

Abstract
Many blockers of Na+ and K+ channels act by blocking the pore from the intracellular side. For Shaker K+ channels, such intracellular blockers vary in their functional effect on slow (C-type) inactivation: Some blockers interfere with C-type inactivation, whereas others do not. These functional differences can be explained by supposing that there are two overlapping “subsites” for blocker binding, only one of which inhibits C-type inactivation through an allosteric effect. We find that the ability to bind to these subsites depends on specific structural characteristics of the blockers, and correlates with the effect of mutations in two distinct regions of the channel protein. These interactions are important because they affect the ability of blockers to produce use-dependent inhibition.
Keywords: potassium channels, quaternary ammonium blockers, use-dependent blockade
Many ion channel inhibitors such as local anesthetics, anticonvulsants, and antiarrhythmics act by binding to the pore of ion channels and occluding current. The most therapeutically valuable blockers have the feature of use-dependence—inhibition increases as the channel is used, during periods of unusually high activity. The traditional explanation for use-dependence is that the blocker binds more tightly to an activated or stimulus-inactivated state of the channel, so that inhibition accumulates with increased stimulation. This explanation is the basis of the “modulated-receptor” and “guarded-receptor” models (1–3).
Recently, a different mechanism of use-dependent inhibition was found to operate in Shaker K+ channels (4). The alkyl triethylammonium compounds are ordinary open-channel blockers (5, 6) that bind and unbind so rapidly that they cannot produce use-dependent inhibition by the traditional mechanism. They do, however, produce use-dependent inhibition by promoting the intrinsic gating process called C-type inactivation.
During prolonged depolarization, Shaker K+ channels inactivate by two mechanisms: a rapid N-type mechanism and a slower C-type mechanism. N-type inactivation occurs when the NH2-terminal part of the channel protein acts as a tethered open-channel blocker, which occludes current through the pore after it opens (7–9). C-type inactivation involves a conformational change at the outer mouth of the pore (10–12).
The rate of C-type inactivation is very sensitive to the presence of K+ ions in the pore (4, 13). When the pore is depleted of K+ ions, it occurs very quickly. Intracellular quaternary ammonium (QA) blockers can speed up the rate of C-type inactivation by blocking the access of intracellular K+ to the pore and starving the pore of K+ ions. Because they increase the rate of C-type inactivation, which is slow to recover, these blockers produce a cumulative inhibition of the K+ channel that appears as use-dependent inhibition.
A second action of the blockers can interfere with this use-dependent inhibition. Some of the blockers interfere with C-type inactivation by a mechanism that does not involve the movement of K+ ions. This “allosteric” effect by tetraethylammonium (TEA) and a few other compounds prevents C-type inactivation and thus prevents the use-dependent inhibition. (The term “allosteric” is used to distinguish this mechanism from those effects that depend on blocker-induced changes in K+ ion movement.) We asked: What inhibitor structure is required for this allosteric effect, and how does it fit with the known properties of the receptor site for intracellularly applied QA blockers?
MATERIALS AND METHODS
Expression and Recording of Cloned Shaker Channels.
Mutants of the Shaker H4:Δ6-46 channel were prepared and expressed as described previously (10, 12, 14). For channel expression, we used the mammalian human embryonic kidney 293 cells (HEK293; ATCC/CRL-1533). Briefly, cell cultures were grown in DMEM-F12 (Life Technologies, Gaithersburg, MD) with 10% fetal bovine serum (Sigma) to ≈50% confluence on the day of transfection. Before transfection, cells were treated with 0.05% trypsin, 0.53 mM EDTA, washed once, and resuspended in Hepes-buffered saline at 2 × 106 cells/ml. Electroporation was done at 300–350 V (Electroporator, Invitrogen) in 0.4-cm cuvettes containing 200 μl of cells, 10–25 μg of channel plasmid, 1 μg of an simian virus 40 T antigen expression plasmid, and 3–5 μg of πH3-CD8, an expression plasmid that encodes the α subunit of the human CD8 lymphocyte surface antigen (kind gift from Brian Seed, Massachusetts General Hospital). Cells were maintained in growth medium at 37°C with 5% CO2 until use.
