

Abstract
Eukaryote cells balance production of reactive oxygen species (ROS) with levels of anti-oxidant enzyme activity to maintain cellular redox homeostasis. Mitochondria are a major source of ROS, while many anti-oxidant genes are regulated by the Nrf2 transcription factor. Keap1, a redox-regulated substrate adaptor for a cullin-based ubiquitin ligase, targets Nrf2 for proteosome-mediated degradation and represses Nrf2-dependent gene expression. We have previously identified a member of the phosphoglycerate mutase family, PGAM5, as a Keap1-binding protein. In this report, we demonstrate that PGAM5 is targeted to the outer membrane of mitochondria by an N-terminal mitochondrial-localization sequence. Furthermore, we provide evidence that PGAM5 forms a ternary complex containing both Keap1 and Nrf2, in which the dimeric Keap1 protein simultaneously binds both PGAM5 and Nrf2 through their conserved E(S/T)GE motifs. Knockdown of either Keap1 or PGAM5 activates Nrf2-dependent gene expression. We suggest that this ternary complex provides a molecular framework for understanding how nuclear anti-oxidant gene expression is regulated in response to changes in mitochondrial function(s).
Keywords: Mitochondrial proteins, Anti-oxidant gene expression, Oxidative stress, ubiquitin ligases, chemoprevention
Introduction
Oxidative stress has been implicated in diverse pathophysiological processes, including cancer, neurodegenerative diseases, and aging [1–4]. Oxidative stress is often viewed as an imbalance between levels of reactive oxygen species (ROS) and the ability of cells to counteract ROS-mediated oxidative damage to biological macromolecules [5]. ROS are also essential molecular components of signal transduction pathways that control cell fate decisions and perturbation of these pathways can contribute to the pathological consequences of oxidative stress [6]. Mitochondria are a major source of endogenous ROS production and perturbation of mitochondrial function has been linked to increased ROS production and, ultimately, adverse pathophysiological consequences [1–9].
In mammals, the bZIP transcription factor Nrf2 is a major regulator of cytoprotective genes which encode enzymes that neutralize reactive molecules, eliminate damaged macromolecules and restore cellular redox homeostasis [10]. Nrf2-dependent gene expression is increased by diverse stimuli, including increased mitochondrial ROS production arising from pharmacological inhibition of electron transport complex I or II [10–13]. Conversely, Nrf2-dependent gene expression is markedly decreased in cells that lack the DJ1 protein, a mitochondrial protein that has been linked to familial Parkinson’s [14]. Nrf2 nullizygous mice develop normally but several indices of oxidative damage are increased in aged female mice, coincident with the development of symptoms that resemble systemic lupus erythromatosus in humans [15–17]. Extensive analysis of Nrf2 nullizygous mice has demonstrated that Nrf2-dependent genes provide protection from ROS generated by diverse cellular stresses [18].
A BTB-Kelch protein termed Keap1 is the major negative regulator of Nrf2 [19–21]. Mice that lack Keap1 contain elevated levels of nuclear Nrf2 protein and increased levels of Nrf2-dependent gene expression [19]. Keap1 is a substrate adaptor for an E3 ubiquitin ligase complex that assembles around a Cul3 scaffold protein [22–25]. The N-terminal BTB and central linker domains of Keap1 bind to Cul3 while the C-terminal Kelch domain of Keap1 binds to Nrf2 [24–27]. This E3 ubiquitin ligase complex directs the covalent attachment of ubiquitin onto specific lysine residues within the N-terminal Neh2 domain of Nrf2 [25]. Cyclical assembly and disassembly of this E3 ubiquitin ligase complex is required for efficient ubiquitination of Nrf2 and repression of Nrf2-dependent gene expression [28]. Electrophilic chemicals modify highly reactive cysteine residues within Keap1 and perturb assembly of a functional ubiquitin ligase complex [25, 29–35], resulting in decreased ubiquitination of Nrf2, increased steady-state levels of Nrf2 and increased transcription of Nrf2-dependent genes. Other mechanisms, including phosphorylation of Nrf2 [36], may also contribute to increased steady-state levels of Nrf2 in cells exposed to electrophilic and reactive chemicals.
Keap1 is a cytoplasmic protein that has been reported to associate with actin filaments [20, 23, 37]. Keap1 has also been localized to specialized adherens junctions and focal adhesions [38]. Several reports have suggested that both Keap1 and Nrf2 shuttle between the nucleus and the cytoplasm [39–42]. Different subcellular compartments have different redox potentials and different oxidant-generating complexes are confined to discrete subcellular locations [43, 44]. As Keap1 is a direct target for ROS and other electrophilic molecules, the molecular and spatial relationship(s) between Keap1 and the subcellular locations where ROS generation occurs is a critical unanswered question.
The human phosphoglycerate mutase family contains at least ten distinct proteins that share a structurally conserved PGAM domain [45]. Most members of this family, including PGAM1, PGAM2, 2,3-bisphosphoglycerate mutase (BPGM), and the four members of the 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB) family, are metabolic enzymes that mediate phosphotransfer via a histidine residue located within a highly conserved motif in the PGAM domain [45]. Several other members of this family, including STS-1, STS-2 and PGAM5, contain a divergent catalytic motif [46]. Neither STS-1 nor STS-2 have phosphoglycerate mutase activity, although STS-1 has phosphotransferase activity towards other substrates [47–50]. PGAM5 was first identified as a Bcl-XL-interacting protein and subsequently identified as a protein that bound to Keap1 [46, 51]. The PGAM5 gene, located on chromosome 12 in humans, encodes two protein isoforms, PGAM5-L (32 kDa) and PGAM5-S (28 kDa), generated by alternative splicing of a 3′ exon. Both isoforms of PGAM5 contain a conserved N-terminal NxESGE motif that is required for binding to Keap1. This motif bears striking resemblance to the DxETGE motif in Nrf2 that binds to a shallow pocket located on the top face of Kelch domain of Keap1 [26, 27]. Indeed, amino acids in Keap1 that make critical contacts with the DxETGE motif from Nrf2 are also required for binding PGAM5 [46].
