

Abstract
The skeletal muscle chloride channel CLC-1 and the ubiquitous volume-activated chloride channel CLC-2 belong to a large gene family whose members often show overlapping expression patterns. CLC-1 and CLC-2 are coexpressed in skeletal and smooth muscle and in the heart. By coexpressing CLC-1 and CLC-2 in Xenopus oocytes, we now show the formation of novel CLC-1/CLC-2 heterooligomers that yield time-independent linear chloride currents with a chloride → bromide → iodide selectivity sequence. Formation of heterooligomeric CLC channels increases the number and possible functions of chloride channels.
Keywords: anion channel, multimers, myotonia, cystic fibrosis, cell volume regulation
CLC chloride channels form an expanding new gene family with at least nine mammalian members known to date (for review, see refs. 1 and 2). In mammals, they play important roles in cell volume regulation (3), control of muscle excitability (4), and possibly transepithelial transport (5, 6). Two human diseases are known to be caused by defective CLC genes: mutations in the muscle channel CLC-1 can cause recessive (Becker) as well as dominant (Thomsen) myotonia (7, 8), whereas X-linked hereditary hypercalciuric nephrolithiasis is due to mutations in CLC-5 (9, 10).
CLC proteins are structurally unrelated to other known ion channels and span the membrane about 12 times (3, 6, 11, 12). First insights into their structure-function relationship have been obtained for the mechanism of swelling activation of CLC-2 (3). The gating mechanism of the Torpedo channel CLC-0 was studied in detail. It was proposed that the channel is intrinsically voltage-independent, being gated by the permeant anion (12). Finally, analysis of dominant negative mutations found in human myotonia (Thomsen disease) suggested that CLC-1 chloride channels function as homomultimers with probably more than two subunits (8, 13).
Several CLC genes are rather broadly expressed. CLC-2, CLC-6, and CLC-7 are present in every cell and tissue examined (14, 15), while CLC-3 and CLC-4 are expressed in brain, heart, kidney, and, depending on the species, several other tissues (1, 16, 17) Thus, many cells will express several members of the CLC gene family in parallel. Since CLC-1 is known to function as a (homo)multimer (8), this raises the possibility that coexpression of different CLC members in the same cell may lead to the formation of functional heterooligomeric channels with novel properties. Such a situation is, for example, found with potassium channels (18–20) or glutamate receptors (21).
We used coexpression of the easily expressible CLC-1 and CLC-2 chloride channels to test for the formation of heteromultimeric CLC channels. This is also of physiological importance, as both channel mRNAs are coexpressed in several tissues (14, 22).
CLC-1 is very predominantly expressed in skeletal muscle, but low transcript levels could also be detected in kidney, heart, and smooth muscle (22). CLC-1 mediates the majority of muscle membrane chloride conductance, which is unusually high in that tissue (≈70–80% of total resting conductance; ref. 23).
In contrast to CLC-1, CLC-2 is ubiquitously expressed. It is normally closed but can be slowly activated by strong hyperpolarization (14). Another activation mechanism is cell swelling, suggesting a role of CLC-2 in regulatory volume decrease (3). Its expression in apical membranes of airway epithelia renders it interesting for cystic fibrosis (24).
We now show that coexpression of CLC-1 and CLC-2 indeed results in the formation of novel heteromultimeric channels. This demonstrates for the first time that subunits encoded by different CLC genes can assemble to form functional heteromeric channels, and thus adds a further level of complexity to the CLC chloride channel family.
MATERIALS AND METHODS
Site-Directed Mutagenesis.
Point mutations were introduced as described (13). PCR-derived fragments were entirely sequenced. For the experiments using CLC-1 carrying the P480L mutant, we used the human CLC-1 as we have constructed this mutant previously (8). For most measurements, rat CLC-1 and CLC-2 were used.
Construction of the Oocyte Expression Vector pTLN.
