

Abstract
In cerebellar Purkinje neurons, γ-aminobutyric acid (GABA)-mediated inhibitory synaptic transmission undergoes a long-lasting “rebound potentiation” after the activation of excitatory climbing fiber inputs. Rebound potentiation is triggered by the climbing-fiber-induced transient elevation of intracellular Ca2+ concentration and is expressed as a long-lasting increase of postsynaptic GABAA receptor sensitivity. Herein we show that inhibitors of the Ca2+/calmodulin-dependent protein kinase II (CaM-KII) signal transduction pathway effectively block the induction of rebound potentiation. These inhibitors have no effect on the once established rebound potentiation, on voltage-gated Ca2+ channel currents, or on the basal inhibitory transmission itself. Futhermore, a protein phosphatase inhibitor and the intracellularly applied CaM-KII markedly enhanced GABA-mediated currents in Purkinje neurons. Our results demonstrate that CaM-KII activation and the following phosphorylation are key steps for rebound potentiation.
Keywords:
Many features of the activity-dependent plasticity of excitatory synapses, thought to be the cellular basis of learning and memory, are well established (1). Much less is known about the plasticity of inhibitory synapses in vertebrates. Only recently it was shown that inhibitory synaptic transmission itself undergoes activity-dependent modification in several regions of the vertebrate brain, i.e., cerebellar Purkinje neurons (2–4), the Mauthner cells of the goldfish (5), deep cerebellar nuclei (6), visual cortex (7), and hippocampus (8, 9). Besides the well-characterized activity-dependent plasticity of excitatory synapses (1), plasticity of inhibitory synapses may also play important roles in learning, memory, and development (10).
In cerebellar Purkinje neurons, the activation of an excitatory synaptic input can induce a long-lasting potentiation of γ-aminobutyric acid (GABA)-mediated inhibitory synaptic currents, a phenomenon that was named rebound potentiation (3). Rebound potentiation is triggered by a transient elevation of the intracellular Ca2+ concentration (3) due to activation of voltage-gated Ca2+ channels (2, 4), but the intracellular mechanisms linking the short-lived Ca2+ transient to the much longer lasting rebound potentiation are unclear. A rise in intracellular Ca2+ concentration is also an important step for triggering activity-dependent plasticity of various excitatory synapses, such as long-term potentiation in the CA1 area of the hippocampus (1) and long-term depression (LTD) of parallel fiber synapses in the cerebellum (11, 12). These Ca2+ signals were shown to trigger various protein kinase cascades and may eventually lead to up- or down-regulation of post synaptic glutamate receptors (13–15). Since protein phosphorylation is a major mechanism for regulating the function of ligand-gated ion channels (16), protein kinase cascades may also play key roles for rebound potentiation of inhibitory synapses.
The present study was undertaken to examine whether rebound potentiation involves Ca2+/calmodulin-dependent protein kinase II (CaM-KII) signal transduction pathway. We used various pharmacological agents with different modes of action to block this cascade, as well as direct injection of activated CaM-KII to Purkinje neurons. Our results strongly suggest that rebound potentiation involves CaM-KII-mediated protein phosphorylation.
MATERIALS AND METHODS
Sagittal cerebellar slices of 200 μm thickness were prepared from the vermis of 10- to 14-day-old rats as described (17, 18). The slices were kept in oxygenated standard saline (composition, see below) at 34°C until use. Whole-cell patch-clamp recording methods (19) were used to record from Purkinje neurons directly visualized through a ×40 water immersion objective in an upright microscope (Zeiss Axioskop) (17, 18). Seal resistances greater than 10 GΩ were obtained using Sylgard-coated (General Electric) patch pipettes with resistances of 2–3 MΩ. In all experiments, compensation of series resistance was performed (2) using the standard procedure of the EPC-7 amplifier (List Electronics, Darmstadt, F.R.G.). The standard internal solution contained 80 mM CsCl, 2 mM MgCl2, 80 mM cesium d-gluconate, 1 mM EGTA, 4 mM NaATP, 0.4 mM NaGTP, and 10 mM Hepes (pH 7.3, adjusted with CsOH). The experimental chamber was continuously perfused with a saline containing 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 20 mM glucose (pH 7.4, when bubbled with 95% O2/5% CO2). Experiments were performed at room temperature (22–24°C), except those with a phosphatase inhibitor, calyculin A (see Fig. 4), which were performed at 32°C.
Figure 4.