In this report, we demonstrate that PGAM5 contains an N-terminal mitochondrial targeting sequence that is necessary and sufficient for localization of PGAM5 to the outer membrane of mitochondria. Co-immunoprecipitation and co-localization experiments revealed the presence of a ternary complex containing Nrf2, Keap1 and PGAM5 that is localized to mitochondria. This ternary complex of PGAM5, Keap1 and Nrf2 contributes to repression of Nrf2-dependent gene expression, as siRNA-mediated knockdown of PGAM5 increased expression of an Nrf2-dependent reporter gene. PGAM5, by anchoring the Keap1-Nrf2 complex to outer membrane of mitochondria, may facilitate coordination between mitochondrial function and regulation of Nrf2-dependent anti-oxidant gene expression.
Materials and Methods
Construction of recombinant DNA molecules
The PGAM5-L cDNA (Accession number BG030775), the PGAM5-S cDNA (Accession number BC008196) and mutant PGAM5-L-E79A/S80A cDNA have been described previously [46]. The deletion mutants in PGAM5-L and PGAM5-S lacking codons 2 to 25 were constructed in pcDNA3 and in pCMV-Tag4A using standard recombinant DNA techniques. The expression vectors for GFP fusion proteins containing N-terminal PGAM5-derived sequences were constructed by cloning PCR-generated fragments into the XhoI/BamHI sites of pEGFP-N1. Plasmid expression vectors for both wt and mutant Keap1 and Nrf2 have been previously described [26]. The Mito-GFP expression vector was a gift from Dr. Seamus J. Martin. The pEGFP-N1 empty vector was a gift from Dr. Marc C. Johnson. All the genes used in this study were sequenced in the context of the expression vectors used for the experiments.
Cell culture, transfections, and reporter gene assays
COS1, HeLa, MDA-MB-231, HEK-293T cells were purchased from ATCC. Cells were maintained in either Dulbecco’s modified Eagle’s medium (DMEM) or Eagle’s minimal essential medium (EMEM) in the presence of 10% fetal bovine serum (FBS). Transfections of plasmid DNA were performed with Lipofectamine 2000 or Lipofectamine Plus (Invitrogen) according to the manufacturer’s instruction. Transfections of small interfering RNA (siRNA) nucleotides were performed with Oligofectamine (Invitrogen) according to the manufacturer’s instructions. All siRNA nucleotides used in this study were obtained from Dharmacon as purified and annealed duplexes. The antioxidant response element (ARE) TATA-Inr luciferase reporter plasmid pARE-Luc and a control plasmid encoding Renilla luciferase for transfection efficiency have been previously described [28]. Reporter gene assays were performed using a Promega dual-light assay system.
Antibodies, immunoprecipitation and immunoblot analysis
The PGAM domain (codons 89 to 289) of PGAM5-L protein was expressed in Escherichia coli as a His6-tagged protein and purified by metal–chelate chromatography. Care was taken to maintain reducing conditions during all stages of the purification and subsequent use of the purified PGAM domain of PGAM5 protein. Antibodies were generated in chickens against the PGAM domain by Alpha Diagnostics (San Antonio, TX) and affinity-purified over a column containing immobilized His-PGAM5 (89–289) protein with reagents obtained from Pierce. Specificity of the anti-PGAM5 (89–289) antibodies was confirmed by immunoblotting, immunoprecipitation, and immunocytochemistry techniques. The affinity-purified anti-PGAM5 antibodies were able to bind both native and denatured PGAM5 isoforms. The affinity-purified anti-Keap1 antibody has been described previously [34]. Antibodies against Cytochrome c (6H2.B4, BD Pharmingen), COXIV (10G8, Molecular Probes), TOM20 (FL-145, Santa Cruz), Tubulin (TU-02, Santa Cruz), the chitin binding domain (New England Biolabs), the FLAG epitope (Sigma), the HA epitope (Covance), and Nrf2 (H-300, Santa Cruz) were purchased from commercial sources.
For detection of protein expression in total cell lysates, cells were lysed in sample buffer (50 mM Tris-HCl [pH 6.8], 2% SDS, 10% Glycerol, 100 mM DTT, 0.1% bromophenol blue) at 24 to 48 hr post-transfection. For immunoprecipitation assays, cell extracts were prepared in RIPA buffer (10 mM sodium phosphate [pH 8.0], 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.2% SDS) containing 1mM dithiothreitol (DTT), 1mM phenylmethylsulfonyl fluoride (PMSF) and protease inhibitor cocktail (Sigma). Soluble cell lysates were incubated with either antibodies immobilized to agarose beads or with affinity-purified antibodies for 2 hr at 4°C, followed by incubation at 4°C with protein A-agarose beads (Sigma) or with anti-chicken IgY-agarose (gallus Immunotech Inc.) for two hours. Unbound proteins were removed by washing four times with lysis buffer. The immunoprecipitated proteins were eluted in sample buffer by boiling for 5 minutes, electrophoresed through SDS-polyacrylamide gels, transferred to nitrocellulose membranes and subjected to immunoblot analysis.