We constructed a vector (pTLN) that uses the Xenopus β-globin untranslated regions to boost expression in ooyctes. Starting from pSP64T (25), we inserted a new polylinker into its unique BglII cloning site. It contains unique NruI, NcoI (providing an optimal sequence for eukaryotic initiation of translation), XbaI, KpnI, EcoRI, ClaI, EcoRV, and BglII sites. These are now flanked by the β-globin 5′ and 3′ untranslated regions downstream of a SP6 RNA polymerase promoter. To provide restriction sites for the linearization of the template, we inserted another polylinker downstream of the 3′ β-globin untranslated region. It has unique RsrII, MluI, HpaI, SnaBI, and SwaI recognition sites. All cDNAs were engineered such that their initiator methionin was covered by an NcoI site for direct ligation into pTLN.
cRNA Synthesis and Electrophysiology.
cRNA preparation, oocyte handling and injection, and two-microlelectrode voltage-clamp of oocytes were performed as described (11, 13). In ion substitution experiments, 96 mM (or 80 mM) chloride of the ND96 solution (11) were replaced by equal concentrations of bromide, glutamate, or iodide. To block CLC-1 homomers, oocytes were superfused for 4 min with 0.2 mM 9-anthracene carboxylic acid (9-AC) solution in ND96.
RESULTS
Electrophysiological Properties of CLC-1 and CLC-2 Homomultimeric Channels.
Typical current traces of CLC-1 expressed in Xenopus oocytes are shown in Fig. 1B. CLC-1 channels are open under resting conditions, and have an inwardly rectifying instantaneous current–voltage (I–V) relationship and a pronounced deactivation (within a 50-ms range) at voltages more negative than −100 mV (22). This gating process leads to a decrease of steady-state currents at very negative potentials (Fig. 2A). CLC-1 has a Cl− > Br− > I− halide selectivity sequence (Fig. 2D) and is blocked by 0.2 mM 9-AC applied in the bath (22). This block is poorly reversible. Human and rat CLC-1 do not differ in either of these properties.
Figure 1.
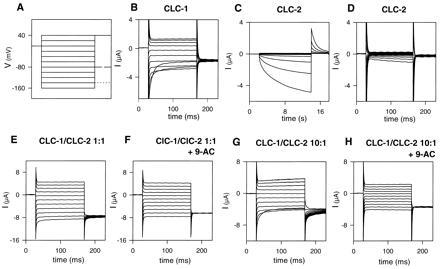
Voltage-clamp traces of oocytes injected with CLC-1 cRNA (B), CLC-2 cRNA (C and D), and coinjected with both RNAs (E and F). (A) Voltage-clamp pulse protocol. Test pulses were applied in −20 mV steps starting at +40 mV (holding potential: −30 mV). The tail pulse was varied depending on the cRNA injected. Solid line, CLC-2; dashed line, CLC-1; dotted line, coinjection. (B) Typical current traces of an oocyte expressing CLC-1, showing the characteristic inward rectification at positive voltages and the deactivation at negative voltages. (C and D) Typical currents of an oocyte expressing CLC-2. Long pulses (C) elicit a slowly activating current at negative voltages, which deactivates relatively fast when stepping to +40 mV. Short pulses (as in B, E, and F) does not significantly activate CLC-2 (D). (E and F) Typical currents of an oocyte coinjected with CLC-1 and CLC-2 cRNA at a 1:1 concentration ratio. Treatment with 0.2 mM 9-AC (F) abolishes a remaining deactivating component at negative voltages, as well as the gating seen in the tail current at a potential of −140 mV. This is more obvious when oocytes injected with an about 10-fold excess of CLC-1 over CLC-2 cRNA (G) are treated with 9-AC (H).
Figure 2.

