boucher W. Neuroscience. 1993;57:861–871. doi: 10.1016/0306-4522(93)90030-j. [DOI] [PubMed] [Google Scholar]
- 15.Vanguri P, Farber J M. J Immunol. 1994;152:1411–1418. [PubMed] [Google Scholar]
- 16.Taub D D, Lloyd K, Wang J M, Ortaldo J R, Harada A, Matsushima D, Kelvin D J, Oppenheim J J. J Exp Med. 1993;177:1809–1814. doi: 10.1084/jem.177.6.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breder C D, Tsujimoto M, Terano Y, Scott D W, Saper C B. J Comp Neurol. 1993;337:543–567. doi: 10.1002/cne.903370403. [DOI] [PubMed] [Google Scholar]
- 18.Scheinman R I, Cogswell P C, Lofquist A K, Baldwin A S., Jr Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 19.Yang-Yen H-F, Chambard J-C, Sun Y-L, Smeal T, Schmidt T J, Drouin J, Karin M. Cell. 1990;62:1205–1215. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 20.Mueller S, Weihe E. Brain Behav Immun. 1991;5:55–72. doi: 10.1016/0889-1591(91)90007-w. [DOI] [PubMed] [Google Scholar]
- 21.Zentel H J, Weihe E. Brain Behav Immun. 1991;5:132–147. doi: 10.1016/0889-1591(91)90012-y. [DOI] [PubMed] [Google Scholar]
- 22.Shankar V, Kao M, Hamir A M, Sheng H, Koprowski H, Dietzschold B. J Virol. 1992;66:992–998. doi: 10.1128/jvi.66.2.992-998.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tso J Y, Sun H-H, Kao T-H, Reece K S, Wu R. Nucleic Acids Res. 1985;13:2485–2501. doi: 10.1093/nar/13.7.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McClure M A, Thibault K J, Hatalski C G, Lipkin W I. J Virol. 1992;66:6572–6577. doi: 10.1128/jvi.66.11.6572-6577.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakai M, Okuda A, Hatayama I, Nishi S, Muramatsu M. Cancer Res. 1989;49:5633–5637. [PubMed] [Google Scholar]
- 26.Curran T, Gordon M B, Rubino K L, Sambucetti L C. Oncogene. 1987;2:79–84. [PubMed] [Google Scholar]
- 27.Xie Q-W, Cho H J, Calavcav J, Mumford R A, Swiderek K M, Lee T D, Ding A, Troso T, Nathan C. Science. 1992;256:215–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 28.Furuto-Kato S, Matsumoto A, Kitamura N, Nakanishi S. J Biol Chem. 1985;260:12054–12059. [PubMed] [Google Scholar]
- 29.Yamagata K, Andreasson K I, Kauffmann W E, Barnes C A, Worley P F. Neuron. 1993;11:321–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 30.Baggiolini M, Dahinden C A. Immunol Today. 1994;15:127–133. doi: 10.1016/0167-5699(94)90156-2. [DOI] [PubMed] [Google Scholar]
- 31.Liang P, Averboukh L, Zhu W, Pardee A. Proc Natl Acad Sci USA. 1994;91:12515–12519. doi: 10.1073/pnas.91.26.12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barnes P J, Adcock I. Trends Pharmacol Sci. 1993;141:436–442. doi: 10.1016/0165-6147(93)90184-l. [DOI] [PubMed] [Google Scholar]
- 33.Fauci A. Ann Intern Med. 1976;84:304–315. doi: 10.7326/0003-4819-84-3-304. [DOI] [PubMed] [Google Scholar]
- 34.Williams T J, Peck M J. Nature (London) 1977;270:215–217. doi: 10.1038/270530a0. [DOI] [PubMed] [Google Scholar]
- 35.Nathan C. FASEB J. 1992;6:3051–3064. [PubMed] [Google Scholar]