Immunofluorescence assays
Cells were grown on glass coverslips on 35-mm-diameter plates. Cells were either non-tranfected or transfected with 1.0 μg of the empty vector, or the indicated expression vectors. Cells were fixed with 3.7% formaldehyde in culture media at 37ºC for 15 min and incubated with 0.1 M glycine in phosphate buffered saline (10 mM sodium phosphate [pH 8.0] and 150 mM NaCl) at room temperature for 5 min to quench autofluorescence. Cells were permeabilized in 0.2% Triton X-100 in phosphate buffered saline at room temperature for 10 min. Fixed cells were incubated at room temperature for 60 min with affinity-purified primary antibodies as indicated in phosphate buffered saline containing 10% (vol/vol) FBS. Coverslips were washed and incubated with anti-chicken, anti-mouse, or anti-rabbit antibodies conjugated with either Alexa Fluor 488 or Alexa Fluor 568 (Molecular Probes), and with Hoechst 33258 for another 60 min. GFP expression was visualized by direct fluorescence. Coverslips were washed and mounted in ProLong Gold (Molecular Probes) on glass slides. Images were obtained with a Zeiss Meta NLO 2-photon laser scanning confocal microscopy system at the MU Cytology Core. The images were captured with LSM 510 META, and transferred to Adobe Photoshop for construction of the figure.
In vivo ubiquitination assays
For detection of ubiquitinated PGAM5 proteins in vivo, cells were lysed by boiling and sonication in a buffer containing 2% SDS, 10 mM N-ethylmaleimide (NEM), 150 mM NaCl and 10 mM Tris-HCl pH [8.0]. This rapid lysis procedure inactivates cellular ubiquitin hydrolases and preserves ubiquitin-substrate protein conjugates present in cells prior to lysis. Protein-protein interactions, including association of Keap1 with PGAM5-FLAG, are also disrupted by this lysis procedure. Lysates were diluted with 4 volumes of a buffer containing 150 mM NaCl, 10 mM Tris-HCl pH [8.0], 1 mM DTT and 1% Triton X-100 prior to immunoprecipitation. The diluted lysates were pre-cleared with protein A-agarose beads (Sigma) and incubated with anti-FLAG M2 agarose (Sigma). HA-ubiquitin conjugates in immunoprecipitated PGAM5-L-FLAG proteins were analyzed by immunoblot analysis with antibodies against the HA epitope.
Subcellular fractionation and proteinase K protection assays
A mitochondrial-enriched subcellular fraction was obtained using a digitonin-permabilization protocol essentially as described [52, 53]. A range of digitonin concentrations (20 to 200 μg/ml) was first tested to identify the optimal digitonin concentration (20 μg/ml) that minimized loss of both PGAM5 and Bcl-XL into the cytosolic fraction. HeLa cells were permeabilized in 250 mM sucrose, 70 mM KCl, 137 mM NaCl, 4.3 mM Na2HPO4 [ph 7.2], 1.4 mM KH2PO4 ([ph 7.2) containing 20 μg/ml digitonin. Cell lysates were centrifuged at 1,000 × g for five minutes at 4°C to obtain cytosolic (supernatant) and a mitochondria-enriched heavy membrane (pellet) fraction. The pellet was resuspended in permeabilization buffer and incubated with proteinase K for 30 minutes on ice. The reactions were boiled in SDS-PAGE sample buffer and cell equivalents of the cytosol and heavy membrane fractions were analyzed by SDS-PAGE and immunoblotting. In some experiments, the heavy membrane was resuspended in permeabilization buffer containing 0.2% Triton X-100 and 0.3% NP-40 followed by centrifugation at 10,000 × g to obtain heavy membrane (soluble) and pellet (insoluble) fractions.
Results
PGAM5 is a nuclear-encoded mitochondria-resident protein
The PGAM5 gene encodes two distinct protein isoforms that differ at their C-termini due to alternative splicing of a 3′ exon (Fig. 1A). A polyclonal antibody generated against amino acids 89–289 of the PGAM5-L protein was used to characterize expression and subcellular localization of the endogenous PGAM5 proteins. As expected, the affinity-purified antibody recognized PGAM5-L and PGAM5-S proteins (Fig. 1B, lanes 3–6). In cell lysates from several different cultured cell lines, the affinity-purified antibody recognized an endogenous protein of 32kDa that co-migrated with the ectopic PGAM5-L isoform (Fig. 1B, lanes 1 and 2, data not shown). This 32 kDa endogenous protein was not recognized by pre-immune antisera (data not shown). The subcellular localization of the endogenous PGAM5-L protein in COS1 cells was determined by indirect immunofluorescence using the affinity-purified anti-PGAM5 antibody. No staining was observed with the pre-immune antisera, while a reticular-tubular pattern of cytoplasmic staining was observed with the anti-PGAM5 antibody (Fig. 1C, compare panels b and e). Pre-incubation of the purified anti-PGAM5 antibody with PGAM5 protein abolished the reticular-tubular staining pattern, confirming the specificity of the antibody (Supplemental Figure 1). Confocal microscopy analysis revealed that PGAM5-L co-localized with several different mitochondrial resident proteins, including cytochrome c (Fig. 1C, panels d–f; data not shown). No colocalization was observed with the endoplasmic reticulum marker, protein disulphide isomerase (data not shown). Colocalization of PGAM5-L with cytochrome c was observed in HeLa, HEK293T, and MDA-MB-231 cells (Fig. 1C, panels g–l, and data not shown).
Fig.1.