I–V relationships and ion selectivity of currents of oocytes expressing CLC-1 (A and D), CLC-2 (B and E), or both (C and F). (Upper A–C) Steady-state I–Vs in ND96 (104 mM Cl). (Lower D–F) Instantaneous I–Vs (open pore properties) with different anions in the bath. Measurements were performed in ND96 having 104 mM Cl (○), or in ND96 where 96 mM Cl were replaced by equal amounts of Br (□), glutamate (▵), or I (▿). This leaves 8 mM Cl in the medium. Data of one representative oocyte each are shown as a plot against the test potential. (A) Steady-state I–V of CLC-1. The current was measured at the end of a 100-ms test pulse. (B) Quasi-steady-state I–V of CLC-2 (measured at the end of a 6-s test pulse). (C) Steady-state I–V of CLC-1/CLC-2 heterooligomers (measured after treatment with 9-AC to block CLC-1 homooligomers). (D) Instantaneous I–V of CLC-1. Currents were extrapolated to the beginning of the test pulse by fitting the sum of two exponential functions. (E) Open-channel I–V of CLC-2. The oocyte was held at −140 mV until the current saturated before 200-ms test pulses were applied. (F) Instantaneous (equivalent to steady-state) I–V of the heterooligomer currents measured after 9-AC treatment.
Under isotonic conditions, CLC-2 is nearly completely closed at physiological voltages (14), but can be slowly activated by strong hyperpolarization as shown in Fig. 1C. Several seconds are required for this activation (compare Fig. 1 C and D). In contrast to the resulting strong inward rectification of steady state currents (Fig. 2B), its open channel I–V is nearly linear (Fig. 2E). CLC-2 shows the same ion permeability sequence as CLC-1. In contrast to CLC-1, however, it is not blocked by bromide (compare Fig. 2 D and E). Also, application of 0.2 mM 9-AC does not affect CLC-2 currents (data not shown). Swelling of the oocyte by prolonged (>10 min) exposure to hypotonic medium results in a dramatic change of CLC-2 properties (3). Currents become time-independent with a linear I–V, which resembles the open channel I–V of CLC-2 under isotonic conditions.
Coinjection of Wild-Type CLC-1 and CLC-2.
Coexpression of CLC-1 and CLC-2 cRNA at about equal cRNA concentrations yielded large, essentially time- and voltage-independent currents (Fig. 1E). This suggests the formation of heteromultimeric channels with novel properties, because the currents cannot be simply explained by a sum of CLC-1 and CLC-2 currents. This is evident at positive voltages where both CLC-1 and CLC-2 steady-state currents are inwardly rectifying (Fig. 2 A and B), whereas the novel current is linear (Fig. 2C). Also with hyperpolarization, the slow activation of CLC-2 cannot compensate for the fast inactivation of CLC-1 to produce the observed linear, essentially time-independent currents. The small, fast deactivating current component at the beginning of hyperpolarizing pulses (Fig. 1E) may be in part due to homomultimeric CLC-1 channels that are also expressed from the CLC-1/CLC-2 cRNA mixture. This component increased with the the ratio of CLC-1 to CLC-2 cRNA (Fig. 1G) and could be abolished by 0.2 mM 9-AC (Fig. 1 F and H), which completely blocks CLC-1 homooligomers (22). 9-AC also inhibits the gating relaxations seen at −140 mV after stepping to this potential from the various test pulses. This gating is probably also (in part) due to a residual CLC-1 component. At this concentration of 9-AC only the linear, time-independent currents elicited by the coinjection remain (Fig. 1 F and H). The open channel properties of the heterooligomer (Fig. 2F) are similar to that of CLC-2, including the linear I–V relationship, the lack of voltage-dependent block by bromide, and the insensitivity (of the time-independent component) to 9-AC. These properties were independent of the concentration ratios of coinjected cRNAs.