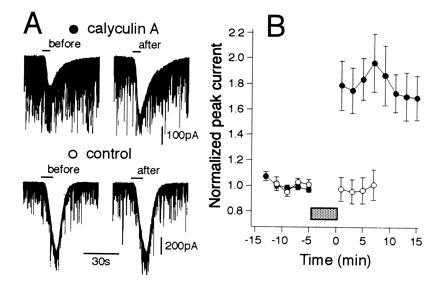
Effects of a protein phosphatase inhibitor, calyculin A, on GABA-induced whole-cell current responses in cerebellar Purkinje neurons. (A) Whole-cell membrane currents induced by bath-applied GABA (5 μM) before (left column) and after (right column) a 5-min perfusion of a saline containing calyculin A (1 μM) (Upper) or that containing vehicle alone (Lower). (B) Changes in peak amplitudes of GABA-induced currents of Purkinje neurons in calyculin A-treated (solid symbols, n = 8) and control (open symbols, n = 7) groups. Data points given in mean ± SEM. Current amplitudes were normalized with respect to the mean value recorded 8 min prior to bath application of the compounds (horizontal bar).
Staurosporine (Calbiochem), calmidazolium (Calbiochem), KN62 (Kamiya Biomedical, Thousand Oaks, CA), and calyculin A (Wako Biochemicals, Osaka) were dissolved in dimethyl sulfoxide (DMSO), added to the saline, and applied extracellularly by perfusion. The final concentrations of DMSO in the saline were always kept below 0.1%. Staurosporine was applied intracellularly (10 μM, included in the internal patch-pipette solution) to the Purkinje neuron and, in addition, the cerebellar slice was incubated for 60 min in a 0.1 μM staurosporine-containing bath solution to ensure its action on the (not well dialyzed) distal dendrites. Calmodulin-binding domain (CBD; Calbiochem) was dissolved in distilled water (100 mM) and added to the internal solution so that the final concentration of CBD was 100 μM. The internal solution used for intracellular application of CaMK-II (see Fig. 5) contained 130 mM CsCl, 5 mM MgCl2, 1 mM CaCl2, 11 mM EGTA, 5 mM NaATP, and 5 mM Hepes (pH 7.3, adjusted with CsOH). Purified CaM-KII was stably activated by autothiophosphorylation as described (14). CaM-KII was incubated for 10 min at 5°C in 50 mM Hepes, pH 7.5/0.5 mM CaCl2/6 mM calmodulin/10 mM magnesium acetate/0.4 mM adenosine 5′-[γ-thio]triphosphate/BSA (1 mg/ml). The autophosphorylated CaM-KII was maintained on ice and diluted 1:10 in the pipette solution just before use. For the control experiments, the CaM-KII was heat-inactivated (10 min at 100°C) before addition to the autothiophosphorylation reaction mixture.
Figure 5.
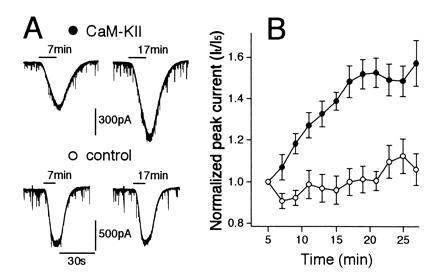
Effects of intracellular injection of activated CaM-KII on GABA-induced whole-cell current responses in cerebellar Purkinje neurons. (A) Whole-cell membrane currents induced by bath-applied GABA (5 μM) 7 min (left column) and 17 min after (right column) initiation of whole cell recording in Purkinje neurons injected with activated (Upper) (200 nM) or heat-inactivated (Lower) (200 nM) CaM-KII. (B) Changes in average peak amplitudes of GABA-induced currents of Purkinje neurons injected with activated CaM-KII (solid symbols, n = 6) and those with heat-inactivated enzyme (open symbols, n = 5). Data points given in mean ± SEM. The ratio of the peak GABA-induced current at indicated time (It) was divided by the peak GABA-induced current attained 5 min after patch formation (I5).
RESULTS
Rebound Potentiation Is Blocked by CaM-KII Inhibitors.
Whole-cell patch-clamp recordings (19) were obtained from Purkinje neurons in cerebellar slices from 10- to 14-day-old rats (17, 18). Voltage-clamped Purkinje neurons displayed spontaneous inhibitory postsynaptic currents (IPSCs) at a high frequency and responded to bath-applied exogenous GABA (2–5 μM, 10 s) with currents of 200- to 1000-pA in amplitude (2, 3). Both, IPSCs and agonist-induced currents were sensitive to bicuculline (10 μM), indicating that they were GABAA-receptor-mediated (2). We have reported (3) that stimulation of excitatory climbing fibers induces a long-lasting rebound potentiation of both IPSCs and current responses to exogenous GABA. Rebound potentiation was blocked by intracellular application of a Ca2+ chelator, bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate (BAPTA) (3). Furthermore, activation of Ca2+ channels by direct depolarization of Purkinje cells resulted in a potentiation of GABAA receptor function with a time course and an amplitude similar to the climbing-fiber-induced rebound potentiation (3). The climbing-fiber-induced rebound potentiation occluded the subsequent induction of long-lasting potentiation by direct depolarization, and vise versa (M.K. and A.K., unpublished observation). These results strongly suggest that climbing-fiber stimulation and direct depolarization activate, through a transient elevation of the intracellular Ca2+ concentration, the common cellular processes that leads to rebound potentiation. Therefore, in the present study, we used depolarizing pulses to −10 mV for 500 ms to induce rebound potentiation. This depolarizing pulse induces a dendritic Ca2+ transient in Purkinje neurons similar to that caused by climbing-fiber stimulation (20).