An N-terminal sequence targets PGAM5 to mitochondria. (A) The two PGAM5 isoforms are depicted. Relevant landmarks include a putative transmembrane helix (amino acids 9–29), a conserved NxESGE motif that binds Keap1 (amino acids 77–82) and the PGAM domain (beginning at amino acid 98). The two isoforms have different C-terminal sequences beginning at amino acid 239. The amino acid sequence of the N-terminus of PGAM5 is shown, along with the amino acids used to make several GFP fusion proteins and the region deleted in the PGAM5-Δ2–25 protein. (B) Hela cells were transfected with an empty vector (lanes 1 and 2), or expression vectors for either of the untagged PGAM5 isoforms as indicated (lanes 3–6). Total lysates were analyzed by immunoblot with affinity-purified anti-PGAM5 antibodies. The relative positions of the PGAM5-L and PGAM5-S proteins are indicated by the arrows on the right side. Two different exposures of the same immunoblot are shown. (C) The cellular localization of the endogenous PGAM5 protein in COS1 (a–f), HeLa (g–i), or HEK-293T (j–l) was determined by indirect immunofluorescence using pre-immune (panel b) or affinity-purified anti-PGAM5 antibodies (panels e, h and k). Mitochondria were visualized with anti-cytochrome c antibodies (panels a, d, g and j). Nuclei were visualized by staining with Hoechst 33258. Merged images are shown on the right (panels c, f, i and l). (D) COS1 cells growing on coverslips were transfected with an expression vector for Mito-GFP along with either an empty vector (bottom panels) or an expression vector for PGAM5-L-Δ2–25 (top panels). The cellular localization of the PGAM5-L-Δ2–25 mutant protein was determined by indirect immunofluorescence with affinity-purified anti-PGAM5 antibodies (panels a and d). Mitochondria were visualized by Mito-GFP fluorescence (panels b and e). The laser intensity of the confocal microscope was reduced in these experiments so that only the strong signal from the ectopically expressed proteins was detected. Nuclei were stained with Hoechst 33258. Merged images were shown on the right (panels c and f). (E) COS1 cells were transfected with an expression vector for GFP (panels ac), or with expression vectors for the indicated GFP fusion proteins (panels d–l). The cellular localization of GFP and the GFP fusion proteins was determined by direct fluorescence. Mitochondria were visualized by indirect immunofluorescence with anti-cytochrome c antibodies (panels b, e, h and k). Nuclei were stained with Hoechst 33258 (all panels). Merged images were shown on the right (panels c, f, i, and l). White bars = 10 microns.
Examination of the amino acid sequence of PGAM5 by MITOPRED revealed a potential mitochondrial targeting sequence, including the presence of a short stretch of amino acids with the potential of forming a transmembrane α-helix (amino acids 9–29) and a candidate matrix processing peptidase (MPP) cleavage site (between amino acids 33 and 34) (Fig. 1A) [54, 55]. The subcellular localization of a mutant PGAM5 protein (PGAM5-L-Δ2–25), containing a deletion of amino acids 2 to 25, was determined by confocal microscopy using reduced laser intensity so that only the strong signal from the ectopic PGAM5-L-Δ2–25 and Mito-GFP proteins was detected (Fig. 1D, compare panels a, b, d and e). The PGAM5-L-Δ2–25 protein displayed diffuse cytoplasmic staining while the Mito-GFP protein was efficiently targeted to mitochondria (Fig. 1D; panels a–c). Fusion of either the first 35 or 45 amino acids of PGAM5 onto GFP was sufficient to target GFP to mitochondria, while a GFP fusion protein containing the first 25 amino acids of PGAM5 was diffusely localized in the cytoplasm and nucleus (Fig. 1E; panels d–l). Taken together, these results demonstrate that the N-terminal 35 amino acids of PGAM5, including the putative transmembrane α-helix (amino acids 9–29), function as a mitochondrial targeting sequence.