To determine the optimal ratio of CLC-1 to CLC-2 cRNA concentrations for the formation of heteromultimeric channels, we systematically varied the ratios of the corresponding cRNAs in coinjection experiments. In these titrations, total RNA concentration was held constant to avoid problems with the nonlinearity of expression in Xenopus oocytes (Fig. 3). We measured the slope conductance at +50 mV. As described above, at this voltage CLC-2 homooligomers are closed and CLC-1 has only a small slope conductance. Hence these measurements should mainly detect the linear heterooligomeric channels. Coinjection of both cRNAs led to a pronounced increase in slope conductance with a maximum at a 1:1 cRNA ratio (Fig. 3A). When CLC-1 homooligomers were blocked by 0.2 mM 9-AC, the effect became even more dramatic (Fig. 3B). These conditions eliminate currents by homooligomers, and only coinjections of CLC-1 and CLC-2 cRNAs yield chloride currents. The current maximum at a 1:1 cRNA concentration ratio is compatible with functional heterooligomers formed by an equal number of CLC-1 and CLC-2 subunits.
Figure 3.
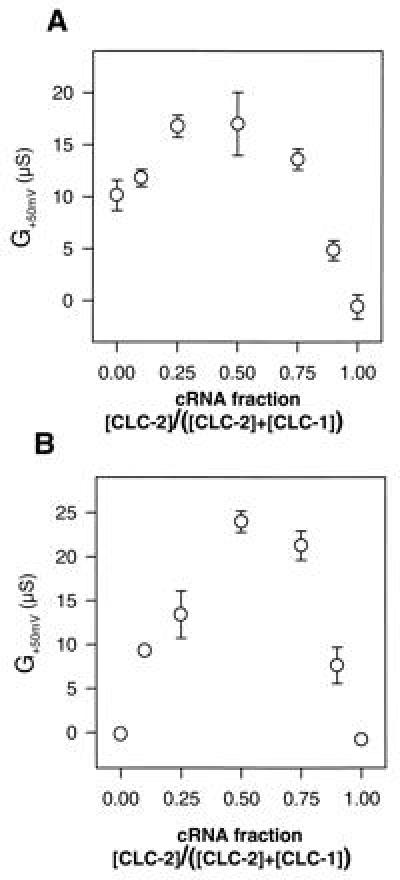
Generation of CLC-1/CLC-2 heteromeric conductance as a function of cRNA concentration ratios. (A) Slope conductance at 50 mV measured in ND96 is plotted vs. the fraction of CLC-2 cRNA in the CLC-1/CLC-2 coinjection. Slope conductance of uninjected oocytes was subtracted. Conductance at +50 mV is predominantly due to heterooligomers. (B) Same as A (different oocytes from the same batch), but measured after incubation in 0.2 mM 9-AC that blocks CLC-1 homooligomers. Error bars = SEM, n = 4–5.
Since CLC-2 is a swelling-activated chloride channel, we sought to determine whether the CLC-1/CLC-2 heteromeric channels also respond to medium osmolarity. Since we know of no blocker specific for CLC-2 (which does not block CLC-1 or the heteromers), we had to use a large excess of CLC-1 to avoid a significant background in swelling-activated CLC-2 homomers. The excess of CLC-1 homomers can again be blocked by 9-AC. Under these conditions, we could not detect a significant activation of CLC-1/CLC-2 heterooligomers by cell swelling. We neither could reduce the currents by hyperosmolarity (data not shown).
Coinjections with Mutant Subunits.
Additional evidence for the formation of heteromeric channels was obtained by coexpression of CLC-2 with a CLC-1 mutant (P480L) found in autosomal dominant myotonia congenita (8). Similar to its dominant action on CLC-1 (8, 13), CLC-2 is strongly suppressed in a 1:1 coexpression (Fig. 4). The same total amount of cRNA was injected to avoid a nonspecific decrease of CLC-2 currents caused by saturation of the oocyte expression system. Thus, for the 1:1 coexpression experiment shown in Fig. 4, the CLC-2 conductance is expected to decrease by ≈50% if there is no interaction between both channels. However, the observed decrease is much stronger (84%, Fig. 4B), demonstrating a dominant negative effect of this CLC-1 mutant on CLC-2. Interestingly, the homologous mutation in CLC-2 (P461L), which by itself was nonfunctional, had no effect neither on CLC-2 nor on CLC-1 in coinjection experiments (data not shown). In CLC-1 the P480L mutation reduces CLC-1 currents in the physiological voltage-range by drastically shifting the voltage-dependence of activation to more positive voltages (13). In contrast, in the coexpression experiments with CLC-2 no qualitative change of the remaining CLC-2 currents was visible (Fig. 4A).