This procedure consistently potentiated whole-cell current responses to bath-applied exogenous GABA (Fig. 1A Top). The potentiation of the current amplitudes consisted of an initial transient component (reaching up to 200% of the control values), followed by a long-lasting (>40 min) plateau-like phase (of about 150% of the control values) (Fig. 1B, open symbols). In the present study, we investigated exclusively the sustained component of rebound potentiation. When staurosporine, a potent but nonselective protein kinase inhibitor was applied to Purkinje neurons both intra- and extracellularly, a conditioning depolarizing pulse induced no long-lasting change of the amplitudes of GABA-mediated currents except for a transient and smaller potentiation (Fig. 1 A Middle and B Upper, solid symbols). The amplitudes of Ca2+ currents activated by depolarizing pulses were virtually not affected by staurosporine [2.05 ± 0.71 nA (mean ± SD) for the control (n = 9) and 1.74 ± 0.70 nA for staurosporine treated cells (n = 5), measured at 50 ms after the onset of the depolarizing pulses; P > 0.05, Mann–Whitney U test]. Therefore, we conclude that staurosporine depresses rebound potentiation by interfering with cellular processes that follow the Ca2+ influx into Purkinje neurons, presumably by blocking phosphorylation by some protein kinases.
Figure 1.
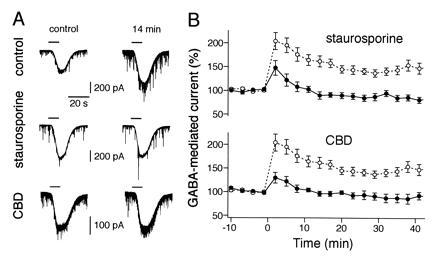
Blocking effect of staurosporine and CBD on rebound potentiation of GABA-induced whole-cell current responses in cerebellar Purkinje neurons. (A) A conditioning depolarizing pulse (500 ms, from −70 mV to −10 mV) induced a long-lasting rebound potentiation of inward currents elicited by the application of 2 μM GABA (Top). Note the effective block of the potentiation by staurosporine (Middle) and CBD (Bottom). Records were taken 4 min before (left column) and 14 min after (right column) the conditioning depolarizing pulse. GABA was applied during the periods indicated by horizontal bars on top of each trace (10 s). (B) Changes in amplitudes of GABA-induced currents of Purkinje neurons for the staurosporine treated (Upper) (solid symbols, n = 6) and CBD injected (Lower) (solid symbols, n = 7) groups plotted against time after the conditioning pulse. The data from the same control group (open symbols, n = 9) are illustrated in both graphs. Data points are the mean ± SEM. Current amplitudes were normalized with respect to the mean value recorded 10 min prior to the depolarizing pulse.
The Ca2+ influx through voltage-gated Ca2+ channels can activate various intracellular processes in Purkinje neurons. For example, Ca2+ may bind to calmodulin and then the Ca2+/calmodulin complex subsequently may activate the multifunctional CaM-KII (21). This enzyme is rich in postsynaptic densities at various sites of the brain (22, 23) being present abundantly also in cerebellar Purkinje neurons (24). It has been shown that CaM-KII may play a key role for the induction of another form of synaptic plasticity, namely long-term potentiation of glutamatergic excitatory synapses in the CA1 region of the hippocampus (25–28) and the developing rat visual cortex (29). To test whether activation of CaM-KII is required for rebound potentiation, we examined the effect of calmodulin binding domain (CBD), a synthetic peptide inhibitor of CaM-KII. CBD inhibits the binding of Ca2+/calmodulin complex to the regulatory domain of CaM-KII and thereby blocks the activation of this kinase (30). Intracellular application of CBD (100 μM) through the recording patch pipette effectively blocked the potentiation of the amplitudes of GABA-mediated currents (Fig. 1 A Bottom and B Lower, solid symbols) to an extent similar to that of staurosporine (Fig. 1 A Middle and B Upper, solid symbols). The amplitudes of Ca2+ currents activated by depolarizing pulses were not affected by CBD [2.05 ± 0.71 nA (mean ± SD) for the control (n = 9) and 1.94 ± 0.47 nA for CBD-treated cells (n = 7)]. Thus these data strongly suggest that activation of calmodulin-dependent protein kinase after Ca2+ influx, presumably CaM-KII, is involved in rebound potentiation.
CaM-KII Activation Occurs Shortly After Conditioning Calcium Transients.