The subcellular localization of PGAM5-L was also determined by subcellular fractionation. In agreement with the immunofluorescence data, the majority of the endogenous PGAM5-L protein was present in a mitochondrial-enriched subcellular fraction, with trace amounts of PGAM5-L present in the soluble cytosolic fraction (Fig. 2A, compare lanes 1 and 2). Addition of proteinase K to this mitochondrial-enriched subcellular fraction resulted in complete digestion of the endogenous PGAM5-L protein (Fig. 2A, lanes 2–4). TOM20, an outer membrane protein, was also susceptible to proteinase K digestion (Fig. 2A, lanes 2–4). In contrast, VDAC (a integral protein of the mitochondrial outer membrane), cytochrome c and AIF (proteins located in the intermembrane space) and COXIV (a mitochondrial inner membrane protein) were all resistant to proteinase K digestion (Fig. 2A, lanes 2–4), confirming that mitochondrial integrity was preserved during fractionation and proteinase K treatment.
Fig. 2.
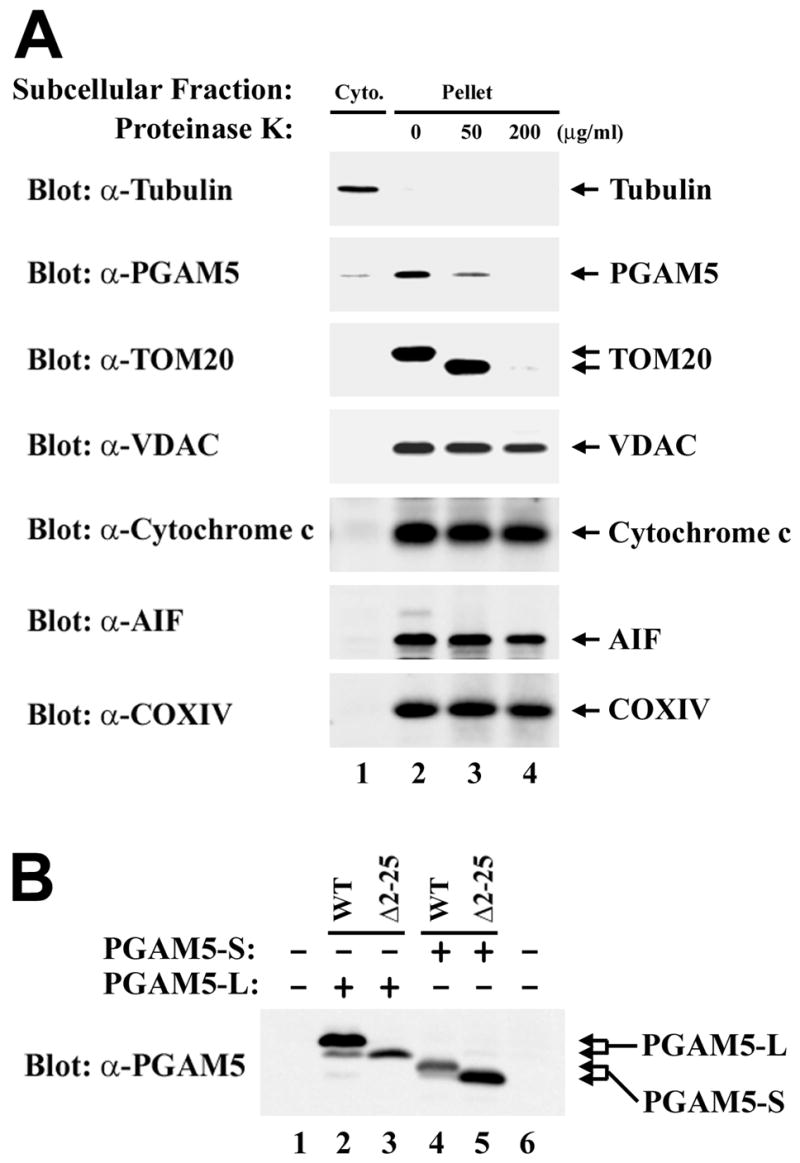
PGAM5 is targeted to the cytoplasmic side of the mitochondrial outer membrane. (A) Hela cells were fractionated into cytosolic (lane 1) and heavy membrane (mostly mitochondria) (lane 2) fractions as described [52, 53]. The heavy membrane fractions were either left untreated (lane 2) or treated with 50 or 200 μg/ml of Proteinase K for 30 minutes on ice. Digestions were terminated by the addition of 2 mM PMSF followed by boiling. Samples were analyzed by immunoblot using antibodies against the indicated proteins. (B) Cell lysates from COS1 cells either mock-transfected (lane 1) or transfected with expression vectors for the indicated proteins (lanes 2–5) were analyzed by immunoblot with anti-PGAM5 antibodies. The wt and mutant PGAM5 proteins are indicated by the arrows on the right side.
To determine if the mitochondrial localization sequence in PGAM5 is cleaved, the mobility of wild-type and mutant PGAM5 proteins were compared using SDS-PAGE. Both PGAM5-L and PGAM5-S migrated as a tight doublet during SDS-PAGE, with the predominant species of the wild-type isoforms migrating slower than the corresponding PGAM5-Δ2–25 deletion mutants (Fig. 2B). Mass spectrometry of tryptic peptides derived from endogenous PGAM5 protein purified from MDA-MB-231 cells identified a peptide that corresponding to amino acids 5–32 of PGAM5 (Table 1). Taken together, the immunolocalization, subcellular fractionation and mass spectrometric data indicate that PGAM5 contains a non-cleaved N-terminal mitochondrial targeting sequence that targets PGAM5 to the cytosolic side of the outer mitochondrial membrane.
Table 1.
| Position and sequence of PGAM5 peptidesa | Mr (calculated)b | Mr (measured)c | |
|---|---|---|---|
| 5–32 | QALQLAACGLAGGSAAVLFSAVAVGKPR | 2682.46 | 2682.50 |
| 65–74 | REPLSLINVR | 1195.70 | 1195.73 |
| 66–74 | EPLSLINVR | 1039.60 | 1039.63 |
| 99–104 | HIFLIR | 797.49 | 797.51 |
| 105–116 | HSQYHVDGSLEK | 1398.65 | 1398.70 |
| 119–125 | TLTPLGR | 756.45 | 756.46 |
| 126–134 | EQAELTGLR | 1015.53 | 1015.56 |
| 135–141 | LASLGLK | 700.45 | 700.46 |
| 153–169 | AIETTDIISRHLPGVCK | 1909.01 | 1908.93 |
| 163–169 | HLPGVCK | 809.42 | 809.51 |
| 170–176 | VSTDLLR | 802.45 | 802.48 |
| 204–209 | IEAAFR | 705.38 | 705.39 |
| 219–235 | QEEDSYEIFICHANVIR | 2121.98 | 2122.04 |
: The position and sequence of PGAM5-derived peptides identified by MALDI-TOF MS. The MOWSE score in this experiment was 74, with a statistical significance cut-off of 63.
: The calculated mass of the indicated PGAM5-derived peptides.
: The observed mass of the indicated PGAM5-derived peptides by mass spectrometry.
Mitochondrial morphology is altered by overexpression of PGAM5 isoforms
Ectopic expression of either PGAM5 isoform revealed that both PGAM5-S and PGAM5-L colocalized with cytochrome c oxidase IV (COXIV) (Fig. 3A). However, profound differences in the morphology of mitochondria were observed in the transfected cells expressing either of the two PGAM5 isoforms. In particular, while mitochondria in mock-transfected COS1 cells displayed a typical reticular-tubular morphology (Fig. 1C), ectopic expression of the PGAM5-S protein resulted in the formation of disconnected punctuate mitochondria distributed throughout the cytoplasm (Fig. 3A, panels a–c). In contrast, ectopic expression of the PGAM5-L protein resulted in the clustering of mitochondria around the nucleus (Fig. 3A, panels d–f). Confocal analysis of COXIV and TOM20 localization in PGAM5-L-expressing cells revealed that the perinuclear mitochondrial aggregates are not giant fused mitochondria but are aggregates of individual mitochondria (Fig. 3B, panels a–f). Similar perturbations of mitochondrial morphology were observed in Hela cells transfected with expression vectors for PGAM5-S or PGAM5-L (data not shown).
Fig. 3.