Figure 4.
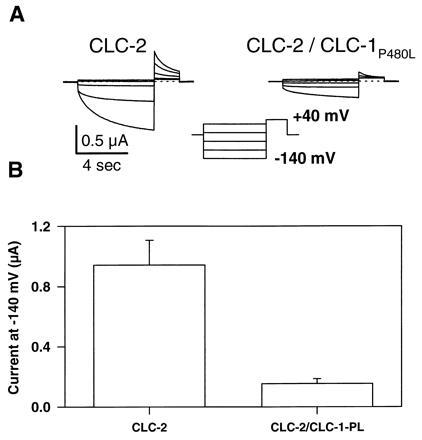
Dominant negative effect of the CLC-1 mutation P480L on wild-type CLC-2. (A) Current traces of an oocyte expressing CLC-2 (Left) or coexpressing CLC-2 and CLC-1P480L (Right). (B) Absolute current at the end of the test pulse to −140 mV (error bar = SEM, n = 9). Background current of noninjected oocytes was subtracted. RNA was coinjected at a 1:1 ratio, with a constant total amount of RNA.
DISCUSSION
Many ion channels function as multimeric complexes. They may be composed of identical subunits (homomultimers) or of subunits belonging to the same gene family, and/or may associate with structurally unrelated subunits.
Channels assumed to be functional as monomers are the cystic fibrosis transmembrane conductance regulator (the cAMP-activated chloride channel defective in cystic fibrosis; ref. 26) and α-subunits of voltage-dependent Na+- and Ca2+-channels. However, these α-subunits have probably arisen from two gene duplications, and can be regarded as tetramers with covalently linked subunits analogous to the tetrameric K+-channel family (27, 28). With voltage-dependent and also inwardly rectifying K+-channels, the formation of homooligomers and of heterooligomers is possible (18, 19, 29). Epithelial Na+-channels require three different homologous subunits (assembled with unknown stoichiometry) for efficient function (30).
In the present study, we demonstrate that CLC-1 and CLC-2 together indeed form novel channels with a linear, almost time-independent I–V relationship. Pore properties of the heteromultimer resemble those of CLC-2.
The I–V relationship of CLC-1/CLC-2 heteromers resembles that of “open phenotype” mutants of CLC-2 (3). Activation of CLC-2 by cell swelling or by hyperpolarization seemed to operate via a “ball-and-chain” type mechanism, in which an N-terminal domain (the “ball”) binds to a receptor on the channel and thereby inactivates it (3). Deletions in the “ball” destroy its swelling-activation and lead to constitutively open channels with a linear I–V relationship. Its pharmacology (insensitivity to 9-AC) and pore properties (lack of block by bromide) resemble the heterooligomeric channel, too. This raises the question whether the pore that is seen in heterooligomeric channels actually is a CLC-2 pore that has lost its control by the “ball-and-chain” mechanism. Such a model is plausible because the homologous channel CLC-0 is thought to be a “double-barreled” channel (31, 32). This model assumes two identical pores, which can open and close independently from each other, but are dependent on a common slow gate. From our experiments we cannot conclude how many subunits would form one pore. Recent experiments with CLC-0 mutants, however, favor the idea that one pore is formed by a single subunit (43, 44).
The alternative model in which both CLC-1 and CLC-2 subunits directly line a common pore, either in a single-barreled or double-barreled channel, cannot be strictly ruled out, however.