To further examine mechanisms of CaM-KII activation in the induction of rebound potentiation, we used a calmodulin antagonist, calmidazolium, and another selective CaM-KII antagonist, KN62. Because both of these agents are membrane permeable, they can be applied by extracellular perfusion.
When calmidazolium was applied to the bath solution (100 nM) prior to and during the depolarization of Purkinje neurons, it effectively blocked rebound potentiation (Fig. 2A Upper) in a manner similar to that seen when the neurons were treated with staurosporine or CBD (Fig. 1). On the other hand, when the application of calmidazolium started 5 min after the depolarizing pulse, responses to bath-applied GABA were potentiated with a time course equivalent to that seen during rebound potentiation of the control group (Fig. 2A Lower). At the low concentration (100 nM) used in these experiments, calmidazolium did not affect the amplitudes of Ca2+ currents [2.05 ± 0.71 nA for the control (n = 9) and 2.33 ± 0.86 nA for calmidazolium treated cells (n = 5); P > 0.05, Mann–Whitney U test]. Calmidazolium also had no effect on the amplitudes of GABA-induced currents and spontaneous IPSCs [101.3 ± 10.8% and 100.9 ± 15.3% (mean ± SD, n = 6), respectively, relative to the control values prior to calmidazolium application, Mann–Whitney U test]. Thus, the blocking effect of calmidazolium on rebound potentiation is not due to a general depression of GABAA receptor function or to a reduced Ca2+ influx during depolarization, but most likely to the inhibition of the calmodulin activation process by Ca2+. The results also indicate that this process takes place within 5 min after the depolarizing pulse.
Figure 2.
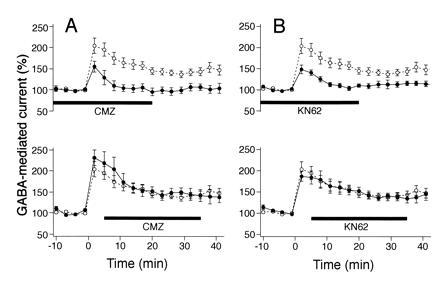
Effects of calmidazolium and KN62 on rebound potentiation of Purkinje neuron current responses to bath-applied GABA. Same experimental design as that illustrated in Fig. 1B. (A) Calmidazolium (100 nM) applied prior to and during the conditioning depolarizing pulse effectively blocks rebound potentiation (Upper) (solid symbols, n = 6). For this and the following three graphs, the open symbols represent the same set of control data as that illustrated also in Fig. 1B, and the thick horizontal bars at bottom indicate the period of application of the CaM-KII inhibitors. The application of calmidazolium (CMZ, 100 nM) that started 5 min after the depolarizing pulse had no effect on rebound potentiation (Lower) (solid symbols, n = 6). (B) KN62 (3 μM) applied prior to and during depolarizing pulse prevented the formation of rebound potentiation (Upper) (solid circles, n = 7). KN62 (3 μM) had no effect on rebound potentiation when given 5 min after the depolarizing pulse (Lower) (solid symbols, n = 6).
Next we examined the effect of KN62, a specific membrane-permeable inhibitor of CaM-KII (31). This compound inhibits the binding of the Ca2+/calmodulin complex to CaM-KII and thereby blocks activation of this enzyme (31). When applied to the bath solution (3 μM) prior to and during depolarization of Purkinje neurons, KN62 strongly suppressed rebound potentiation (Fig. 2B Upper) to an extent similar to that observed in neurons treated with staurosporine, CBD, or calmidazolium (Figs. 1 and 2A Upper). By contrast, when the application of KN62 started 5 min after the conditioning depolarizing pulse, the rebound potentiation was virtually identical to that registered in the control experiments (Fig. 2B Lower). Control experiments indicated that KN62 had no significant effect on the amplitudes of GABA-induced currents or the spontaneous IPSCs (103.2 ± 18.6% and 102.0 ± 16.7%, n = 5, respectively, relative to the control values prior to the KN62 application) or on the amplitudes of Ca2+ currents produced by deporalizating voltage steps [2.05 ± 0.71 nA for the control (n = 9) and 1.65 ± 0.32 nA for KN62 treated cells (n = 7); P > 0.05, Mann–Whitney U test]. These results suggest that KN62 blocks the kinase activity of CaM-KII after its interaction with the Ca2+/calmodulin complex.
Effects of CaM-KII Inhibitors on Spontaneous IPSCs.