Ectopic expression of PGAM5 isoforms alter mitochondrial morphology. (A) COS1 were transfected with expression vectors for PGAM5-S or PGAM5-L as indicated. The cellular localization of the overexpressed PGAM5 proteins was determined by indirect immunofluorescence using anti-PGAM5 antibodies (panels b, e and h). Mitochondria were visualized by indirect immunofluorescence with anti-COXIV antibodies (panels a, d and g). Nuclei were stained with Hoechst 33258. Merged images were shown on the right (panels c, f and i). The region within the white squares in panels d–f are shown under higher magnification in panels g–i. (B) Spatial relation between inner mitochondrial membranes (IMM) and outer mitochondrial membranes (OMM). COS1 cells transfected with an expression vector for PGAM5-L (panels a–f) or mock transfected (panels g–l) were analyzed by double-labeled indirect immunofluorescence with anti-COXIV and anti-TOM20 antibodies. The region within the white squares in panels a–c and g–i are shown under higher magnification in panels d–f and j–l, respectively. Nuclei were stained with Hoechst 33258. Merged images are shown on the right. (C) Tubular, intermediate, fragmented and aggregated mitochondria in PGAM5-expressing COS1 cells were shown by the distribution of Mito-GFP (panels a–d). Bright-field microscopic images were shown in the middle (panels e–h). Merged images were shown on the bottom (panels i–l). (D) Percentage of cells displaying each of the four different types of mitochondrial morphology in COS1 cells transfected with an empty vector (Mock) or expression vectors for PGAM5-S or PGAM5-L. At least 200 cells were evaluated in each experiment. The mean and standard deviation from the mean for three independent experiments are shown. White bars = 10 microns.
These dramatic alterations in mitochondrial morphology caused by overexpression of the PGAM5 isoforms were quantified in COS1 cells transiently transfected with the Mito-GFP expression vector along with expression vectors for either of the PGAM5 isoforms. The co-transfected cells were scored for their mitochondrial morphology as being either tubular, intermediate (both tubular and fragmented), fragmented or aggregated (see Fig. 3C for representative images of each morphology). More than 95% of COS1 cells that only expressed the Mito-GFP protein displayed either tubular or intermediate mitochondria (Fig. 3D). In contrast, more than 80% of the cells that expressed the PGAM5-S protein displayed exclusively fragmented mitochondria, while more than 80% of the cells that expressed the PGAM5-L protein displayed perinuclear aggregates of mitochondria (Fig. 3D). Using siRNA molecules that reduced steady-state levels of the endogenous PGAM5 protein by approximately 50%, increased numbers of cells with an intermediate (both tubular and fragmented) morphology was observed (data not shown). Despite these alterations in mitochondrial morphology resulting from increased or decreased expression of the PGAM5 proteins, mitochondrial membrane potential is preserved and cytochrome c is not released (data not shown). We suggest that the PGAM5 isoforms may be novel regulators of mitochondrial dynamics, a possibility that is currently under active investigation.
PGAM5 tethers Keap1 to mitochondria
We have previously characterized PGAM5 as a Keap1-interacting protein and a candidate substrate for a Keap1-dependent E3 ubiquitin ligase complex [46]. To confirm physical interactions between the endogenous Keap1 and PGAM5 proteins, anti-Keap1 immunoprecipitates from HeLa cell lysates were subjected to immunoblot analysis with anti-PGAM5 antibodies. The endogenous PGAM5 protein was present in anti-Keap immunoprecipitates but not in parallel immunoprecipitates performed with pre-immune rabbit sera (Fig. 4A, lanes 1 and 2). Likewise, the endogenous Keap1 protein was specifically detected in anti-PGAM5 immunoprecipitates from HEK293T cell lysates (Fig. 4A, lanes 3 and 4). Confocal microscopy analysis of HeLa cells using anti-Keap1 antibodies revealed a reticulo-tubular staining pattern that colocalized with both cytochrome c and PGAM5 (Fig. 4B, panels d and h). Large punctate bodies that did not co-localize with mitochondria or PGAM5 were also observed with the anti-Keap1 antibody (Fig. 4B, panels d and h). Taken together, the co-immunoprecipitation and co-localization experiments indicate that a significant fraction of the endogenous Keap1 protein co-localizes with the endogenous PGAM5-L protein at mitochondria.
Fig. 4.

PGAM5 recruits Keap1 to mitochondria. (A) Anti-Keap1 immunoprecipitates from 6 mg of Hela cell lysates (lane 2) and anti-PGAM5 immunoprecipitates from 8 mg HEK-293T cell lysates (lane 4) were subjected to immunoblot analysis with affinity-purified anti-PGAM5 (top panel) and anti-Keap1 antibodies (bottom panel). Pre-immune rabbit and chicken serum were used as negative controls for the anti-Keap1 and anti-PGAM5 antibodies, respectively (lanes 1 and 3). The asterik indicates the presence of rabbit IgG in lanes 1 and 2. (B) Hela cells were grown on coverslips in 24-well plates. The cellular localization of the endogenous Keap1 protein was determined by indirect immunofluorescence with affinity-purified anti-Keap1 antibodies against the full-length Keap1 (panels a and e). Mitochondria were visualized by indirect immunofluorescence using anti-Cytochrome c antibodies (panel b). PGAM5 was detected by indirect immunofluorescence using affinity-purified anti-PGAM5 antibodies (panel f). Nuclei were stained with Hoechst 33258. Merged images were shown on the right (panels d and h). (C) COS1 cells were transfected with expression vectors for Keap1, untagged wild-type PGAM5-L, mutant PGAM5-L-E79A/S80A and Mito-GFP as indicated. The cellular localization of the ectopic Keap1 and PGAM5-L proteins was determined by indirect immunofluorescence with anti-Keap1 (panels a, d, and g) and anti-PGAM5 antibodies (panels b, e, h and k). Mito-GFP was visualized by direct fluorescence (panel j). Nuclei were stained with Hoechst 33258. Merged images are shown on the right (panels c, f, i and l). (D) COS1 cells were transfected with expression vectors for HA-Ub (0.4 μg), Keap1 (0.4 μg), and C-terminal FLAG-tagged wild-type or mutant PGAM5-L proteins (0.2 μg) as indicated. Anti-FLAG immunoprecipitates (IP) were analyzed by immunoblot with anti-HA antibodies (top panel). Total lysates were analyzed by immunoblot with anti-FLAG and anti-Keap1 antibodies (bottom two panels). (E) Hela cells were transfected with expression vectors for Keap1 (0.5 μg), and C-terminal FLAG-tagged wild-type or mutant PGAM5-L proteins (0.5 μg) as indicated. Total lysates were analyzed by immunoblot with anti-FLAG, anti-Keap1, and anti-Tubulin antibodies. White bars = 10 microns.