Cells were incubated a few minutes before use with polystyrene microbeads precoated with anti-CD8 antibody (Dynabeads M-450 CD8, Dynal, Great Neck, NY). Beading occurred preferentially on cells that expressed the CD8 antigen. Most of the cells that were beaded also had channel expression (15). Only beaded cells were used for electrophysiological recording. Cells typically expressed functional channels within 15–20 h after transfection. Currents from excised inside-out patches were recorded using standard patch-clamp methods (21). We used a computer-controlled solenoid switch for fast perfusion of solutions, as described by Liu and Dilger (16).
Quaternary Ammonium Compounds.
C3-TEA, C4-TEA, C8-TEA, and C10-TEA were prepared by Ashok Khatri (Massachusetts General Hospital Biopolymer facility) by reacting the appropriate 1-bromoalkane with triethylamine (0.1 M each in absolute ethanol; reflux for 18 h) (17). Diethylmethylpropylammonium (DMPA) was made by reacting 1-bromopropane with diethylmethylamine. Other compounds were available commercially: C6-TEA (Aldrich) and QX314 (Research Biochemicals International, Natick, MA). QX222 was a gift from G. Strichartz (Brigham and Women’s Hospital, Boston).
RESULTS
C-Type Inactivation Is Allosterically Inhibited by Internal TEA.
The allosteric effect of blockers on C-type inactivation was best seen in the complete absence of K+ ions, to eliminate the effect of blockers on K+ occupancy of the pore. To measure the rate of C-type inactivation in the absence of K+, we used rapid switching of the solution bathing the intracellular face of an inside-out patch, with zero [K+] solution in the pipette. We maintained the patch in normal intracellular solution with 160 mM K+, but then switched to a zero [K+] solution. While the patch was bathed with zero [K+] solution, we depolarized the membrane potential to open and inactivate the channels. The state of the channels was monitored by rapidly restoring the intracellular [K+] to 160 mM and observing the outward current (Fig. 1A). The envelope of these initial outward currents showed the time course of inactivation. Under these conditions, C-type inactivation was quite fast, with a rate of ≈12 s−1.
Figure 1.
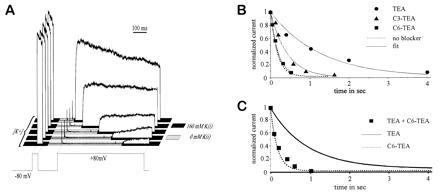
Allosteric inhibition of C-type inactivation by internal blockers. (A) Procedure for measuring C-type inactivation in the absence of potassium. A solenoid-controlled perfusion switch is used to change the solution bathing the intracellular (bath) side of an inside-out patch. The [K+]i is stepped from 160 mM to 0 mM, ≈100 ms before the start of a prolonged depolarization to +80 mV. Inactivation starts at the time of depolarization, and the extent of inactivation is assayed at various times by rapidly restoring [K+]i to 160 mM, as indicated by the shaded bars for the four sample traces. The time course is given by the envelope of these measurements. The brief test pulse to +80 mV before each measurement ensures that complete recovery from inactivation has occurred. (B) Inactivation in the absence of K+ (measured as in A), with blockers added to the internal solution. The peak outward currents after restoring [K+]i are normalized and plotted. The solid lines represent monoexponential fits to the data points, and the dotted line indicates the inactivation time course in the absence of blocker. The concentrations of the internal blockers were: [TEA] = 2 mM; [C3-TEA] = 6 mM; [C6-TEA] = 0.45 mM. (C) Inactivation in the absence of K+, with both 2 mM TEA (5·K1/2) and 5 mM C6-TEA (50·K1/2) added to the internal solution (▪). The lines indicate inactivation time courses in the presence of either TEA alone (solid line) or C6-TEA alone (broken line), for reference.