Single-channel studies are desirable to shed more light on the properties of the heteromeric channels. Unfortunately, the single-channel conductances of CLC-1 (1pS; ref. 33) and CLC-2 (2–3pS; M.P. and T.J.J., unpublished work) are too small to be easily directly visualized. Preliminary noise analysis indicates that also heteromeric CLC-1/CLC-2 channels have a conductance not larger than 2 pS (M.P. and T.J.J., unpublished work). Thus, it will be very difficult to directly demonstrate a possible double-barreled structure of CLC-1/CLC-2 heteromers.
In skeletal muscle, CLC-1 homomeric channels represent the dominant chloride conductance (4, 22, 34) and CLC-1/CLC-2 heterooligomers probably do not play a significant physiological role under normal conditions. However, these multimers could be of medical importance with some CLC-1 mutations causing myotonia. In contrast to homomeric CLC-2, the heteromer will contribute to the resting conductance, performing a role similar to CLC-1. Some CLC-1 mutations [e.g., G230E (35) or R894X (36)] have a weak dominant negative effect (8, 36). In patients heterozygous for these mutations, the chloride conductance is at the limit where it begins to cause myotonia, leading to dominant inheritance in some, and recessive inheritance in other families. In such borderline situations, variations in the CLC-2 gene could modify the phenotype (e.g., the penetrance).
CLC-1 expression is quite tissue specific, so CLC-2 homomeric currents may be observed in other tissues like the salivary gland (37, 38), neuronal cells (39) or osteoclasts (40). The heteromultimeric channels described here may contribute significantly to resting chloride conductance in organs with a minor, but detectable CLC-1 expression like the heart or smooth muscle. In these tissues, chloride conductance is important for the modulation of electrical excitability (41).
The dominant-negative action of the CLC-1 mutation P480L on CLC-2 constitutes additional evidence for the formation of heteromeric channels. In contrast to the effect of P480L on wild-type CLC-1, where the mutation drastically shifts the open probability to more positive voltages (13), we have no clue to the mechanism of the reduction of CLC-2, as the remaining CLC-2 currents are qualitatively identical to wild-type CLC-2. Nevertheless, this dominant negative effect could be used in transgenic animals to down-regulate CLC-2 channels. Compared with a CLC-2 “knock-out,” this approach would have the advantage that tissue-specific or inducible promoters could be used.
It is an important question whether other members of the CLC gene family will also form heterooligomeric channels in various combinations. This would add another level of complexity to the possible number of different chloride channels. In preliminary experiments, we could show (unphysiological) heteromeric channels formed by CLC-0 and CLC-1 (unpublished results). So far, we could not demonstrate functional interactions between some of the newer putative channel proteins (CLC-3, CLC-4, and CLC-5) and other members of the family, including the easily expressible ones (1, 15, 42). Analysis of the formation of functional heteromers with different members of this expanding gene family is an important and challenging task for future research.
Acknowledgments
This work was supported, in part, by the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, and the U.S. Muscular Dystrophy Association.
Footnotes
Abbreviations: 9-AC, 9-anthracene carboxylic acid; I–V, current–voltage.