PGAM5 contains a conserved NxESGE motif (amino acids 77–82) that is required for binding to Keap1 (Fig. 1A) [46]. To determine if localization of Keap1 to mitochondria requires binding to PGAM5, the localization of Keap1 in the absence or presence of either wt or mutant PGAM5-L proteins was determined. Ectopic Keap1 was diffusely localized throughout the cytoplasm but was completely relocalized to perinuclear mitochondria clusters when co-expressed with the PGAM5-L protein (Fig. 4C, panel a–f). In contrast, Keap1 remains distributed throughout the cytoplasm when co-expressed with the mutant PGAM5-L-E79A/S80A protein that is localized to mitochondria but is defective in binding to Keap1 (Fig. 4C, panels g–l).
To determine if Keap1 is able to direct ubiquitination onto PGAM5 at mitochondria, an in vivo ubiquitinatin analysis was performed, in which COS1 cells were transfected with expression vectors for HA-ubiquitin, Keap1, and C-terminal FLAG-tagged PGAM5-L. The C-terminal FLAG-tagged PGAM5-L protein, like the untagged PGAM5-L protein, was localized to mitochondria (data not shown). Ubiquitin conjugation onto the wt PGAM5-L protein was observed in the presence of co-expressed Keap1 (Fig. 4D, lane 3). However, co-expression of Keap1 did not reduce steady-state levels of the wt PGAM5-L protein (Fig. 4E, lane 2). In contrast, steady-state abundance of the cytosolic PGAM5-L-Δ2–25 protein, which was also a substrate for Keap1-dependent ubiquitination (Fig. 4D, lane 6), was markedly reduced by co-expression of Keap1 (Fig. 4E, lane 4). Similar results were obtained with the untagged wt and mutant PGAM5-L proteins (data not shown). Taken together, these results demonstrate that mitochondrial-localized PGAM5 is a substrate for a Keap1-dependent ubiquitin ligase complex but is not efficiently targeted for proteosome-dependent degradation.
Keap1 mediates assembly of a mitochondria complex that contains both PGAM5 and Nrf2
Keap1 is a BTB-Kelch protein, and has been shown to form a homo-dimeric complex through its N-terminal BTB domain [56–58]. Crystallographic analysis has demonstrated that an isolated Kelch domain can bind an Nrf2-derived peptide containing the DxETGE motif [26, 27]. Mutational analysis has confirmed the functional importance of amino acid contacts observed in the crystal structure for Keap1 and Nrf2 interactions in vivo [26]. A Keap1 dimer is able to bind two different Nrf2 molecules at the same time, consistent with a 2:2 stoichiometry for the Keap1:Nrf2 complex [26]. However, Nrf2 has also been reported to form a 2:1 complex with Keap1, using both the DLG motif (amino acid residues 17–32) and the DxETGE motif (amino acids 77–82) to bind to Keap1 [58–60].
We first determined if PGAM5-L, like Nrf2, is able to bind to a Keap1 heterodimer containing one functional and one mutant Kelch domain. A chitin-binding domain (CBD) was appended onto the C-terminus of mutant and wt Keap1 proteins. The presence of the CBD tag did not perturb formation of a heterodimeric complex with untagged wt Keap1 (Fig. 5A, lane 7). Two mutant Keap1 proteins (Y334A and Q337A) containing single amino acid substitutions were used in this experiment, only one of which (Y334A) is impaired in binding to PGAM5-L (Fig. 5A, compare lanes 2, 4 and 6). Co-expression of the untagged wild-type Keap1 protein with the mutant CBD-tagged Keap1-Y334A protein markedly increased the level of the PGAM5-L protein that was pulled down with chitin beads (Fig. 5A, lanes 2 and 3). Thus PGAM5-L, like Nrf2, binds to a dimeric Keap1 complex.
Fig. 5.

Nrf2, Keap1 and PGAM5 form a ternary complex at mitochondria. (A) HEK-293T cells were transfected with expression vectors for PGAM5-L (0.2 μg), untagged wild-type Keap1 (0.4 μg), and wild-type or mutant Keap1-CBD proteins (0.4 μg) as indicated. Cell lysates were incubated with chitin beads and proteins that remained bound after extensive washing were subjected to immunoblot analysis with anti-PGAM5 and anti-Keap1 antibodies (top two panels). Total cell lysates were analyzed by immunoblot with anti-PGAM5 and anti-Keap1 antibodies (bottom two panels). (B) COS1 cells were transfected with expression vectors for HA-Nrf2 (1.6 μg), Keap1 (1.6 μg), and PGAM5-L (0.8 μg) as indicated. The transfected cells were treated with 10 μM MG132 or DMSO (0.1%) for 5 h prior to lysis. Anti-HA immunoprecipitates (IP) were analyzed by immunoblot with anti-PGAM5 and anti-Keap1 antibodies (top two panels). Total lysates were analyzed by immunoblot with anti-HA, anti-Keap1, and anti-PGAM5 antibodies (bottom three panels). IgG bands from the immunoprecipitation are indicated by the asterik (second panel from top), while a non-specific band in the anti-Keap1 immunoblot (second panel from bottom) is indicated as NS. (C) COS1 cells were transfected with an expression vector for Mito-GFP (0.15 μg), along with expression vectors for HA-Nrf2 (0.3 μg), Keap1 (0.3 μg), and PGAM5-L (0.15 μg) as indicated. The cellular localization of HA-Nrf2 was determined by indirect immunofluorescence with anti-HA antibodies (panels b, e, h and k). Mitochondria were visualized by distribution of Mito-GFP (panels a, d, g and j). Nuclei were stained with Hoechst 33258. Merged images are shown on the right (panels c, f, i and l). (D) COS1 cells transfected with expression vectors for the indicated proteins were fractionated into cytosol and heavy membrane fractions. The heavy membrane fraction was further fractionated into a Triton X-100 soluble and insoluble fractions. These subcellular fractions were analyzed for the indicated proteins by immunoblot.