Application of intracellular TEA slowed this rate substantially. We added 2–60 mM TEA to the zero [K+] intracellular solution; in the presence of normal intracellular [K+], this would block the current by 85–99% (and the blockade is probably even more complete in the absence of K+). Because most of the channels were blocked, and because TEA blockade was at rapid equilibrium compared with the rate of inactivation, our measurement should tell us the rate of inactivation for blocked channels. We found that TEA-blocked channels inactivate approximately 5-fold slower than unblocked channels (Fig. 1B).
The Allosteric Effect Disappears for Long-Chain Alkyl Derivatives of TEA.
To investigate the structural requirements for this allosteric slowing of inactivation, we used a series of alkyl triethylammonium blockers (6, 14). The hydrophobic alkyl chain improves the binding of these blockers relative to TEA, with better binding at longer chain lengths. We measured the allosteric effect of these blockers on C-type inactivation in the absence of K+, using a saturating concentration of each blocker. Of this series, only C3-TEA showed an allosteric effect, which was about half that of TEA (Figs. 1B and 3A Left). The other blockers showed no effect, or even a small positive effect on the rate of inactivation (in the case of C8-TEA and C10-TEA).
Figure 3.
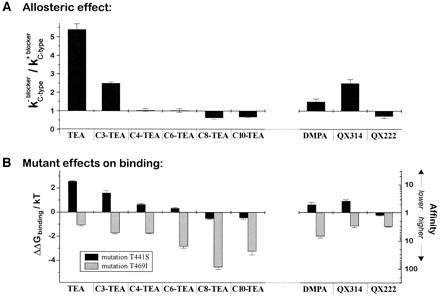
The allosteric effect correlates with binding to the head site. (A) Allosteric effects of the blockers on C-type inactivation. The inactivation rate in zero [K+] was measured as described in Fig. 1. The ratio of the rate without blocker to the rate with blocker is plotted. (B) The half-blocking concentration for each blocker was measured for ShakerΔ wild-type (WT) and for the two mutants T441S and T469I. The fold change in affinity, relative to wild type, is plotted for each of the two mutations. The left axis shows the corresponding change (ΔΔG) in ΔGbinding/kT. (ΔΔG = kT · ln (K1/2{mutant}/K1/2{wild type}).
The Long-Chain Derivatives Occlude the Allosteric Effect of TEA.
To rule out the possibility that the long-chain blockers simply block at a completely different site than TEA, we looked at the interaction between the effects of TEA (which had the 5-fold allosteric effect) and C6-TEA (which had no allosteric effect). If the blockers bind independently, we would expect that TEA would exert its allosteric effect even in the presence of a large excess of C6-TEA. This was not the case: Excess C6-TEA abolished the slowing that would normally be produced by TEA (Fig. 1C), indicating that allosteric and nonallosteric blockers act at exclusive or overlapping sites.
A Mutation Known to Preferentially Affect the Binding of TEA Abolishes the Allosteric Effect.
There is independent evidence from mutagenesis to support the idea that TEA and the long-chain derivatives bind to overlapping but partially separate subsites. Choi et al. (14) found that mutation of a site in the P-loop of the Shaker channel, at position 441, had substantial effects on the binding of intracellular TEA, but negligible effects on the affinity of the long-chain derivatives. By contrast, mutation at position 469 in the S6 segment preferentially affected the binding of the long-chain compounds. These results could be interpreted by supposing that there are two “subsites” on the channel for binding of the QA compounds: a “head” and a “tail” site (referring to the structural triethylamino head and alkyl tail of the QA compounds). TEA, which has no tail, would bind almost exclusively to the head site. The long-chain compounds would have a strong interaction with the hydrophobic tail site, which would pull them away from the head site and reduce the strength of their interaction with it (hence the relatively small effect of 441 head-site mutations on affinity of the long-chain compounds).