The data presented so far demonstrate that inhibition of CaM-KII strongly suppresses the potentiation of the current responses produced by bath-applied exogenous GABA. To examine whether the same treatments also affect the rebound potentiation of IPSCs, we averaged for each cell tested 200–400 events of spontaneously occurring consecutive IPSCs before and an equivalent number of events 15 min after the conditioning pulse. The control experiments demonstrated a potentiation of the IPSC amplitudes with no detectable effect on their time course (Fig. 3A, control traces). The average value of the potentiation calculated from nine experiments was 152 ± 10.4% (mean ± SEM) (Fig. 3B, control bar). By contrast, no potentiation of IPSCs was observed in Purkinje neurons treated with staurosporine (106.3 ± 7.5%, Fig. 3B, staurosporine bar), CBD (105.6 ± 10.7%, Fig. 3B, CBD bar), and in those treated with calmidazolium (107.3 ± 18.6%, Fig. 3B, CMZ-before bar) or KN62 (98.0 ± 7.5%, Fig. 3 A, KN62 trace, and B, KN62-before bar) prior to depolarization. However, when calmidazolium or KN-62 were applied 5 min after the depolarizing pulse, the rebound potentiation of IPSCs had essentially the same time course and extent as in the control experiments (152.5 ± 12.1% and 156.6 ± 10.3%, Fig. 3B, CMZ-after and KN62-after bars). It is noteworthy that rebound potentiation of IPSCs is affected by inhibitors of the CaM-KII signal transduction pathway in a manner quantitatively similar to that observed for the potentiation of current responses produced by bath-applied GABA. This strongly suggests that CaM-KII-dependent up-regulation of postsynaptic GABAA receptor function underlies the potentiation of IPSCs.
Figure 3.
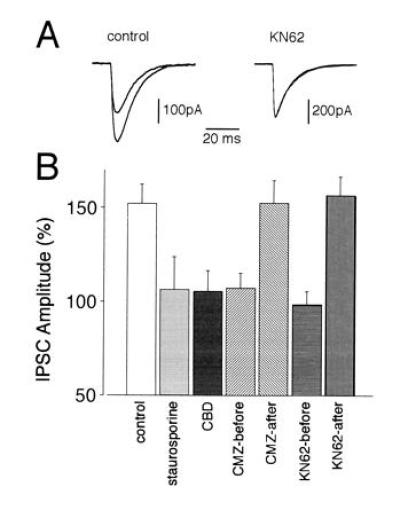
Effects of staurosporine, CBD, calmidazolium, and KN62 on the rebound potentiation of spontaneous IPSCs. (A) Averaged traces (each from 50 consecutive spontaneous IPSCs) recorded in two representative Purkinje neurons from the control and KN62-treated groups, respectively. (Left) Control. Superposition of an averaged trace taken before (small amplitude) and a potentiated averaged IPSC taken 15 min after the depolarizing pulse. The averaged two traces recorded with the same protocol in the presence of KN62 (Right) are almost perfectly superimposed. Note that neither during control nor in the presence of KN62 is the time course of the IPSCs affected. (B) Average changes in IPSC amplitudes induced by the following experimental manipulations: control (n = 9), staurosporine (n = 6), CBD (n = 7), calmidazolium applied prior to and present during depolarization (CMZ-before, n = 6), calmidazolium started 5 min after depolarization (CMZ-after, n = 5), KN62 applied prior to and present during depolarization (KN62-before, n = 7), and KN62 started 5 min after depolarization (KN62-after, n = 6). Values shown represent changes of amplitudes (mean ± SEM) of 200–400 consecutive IPSCs measured 15 min after the conditioning depolarizing pulse normalized with respect to the control values before the depolarization.
Phosphorylation by CaM-KII Potentiates GABA-Mediated Currents.
To demonstrate that CaM-KII-mediated phosphorylation underlies the potentiation of GABA-induced currents in Purkinje neurons, we examined the effects of calyculin A, an inhibitor of protein phosphatase 1 and 2A. The activation state of CaM-KII can be enhanced by phosphatase inhibitors, as reported (32). Thus, bath application of calyculin A (1 μM, 5 min) markedly enhanced GABA-induced currents in Purkinje neurons (Fig. 4 A Upper and B, solid symbols), while vehicle alone had no effect (Fig. 4 A Lower and B, open symbols).
More direct evidence was obtained in experiments in which purified CaM-KII was applied intracellularly to Purkinje neurons (Fig. 5). CaM-KII was autophosphorylated in vitro in the presence of Ca2+, calmodulin, and adenosine 5′-[γ-thio]triphosphate, as reported (14). When activated CaM-KII was included in the pipette (200 nM), amplitudes of GABA-induced currents became larger with time (Fig. 5A Upper) and reached about 150% of the initial amplitudes (Fig. 5B, solid symbols). On the other hand, when heat-inactivated CaM-KII was injected, no significant enhancement of GABA-mediated currents were induced (Fig. 5 A, Lower and B, open symbols). The average amplitudes of spontaneous IPSCs also increased in Purkinje neurons to which active CaM-KII was injected. The value measured around 20 min after whole-cell recording was 166.9% ± 29.8% (mean ± SEM, n = 5) of the initial value. On the other hand, no significant increase was seen in control experiments with heat-inactivated CaM-KII (89.6 ± 11.3%, n = 5). These data clearly indicate that phosphorylation by CaM-KII potentiates GABA-mediated currents in Purkinje neurons.