References
- 1.Jentsch T J, Günther W, Pusch M, Schwappach B. J Physiol (London) 1995;482.P:19S–25S. doi: 10.1113/jphysiol.1995.sp020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jentsch T J. Curr Opin Neurobiol. 1996;6:303–310. doi: 10.1016/s0959-4388(96)80112-7. [DOI] [PubMed] [Google Scholar]
- 3.Gründer S, Thiemann A, Pusch M, Jentsch T J. Nature (London) 1992;360:759–762. doi: 10.1038/360759a0. [DOI] [PubMed] [Google Scholar]
- 4.Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch T J. Nature (London) 1991;354:304–308. doi: 10.1038/354304a0. [DOI] [PubMed] [Google Scholar]
- 5.Uchida S, Sasaki S, Furukawa T, Hiraoka M, Imai T, Hirata Y, Marumo F. J Biol Chem. 1993;268:3821–3824. [PubMed] [Google Scholar]
- 6.Kieferle S, Fong P, Bens M, Vandewalle A, Jentsch T J. Proc Natl Acad Sci USA. 1994;91:6943–6947. doi: 10.1073/pnas.91.15.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch M C, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann-Horn F, Grzeschik K-H, Jentsch T J. Science. 1992;257:797–800. doi: 10.1126/science.1379744. [DOI] [PubMed] [Google Scholar]
- 8.Steinmeyer K, Lorenz C, Pusch M, Koch M C, Jentsch T J. EMBO J. 1994;13:737–743. doi: 10.1002/j.1460-2075.1994.tb06315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fisher S E, Black G C M, Lloyd S E M, Hatchwell E, Wrong O, Thakker R V, Craig I W. Hum Mol Genet. 1994;3:2053–2059. [PubMed] [Google Scholar]
- 10.Lloyd S E, Pearce S H S, Fisher S E, Steinmeyer K, Schwappach B, Scheinmann S J, Harding B, Bolino A, Devoto M, Goodyer P, Rigden S P A, Wrong O, Jentsch T J, Craig I W, Thakker R V. Nature (London) 1996;379:445–449. doi: 10.1038/379445a0. [DOI] [PubMed] [Google Scholar]
- 11.Jentsch T J, Steinmeyer K, Schwarz G. Nature (London) 1990;348:510–514. doi: 10.1038/348510a0. [DOI] [PubMed] [Google Scholar]
- 12.Pusch M, Ludewig U, Rehfeldt A, Jentsch T J. Nature (London) 1995;373:527–531. doi: 10.1038/373527a0. [DOI] [PubMed] [Google Scholar]
- 13.Pusch M, Steinmeyer K, Koch M C, Jentsch T J. Neuron. 1995;15:1455–1463. doi: 10.1016/0896-6273(95)90023-3. [DOI] [PubMed] [Google Scholar]
- 14.Thiemann A, Gründer S, Pusch M, Jentsch T J. Nature (London) 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- 15.Brandt S, Jentsch T J. FEBS Lett. 1995;377:15–20. doi: 10.1016/0014-5793(95)01298-2. [DOI] [PubMed] [Google Scholar]
- 16.Kawasaki M, Uchida S, Monkawa T, Miyawaki A, Mikoshiba K, Marumo F, Sasaki S. Neuron. 1994;12:597–604. doi: 10.1016/0896-6273(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 17.van Slegtenhorst M A, Bassi M T, Borsani G, Wapenaar M C, Ferrero G B, de Conciliis L, Rugarli E I, Grillo A, Franco B, Zoghbi H Y, Ballabio A. Hum Mol Genet. 1994;3:547–552. doi: 10.1093/hmg/3.4.547. [DOI] [PubMed] [Google Scholar]
- 18.Isacoff E Y, Jan Y N, Jan L Y. Nature (London) 1990;345:530–534. doi: 10.1038/345530a0. [DOI] [PubMed] [Google Scholar]
- 19.Ruppersberg J P, Schröter K H, Sakmann B, Stocker M, Sewing S, Pongs O. Nature (London) 1990;345:535–537. doi: 10.1038/345535a0. [DOI] [PubMed] [Google Scholar]
- 20.Christie M J, North R A, Osborne P B, Douglass J, Adelman J P. Neuron. 1990;2:405–411. doi: 10.1016/0896-6273(90)90052-h. [DOI] [PubMed] [Google Scholar]