We wondered whether the dichotomy between blockers affected by the head-site mutations and those not affected might also explain the ability of blockers to produce the allosteric effect. In other words, might the ability to affect C-type inactivation depend on a compound’s interaction with the head site? We first tested this idea by mutating the head site, to see if it would disrupt the allosteric effect.
Mutation of the threonine at position 441 to serine reduces the affinity for intracellularly applied TEA by approximately 10-fold (18). To assess the allosteric effect, we monitored inward currents through the channel in the presence of 10 mM extracellular K+ with no intracellular K+ (this was technically less demanding than the envelope experiments and gave essentially the same information). For wild-type channels, application of 560 μM TEA to the intracellular face of an inside-out patch slowed the inactivation by approximately 1.7-fold (Fig. 2A; compare the TEA trace with the scaled-down control trace). This effect was less than that seen before, because the current was blocked only ≈50% of the time; it is quantitatively consistent with half of the channels inactivating at the normal rate and half of the channels inactivating 5-fold slower than normal. By contrast, the mutant T441S channel showed no effect of TEA blockade on the inactivation rate: A similar degree of blockade, which required 9 mM TEA, produced no slowing of inactivation (Fig. 2B).
Figure 2.
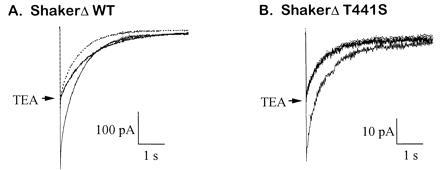
The mutation T441S abolishes the allosteric effect of TEA. (A) Allosteric effect of TEA on wild-type ShakerΔ. The time course of C-type inactivation was measured for inward currents at 0 mV with 0 mM [K+]i and 10 mM [K+]o. A half-blocking concentration of internal TEA (0.56 mM) slowed down the inactivation rate 1.7-fold, from 1.5 ± 0.1 s−1 to 0.9 ± 0.1 s−1. For better comparison, the unblocked trace (dotted) is also shown scaled down 2-fold. (B) Inward current with the T441S mutant was 50% blocked by internal application of 9 mM TEA, but there was no effect on the time course of inactivation (1.7 ± 0.2 and 1.8 ± 0.2, with and without 9 mM TEA) because both traces superimpose when scaled to the same amplitude.
The Allosteric Effect Correlates with TEA-Headgroup-Dominated Binding.
For the alkyl triethylammonium series, we compared the ability of each compound to produce an allosteric effect with its apparent interaction with the head and tail sites. We assessed this interaction using the two mutants studied by Choi et al. (14): T441S and T469I. According to the original work, the main effects are that 441S reduces the binding of head-site compounds, whereas 469I increases the binding of tail-site compounds. Fig. 3B shows this trend for the alkyl-TEA compounds from TEA through C10-TEA. For each compound, the affinity change for each of the two mutants is plotted on a logarithmic scale (so that the length of the bar is proportional to the change in the free energy of binding). There is a clear trend from head-dominated binding to tail-dominated binding, and this correlates well with the size of the allosteric effect on C-type inactivation (Fig. 3A).
To test the validity of this correlation, we extended our analysis to quaternary ammonium compounds outside the alkyl-TEA series. We used a chemical isomer of TEA, DMPA, and two quaternary lidocaine derivatives: QX314 (with a triethylammonium group) and QX222 (with a trimethylammonium group). The three compounds give a similar pattern; significant interaction with the head-site, as measured by the two mutants (Fig. 3B Right), is a good predictor of the allosteric effect (Fig. 3A Right). The dependence on the structure of the compounds is also informative. QX314, which has an aromatic group attached to a triethylammonium headgroup, has a moderate negative allosteric effect (i.e., slowing of inactivation) like that of C3-TEA. Changing the headgroup to trimethylammonium (giving QX222) abolishes the negative allosteric effect. Altering TEA by rearranging the alkyl groups to give DMPA eliminates the triethylammonium structure and substantially reduces the allosteric effect.