DISCUSSION
The present data clearly indicate that Ca2+/calmodulin-dependent activation of CaM-KII and subsequent phosphorylation is a requisite step for the induction of rebound potentiation. Our data indicate that this process is completed within 5 min after the conditioning Ca2+ transients produced by the activation of voltage-gated Ca2+ channels (Fig. 2). Therefore, the maintenance of rebound potentiation requires the contribution of some Ca2+/calmodulin-independent mechanism. Biochemical data indicate that after a calmodulin-dependent autophosphorylation process, CaM-KII no longer requires calmodulin to maintain its kinase activity (33, 34). Thus, the autophosphorylated CaM-KII can be active for a period much longer than the life time of the Ca2+/calmodulin complex. One possibility is that GABAA receptors or related proteins are kept phosphorylated by a persistently active calmodulin-independent form of CaM-KII and that this serves as a mechanism for maintaining the potentiated GABAA receptor function. It was recently reported that the β1 and γ2 subunits of GABAA receptors are directly phosphorylated by CaMK-II (35, 36). By using purified fusion proteins of the major intracellular domain of GABAA receptor subunits, specific sites of phosphorylation for CaMK-II have been identified. Thus, it is possible that persistent phosphorylation of GABAA receptors by CaM-KII is the cause for the maintenance of rebound potentiation. Another possibility is that phosphorylation by CaM-KII triggers an intracellular cascade that persistently modifies the GABAA receptor function in a calmodulin-independent manner. In fact, activation of cAMP-dependent protein kinase (PKA) potentiates both IPSCs and current responses to exogenous GABA with a time course similar to rebound potentiation (37). Although there is no direct evidence available yet suggesting that CaM-KII activates PKA in Purkinje neurons, this could be an interesting mechanism underlying rebound potentiation.
The GABAA receptor function is modulated by Ca2+ and protein kinases in various cell types in the brain and cell expression systems (16). At the molecular level, consensus sites for phosphorylation are identified within the large intracellular domains of various GABAA receptor subunits. For example, all β subunits (β1–4) have a conserved site for phosphorylation by PKA (38, 39). Moreover, an alternatively spliced version of the γ2 subunit, termed γ2L, contains a consensus sequence for phosphorylation by protein kinase C (PKC) (40, 41). Biochemical data indicate that GABAA receptor subunits are directly phosphorylated not only by CaMK-II (35, 36) but by PKA (42, 43), PKC (43), and cGMP-dependent protein kinase (36). It should be noted that both the Ca2+ elevation and the PKA activation depress GABAA receptor functions in most of the native cell types and cell expression systems so far examined (16). Moreover, also PKC-dependent phosphorylation caused a reduction of the GABA-induced currents (44). The cerebellar Purkinje neuron is so far the only example whose GABAA-receptor function is up-regulated by both Ca2+ elevation and PKA activation. This suggests either that Purkinje neurons possess unique GABAA receptors that undergo modulation in an opposite way to that seen in other cell types or that the GABAA receptor function is regulated by an as yet unknown mechanism that is influenced by Ca2+ or PKA in different ways in different types of neurons. Recently, Wang et al. (45) reported that GABA-mediated currents in the spinal dorsal horn and hippocampal neurons are enhanced by intracellularly applied α subunit of CaMK-II (45). This is in accordance with our present results in Purkinje neurons. However, PKA has been shown to produce depression of GABA-mediated currents in spinal neurons (46), an effect that is opposite to that found in Purkinje neurons (37). Such differential effects of CaMK-II and PKA further suggest that there are distinct mechanims of GABAA receptor modulation in different cell types.
We showed that application of calyculin A, an inhibitor of protein phosphatase 1 and 2A, enhances GABA-mediated currents in Purkinje neurons (Fig. 4). This is consistent with results obtained in spinal dorsal horn neurons (45). These results suggest that GABAA receptors are depressed by endogenous protein phosphatases. The functional state of GABAA receptors may be dependent on the balance between the phosphorylation by CaM-KII and the dephosphorylation by protein phosphatase 1 or 2A. Interestingly, application of calyculin A induces depression of both α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) responses and parallel-fiber-mediated excitatory synaptic transmission in Purkinje neurons (13, 47). These findings suggest that endogenous protein phosphatases may differentially regulate the functions of GABAA and AMPA receptors in Purkinje neurons.