- 21.Nakanishi N, Shneider N A, Axel R. Neuron. 1990;5:569–581. doi: 10.1016/0896-6273(90)90212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinmeyer K, Ortland C, Jentsch T J. Nature (London) 1991;354:301–304. doi: 10.1038/354301a0. [DOI] [PubMed] [Google Scholar]
- 23.Bretag A H. Physiol Rev. 1987;67:618–724. doi: 10.1152/physrev.1987.67.2.618. [DOI] [PubMed] [Google Scholar]
- 24.Murray C B, Morales M M, Flotte T R, McGrathmorrow S A, Guggino W B, Zeitlin P L. Am J Respir Cell Mol Biol. 1995;12:597–604. doi: 10.1165/ajrcmb.12.6.7766424. [DOI] [PubMed] [Google Scholar]
- 25.Krieg P A, Melton D A. Nucleic Acids Res. 1984;12:7057–7069. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall J, Fang S, Ostedgaard L S, O’Riordan C R, Ferrara D, Amara J F, Hoppe H, IV, Scheule R K, Welsh M J, Smith A E, Cheng S H. J Biol Chem. 1994;269:2987–2995. [PubMed] [Google Scholar]
- 27.MacKinnon R. Nature (London) 1991;350:232–235. doi: 10.1038/350232a0. [DOI] [PubMed] [Google Scholar]
- 28.Li M, Unwin N, Stauffer K A, Jan Y-N, Jan L Y. Curr Biol. 1994;4:110–115. doi: 10.1016/s0960-9822(94)00026-6. [DOI] [PubMed] [Google Scholar]
- 29.Krapivinsky G, Gordon E A, Wickman K, Velimirovic B, Krapivinsky L, Clapham D E. Nature (London) 1995;374:135–141. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 30.Canessa C M, Schild L, Buell G, Thorens B, Gautschi I, Horisberger J-D, Rossier B C. Nature (London) 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 31.Miller C. Philos Trans R Soc London B. 1982;299:401–411. doi: 10.1098/rstb.1982.0140. [DOI] [PubMed] [Google Scholar]
- 32.Bauer C K, Steinmeyer K, Schwarz J R, Jentsch T J. Proc Natl Acad Sci USA. 1991;88:11052–11056. doi: 10.1073/pnas.88.24.11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pusch M, Steinmeyer K, Jentsch T J. Biophys J. 1994;66:149–152. doi: 10.1016/S0006-3495(94)80753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palade P T, Barchi R L. J Gen Physiol. 1977;69:325–342. doi: 10.1085/jgp.69.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.George A L, Jr, Crackower M A, Abdalla J A, Hudson A J, Ebers G C. Nat Genet. 1993;3:305–310. doi: 10.1038/ng0493-305. [DOI] [PubMed] [Google Scholar]
- 36.Meyer-Kleine C, Steinmeyer K, Ricker K, Jentsch T J, Koch M C. Am J Hum Genet. 1995;57:1325–1334. [PMC free article] [PubMed] [Google Scholar]
- 37.Dinudom A, Young J A, Cook D I. J Membr Biol. 1993;135:289–295. doi: 10.1007/BF00211100. [DOI] [PubMed] [Google Scholar]
- 38.Komwatana P, Dinudom A, Young J A, Cook D I. Pflügers Arch. 1994;428:641–647. doi: 10.1007/BF00374588. [DOI] [PubMed] [Google Scholar]
- 39.Smith R L, Clayton G H, Wilcox C L, Escudero K W, Staley K J. J Neurosci. 1995;15:4057–4067. doi: 10.1523/JNEUROSCI.15-05-04057.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chesnoy-Marchais D, Fritsch J. J Membr Biol. 1994;140:173–188. doi: 10.1007/BF00233706. [DOI] [PubMed] [Google Scholar]
- 41.Ackermann M J, Clapham D E. Trends Cardiovasc Med. 1993;3:23–28. doi: 10.1016/1050-1738(93)90024-Z. [DOI] [PubMed] [Google Scholar]
- 42.Steinmeyer K, Schwappach B, Bens M, Vandewalle A, Jentsch T J. J Biol Chem. 1995;270:31172–31177. doi: 10.1074/jbc.270.52.31172. [DOI] [PubMed] [Google Scholar]
- 43.Ludewig U, Pusch M, Jentsch T J. Nature (London) 1996;383:340–343. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]
- 44.Middleton R E, Pheasant D J, Miller C. Nature (London) 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]