DISCUSSION
Structural Requirements for Inducing the Allosteric Effect.
We propose that the ability of various QA compounds to slow down C-type inactivation by an allosteric effect depends on their interaction with one of two subsites at the intracellular mouth of the channel (Fig. 4). If a compound binds to the head site (defined by the effect of the mutation T441S), it slows down inactivation; if it binds preferentially to the tail-site, it does not. Binding to the head site depends, in turn, on two structural features. One is the presence of a triethylammonium headgroup, which allows a substantial interaction with the head binding subsite at the intracellular mouth. The second is the absence of a hydrophobic moiety, such as a long alkyl chain, which can reduce binding to the head subsite by favoring an alternative mode of binding to the tail subsite. Either by eliminating the triethylammonium headgroup or by adding a hydrophobic moiety (Fig. 4B), it is possible to reduce substantially the allosteric effect and thus to favor the development of use-dependent blockade.
Figure 4.
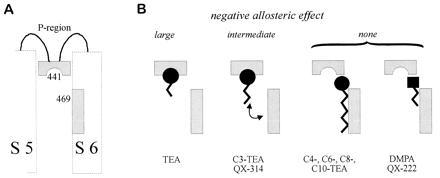
Two functionally distinct subsites for the binding of internal blockers. A model to explain the allosteric effects allows the blockers to bind alternatively to a head site (related to position 441) or a tail site (related to position 469). Only binding to the head site causes the allosteric effect. (A) The two sites are indicated in the context of a partial hypothetical transmembrane folding pattern for a single Shaker channel subunit. Of the six transmembrane segments (S1–S6), only S5 and S6 are shown. The S5–S6 linker (P-region) and S6 are implicated in contributing to the channel pore. (B) The internal blockers fall into three groups depending on their allosteric effects. The first group includes only TEA, which has the largest allosteric effect and binds preferentially to the head site. Blockers in the second group exhibit an intermediate allosteric effect because they bind equally well to the head and tail sites. Blockers in this group possess a triethylammonium headgroup and a moderately hydrophobic tail structure (C3-TEA, QX314). Blockers in the third group exhibit no negative allosteric effect, either because they have a strong hydrophobic element (C4-TEA through C10-TEA), which highly favors binding to the tail site, or because they have no triethylammonium headgroup (QX222, DMPA), which is required for the interaction with the head site.
The different functional effects and the differences in sensitivity to mutations at position 441 suggest that the triethylammonium headgroups of TEA and of (for instance) C6-TEA bind in different positions. Nevertheless, this difference is probably rather small. The two compounds can compete with each other for binding (that is, their binding is exclusive), and they bind with a similar small voltage dependence (14).
For the squid axon K+ channel, TEA and its alkyl derivatives differ also in their interaction with another type of gating. The alkyl triethylammoniums interfere with voltage-dependent closing (deactivation gating) (5, 6, 19), but TEA itself does not (19). This distinction is not seen in the Shaker K+ channel, where all these blockers inhibit deactivation (14).
Alternative modes of blockade, which depend on the structure of the blocker, have been shown also for local anesthetic inhibition of Na+ channels (20). Lidocaine can produce either open-channel blockade or a longer-lasting inhibition that depends on binding to the inactivated state. The open-channel blockade is mimicked by small amines and by quaternary lidocaine derivatives, whereas the slow inhibition is produced by the uncharged form or even by phenol, which resembles the hydrophobic tail of lidocaine. Although the details of this two-part action are different from the allosteric effect on K+ channels, the pattern of alternative head and tail effects is quite similar (and presumably, it too has its basis in alternative binding sites on the channel protein).
Several of the compounds we tested showed a small positive allosteric effect: They made C-type inactivation faster. We do not understand the mechanism for this effect. It does appear to correlate with a small positive effect of the 441S mutation on the affinity for these compounds (see Fig. 3).