Besides rebound potentiation of GABAA-receptor-mediated inhibitory inputs, climbing-fiber stimulaton can induce LTD of excitatory parallel fiber synapses when these two distinct excitatory inputs are activated conjunctively, a phenomenon presumed to be the cellular basis for motor learning in the cerebellum (11, 48). Down-regulation of AMPA receptors at parallel fiber synapses is presumed to be the mechanism reponsible for LTD (13, 49, 50). Both PKC (51) and cGMP-dependent protein kinase (52) were suggested to be essentially involved in the induction of LTD. However, there is no report so far that CaM-KII is involved in LTD. Thus, it is possible that rebound potentiation of GABAA-receptor-mediated inhibition and LTD of AMPA-receptor-mediated excitation are mediated by different kinase systems, although the initial trigger is in both cases a transient Ca2+ influx produced by climbing fiber activity (3, 53).
Acknowledgments
We thank Drs. O. Garaschuk, F. Tempia, and J. Lisman for critically reading earlier versions of the manuscript. This work was supported by grants from the Bundesministerium für Forschung und Technologie, the Deutsche Forschungsgemeinschaft, the Human Frontiers Science Program, and the European Community to A.K. and grants from Japanese Ministry of Education, Science and Culture (Grants 05454677, 05260221, 05267242, 06253216, and 06260237), Uehara Foundation, and Ciba Geigy Foundation and Fellowships of the Alexander von Humboldt Foundation and the Human Frontiers Science Program to M.K.
Footnotes
Abbreviations: CaM-KII, Ca2+/calmodulin-dependent protein kinase II; GABA, γ-aminobutyric acid; GABAA receptor, type A GABA receptor; LTD, long-term depression; CBD, calmodulin-binding domain; PKA, cAMP-dependent protein kinase; PKC, protein kinase C; IPSC, inhibitory postsynaptic current; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid.
References
- 1.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 2.Llano I, Leresche N, Marty A. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- 3.Kano M, Rexhausen U, Dreessen J, Konnerth A. Nature (London) 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- 4.Vincent P, Armstrong C M, Marty A. J Physiol (London) 1992;456:453–471. doi: 10.1113/jphysiol.1992.sp019346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korn H, Oda Y, Faber D S. Proc Natl Acad Sci USA. 1992;89:440–443. doi: 10.1073/pnas.89.1.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morishita W, Sastry B R. NeuroReport. 1993;4:719–722. doi: 10.1097/00001756-199306000-00030. [DOI] [PubMed] [Google Scholar]
- 7.Komatsu Y, Iwakiri M. NeuroReport. 1993;4:907–910. doi: 10.1097/00001756-199307000-00017. [DOI] [PubMed] [Google Scholar]
- 8.Stelzer A, Slater N T, Ten Bruggencate G. Nature (London) 1987;326:698–701. doi: 10.1038/326698a0. [DOI] [PubMed] [Google Scholar]
- 9.Otis T S, De Koninck Y, Mody I. Proc Natl Acad Sci USA. 1994;91:7698–7702. doi: 10.1073/pnas.91.16.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kano M. Neurosci Res. 1995;21:177–182. doi: 10.1016/0168-0102(94)00860-i. [DOI] [PubMed] [Google Scholar]
- 11.Ito M. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- 12.Linden D J. Neuron. 1994;12:457–472. doi: 10.1016/0896-6273(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 13.Ito M, Karachot L. Neurosci Res. 1992;14:27–38. doi: 10.1016/s0168-0102(05)80004-5. [DOI] [PubMed] [Google Scholar]
- 14.McGlade-McCulloh E, Yamamoto H, Tan S-E, Brickey D A, Soderling T R. Nature (London) 1993;362:640–642. doi: 10.1038/362640a0. [DOI] [PubMed] [Google Scholar]
- 15.Kolaj M, Cerne R, Cheng G, Brickey D A, Randic M. J Neurophysiol. 1994;72:2525–2531. doi: 10.1152/jn.1994.72.5.2525. [DOI] [PubMed] [Google Scholar]
- 16.Raymond L A, Blackstone C D, Huganir R L. Trends Neurosci. 1993;16:147–153. doi: 10.1016/0166-2236(93)90123-4. [DOI] [PubMed] [Google Scholar]