Implications for the Conformational Change Involved in C-Type Inactivation.
Contrary to our original understanding of the interaction between TEA blockade and C-type inactivation (10), we find that both external and internal TEA can slow the rate of C-type inactivation. Under normal ionic conditions, only the effect of external TEA (which apparently prohibits C-type inactivation in Shaker channels) is apparent (10). Internal TEA binding has two contrary effects that cancel: It slows down C-type inactivation by an allosteric effect but speeds it up by blocking K+ efflux and depleting the pore of K+ ions. Only when internal K+ is removed can the allosteric effect be seen clearly.
Thus, C-type inactivation can be inhibited by binding of ligands to three distinguishable sites on the pore: external TEA to the outer mouth of the pore (at an electrical distance of 0.2 from the outside), internal TEA to the inner mouth of the pore (at an electrical distance of 0.2 from the inside), and K+ to a site within the pore (at an electrical distance of 0.5). Alternatively stated, we suppose that binding of any of these compounds to the pore stabilizes the open state structure, and this stabilization slows or prevents the conformational change corresponding to C-type inactivation.
Acknowledgments
We thank Yi Liu, Miguel Holmgren, Stuart Forman, Paula Smith, and Mark Jurman for stimulating discussions. We also thank Mark Jurman for supplying the transfected cells, Ashok Khatri of the Massachusetts General Hospital Biopolymer Laboratory for chemical syntheses, and Gary Strichartz for sharing his dwindling supply of QX222. This work was supported by National Institutes of Health Grant NS29693 to G.Y., and by a stipend to T.B. from the Gottlieb Daimler–Karl Benz Foundation.
Footnotes
Abbreviations: TEA, tetraethylammonium; Cn-TEA, alkyl (CnH2n+1) triethylammonium; QA, quaternary ammonium; DMPA, diethylmethylpropylammonium.
References
- 1.Hille B. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butterworth J F, Strichartz G R. Anesthesiology. 1990;72:711–734. doi: 10.1097/00000542-199004000-00022. [DOI] [PubMed] [Google Scholar]
- 3.Starmer C F, Grant A O, Strauss H C. Biophys J. 1984;46:15–27. doi: 10.1016/S0006-3495(84)83994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baukrowitz T, Yellen G. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong C M. J Gen Physiol. 1969;54:553–575. doi: 10.1085/jgp.54.5.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong C M. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoshi T, Zagotta W N, Aldrich R W. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 8.Zagotta W N, Hoshi T, Aldrich R W. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- 9.Demo S D, Yellen G. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- 10.Choi K L, Aldrich R W, Yellen G. Proc Natl Acad Sci USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yellen G, Sodickson D, Chen T-Y, Jurman M E. Biophys J. 1994;66:1068–1075. doi: 10.1016/S0006-3495(94)80888-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Jurman M E, Yellen G. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 13.Baukrowitz T, Yellen G. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 14.Choi K L, Mossman C, Aubé J, Yellen G. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- 15.Jurman M E, Boland L M, Liu Y, Yellen G. BioTechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- 16.Liu Y, Dilger J P. Biophys J. 1991;60:424–432. doi: 10.1016/S0006-3495(91)82068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kellett J C, Jr, Doggett W C. J Pharm Sci. 1966;55:414–417. doi: 10.1002/jps.2600550413. [DOI] [PubMed] [Google Scholar]
- 18.Yellen G, Jurman M E, Abramson T, MacKinnon R. Science. 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]
- 19.Clay J R. J Membr Biol. 1995;147:23–34. doi: 10.1007/BF00235395. [DOI] [PubMed] [Google Scholar]
- 20.Zamponi G W, French R J. Biophys J. 1993;65:2335–2347. doi: 10.1016/S0006-3495(93)81292-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamill O P, Marty A, Nehar E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]