- 17.Edwards F, Konnerth A, Sakmann B, Takahashi T. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 18.Konnerth A, Llano I, Armstrong C M. Proc Natl Acad Sci USA. 1990;87:2662–2665. doi: 10.1073/pnas.87.7.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 20.Kano M, Garaschuk O, Verkhratsky A, Konnerth A. J Physiol (London) 1995;487:1–16. doi: 10.1113/jphysiol.1995.sp020857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanson P I, Schulman H. Annu Rev Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- 22.Kennedy M B, Bennett M K, Erondu N E. Proc Natl Acad Sci USA. 1983;80:7357–7361. doi: 10.1073/pnas.80.23.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelly P T, McGuinnes T L, Greengard P. Proc Natl Acad Sci USA. 1984;81:945–949. doi: 10.1073/pnas.81.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walaas S I, Yvonne L, Gorelick F S, DeCamilli P, Moretti M, Greengard P. Mol Brain Res. 1988;4:233–242. doi: 10.1016/0169-328x(88)90029-0. [DOI] [PubMed] [Google Scholar]
- 25.Malenka R C, Kauer J A, Perkel D J, Mauk M D, Kelly P T, Nicoll R A, Waxham M N. Nature (London) 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 26.Malinow R, Schulman H, Tsien R W. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 27.Silva A J, Stevens C F, Tonegawa S, Wang Y. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 28.Fukunaga K, Stoppini L, Miyamoto E, Muller D. J Biol Chem. 1993;268:7863–7867. [PubMed] [Google Scholar]
- 29.Funaichi M, Tsumoto T, Nishigori A, Yoshimura Y, Hidaka H. NeuroReport. 1992;3:173–176. doi: 10.1097/00001756-199202000-00013. [DOI] [PubMed] [Google Scholar]
- 30.Payne M E, Fong Y-L, Ono T, Colbran R J, Kemp B E, Soderling T R, Means A R. J Biol Chem. 1988;263:7190. [PubMed] [Google Scholar]
- 31.Tokumitsu H, Chijiwa T, Hagiwara M, Mizutani A, Terasawa M, Hidaka H. J Biol Chem. 1990;265:4315–4320. [PubMed] [Google Scholar]
- 32.Fukunaga K, Soderling T R. Mol Cell Neurosci. 1990;1:133–138. doi: 10.1016/1044-7431(90)90017-x. [DOI] [PubMed] [Google Scholar]
- 33.Saitoh T, Schwartz J H. J Cell Biol. 1985;100:835–842. doi: 10.1083/jcb.100.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller S G, Kennedy M B. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 35.Machu T K, Firestone J A, Browning M D. J Neurochem. 1993;61:375–377. doi: 10.1111/j.1471-4159.1993.tb03582.x. [DOI] [PubMed] [Google Scholar]
- 36.McDonald B J, Moss S J. J Biol Chem. 1994;269:18111–18117. [PubMed] [Google Scholar]
- 37.Kano M, Konnerth A. NeuroReport. 1992;3:563–566. doi: 10.1097/00001756-199207000-00004. [DOI] [PubMed] [Google Scholar]
- 38.Ymer S, Schofield P R, Draguhn A, Werner P, Kohler M, Seeburg P H. EMBO J. 1989;8:1665–1670. doi: 10.1002/j.1460-2075.1989.tb03557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swope S L, Moss S J, Blackstone C D, Huganir R L. FASEB J. 1992;6:2514–2523. [PubMed] [Google Scholar]
- 40.Whiting P, McKernan R M, Iversen L L. Proc Natl Acad Sci USA. 1990;87:9966–9970. doi: 10.1073/pnas.87.24.9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moss S J, Doherty C A, Huganir R L. J Biol Chem. 1992;267:14470–14476. [PubMed] [Google Scholar]
- 42.Kirkness E F, Bovenkerk C F, Ueda T, Turner A J. Biochem J. 1989;259:613–616. doi: 10.1042/bj2590613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Browning M D, Bureau M, Dudek E M, Olsen R W. Proc Natl Acad Sci USA. 1990;87:1315–1318. doi: 10.1073/pnas.87.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krishek B J, Xie X, Blackstone C, Huganir R L, Moss S J, Smart T G. Neuron. 1994;12:1081–1095. doi: 10.1016/0896-6273(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 45.Wang R A, Cheng G, Kolaj M, Randic M. J Neurophysiol. 1995;73:2099–2106. doi: 10.1152/jn.1995.73.5.2099. [DOI] [PubMed] [Google Scholar]
- 46.Porter N M, Twyman R E, Uhler M D, MacDonald L. Neuron. 1990;5:789–796. doi: 10.1016/0896-6273(90)90338-g. [DOI] [PubMed] [Google Scholar]
- 47.Ajima A, Ito M. NeuroReport. 1995;6:297–300. doi: 10.1097/00001756-199501000-00018. [DOI] [PubMed] [Google Scholar]
- 48.Aiba A, Kano M, Chen C, Stanton M E, Fox G, Herrup K, Zwingman T A, Tonegawa S. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 49.Kano M, Kato M. Nature (London) 1987;325:276–279. doi: 10.1038/325276a0. [DOI] [PubMed] [Google Scholar]
- 50.Linden D J, Dickinson M H, Smeyne M, Connor J A. Neuron. 1991;7:81–89. doi: 10.1016/0896-6273(91)90076-c. [DOI] [PubMed] [Google Scholar]
- 51.Linden D J, Connor J A. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- 52.Shibuki K, Okada D. Nature (London) 1991;349:326–328. doi: 10.1038/349326a0. [DOI] [PubMed] [Google Scholar]
- 53.Konnerth A, Dreessen J, Augustine G J. Proc Natl Acad Sci USA. 1992;89:7051–7055. doi: 10.1073/pnas.89.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]