As both Nrf2 and PGAM5 utilize similar E(S/T)GE-containing motifs to bind the Kelch domain of Keap1, we considered the possibility that a Keap1 dimer might bind both Nrf2 and PGAM5 to form a ternary complex. To examine this possibility, HA-Nrf2 was expressed in COS1 cells along with PGAM5 and Keap1, and the presence of PGAM5 and Keap1 in anti-HA immunoprecipitates was determined. Both PGAM5 and Keap1 were readily detected in anti-HA immunoprecipitates following ectopic expression of all three proteins (Fig. 5B, lane 6). PGAM5 was also detected, albeit at reduced levels, in anti-HA immunoprecipitates in the absence of ectopic Keap1, presumably due to ability of endogenous Keap1 to bridge HA-Nrf2 and PGAM5 (Fig. 5B, lane 5). Taken together, these results demonstrate that PGAM5, Keap1 and Nrf2 can assemble into a ternary complex.
To determine if the ternary complex of Keap1, PGAM5 and Nrf2 is localized to mitochondria, COS1 cells were transfected with different combinations of expression vectors for Keap1, PGAM5-L and Nrf2 and the subcellular localization of the proteins was determined by confocal microscopy. Mitochondria were visualized by co-expression of Mito-GFP, and nuclei were visualized by DAPI staining. The HA-Nrf2 protein, when expressed alone, was predominantly nuclear and was shifted to the cytoplasm by co-expression of Keap1 (Fig. 5C, panels a–f). Co-expression of HA-Nrf2 and PGAM5-L did not alter either the predominant nuclear localization of HA-Nrf2 or the localization of PGAM5-L to perinuclear clusters of mitochondria (Fig. 5C, panels g–i). However, coexpression of Keap1 with both PGAM5-L and HA-Nrf2 dramatically shifted the localization of HA-Nrf2 from the nucleus to perinuclear mitochondrial clusters (Fig. 5C, panels j–l). Taken together, these results indicate that PGAM5-L, Keap1 and Nrf2 form a ternary complex at mitochondria.
The formation of a ternary complex containing PGAM5-L, Keap1 and Nrf2 at mitochondria was confirmed by subcellular fractionation. COS1 cells transfected with expression vectors for the indicated proteins were fractionated into cytosol and a mitochondria-containing heavy membrane fraction. The mitochondrial pellet was further fractionated into Triton X-100 soluble and insoluble fractions. As expected, tubulin was present only in the cytosolic fraction, while COXIV was present in the Triton X-100 soluble heavy membrane fraction (Fig. 5D). TOM20 was present in both the Triton X-100 soluble and insoluble fractions, consistent with a previous reports indicating the presence of Triton X-100 soluble and insoluble mitochondrial compartments [61–65]. In the absence of PGAM5-L, both Keap1 and Nrf2 were found predominantly in the cytosolic fraction (Fig. 5D, lane 1). However, in the presence of co-expressed PGAM5-L, both Keap1 and Nrf2 were found predominantly in the Triton X-100 insoluble fraction (Fig. 5D, lane 6). As expected, PGAM5, Keap1 and Nrf2 were accessible to Proteinase K in either the absence or presence of Triton X-100, consistent with their localization to the cytosolic face of the mitochondrial outer membrane (data not shown). In contrast, both COXIV and AIF were only digested by proteinase K in the presence of Triton X-100, confirming that the mitochondrial membrane remained intact during the fractionation and proteinase K digestion (data not shown).
To confirm the presence of an endogenous complex containing both PGAM5 and Nrf2, anti-Nrf2 immunoprecipitates or control immunoprecipitates were subjected to immunoblot analysis using affinity-purified antibodies against PGAM5. The endogenous PGAM5-L protein was present in anti-Nrf2 immunoprecipitates but not control immunoprecipitates (Fig. 6A, lanes 1 and 2). Reporter gene assays were performed to determine if PGAM5 participates in regulation of Nrf2-dependent gene expression. A luciferase reporter gene controlled by a minimal promoter containing four copies of an ARE was transfected into Hela cells in which expression of endogenous Nrf2, Keap1, or PGAM5 were individually reduced by siRNA molecules. Three different siRNAs against PGAM5 were used, each of which reduced steady-state levels of the endogenous PGAM5 protein by 40–60% (data not shown). A low level of ARE-dependent reporter gene expression was observed in cells transfected with only the reporter construct, which was further reduced by siRNA against Nrf2. As expected, reporter gene expression was increased by an anti-Keap1 siRNA (Fig. 6B). Reporter gene expression was also increased by any of three different anti-PGAM5 siRNA molecules (Fig. 6B and data not shown). Cotransfection of siRNA against both Nrf2 and PGAM5 reduced reporter gene expression to baseline levels, confirming that Nrf2 is responsible for activation of ARE-dependent gene expression following siRNA-mediated knockdown of PGAM5 (Fig. 6B). Taken together, these results demonstrate that PGAM5 contributes to repression of Nrf2-dependent gene expression.
Fig. 6.
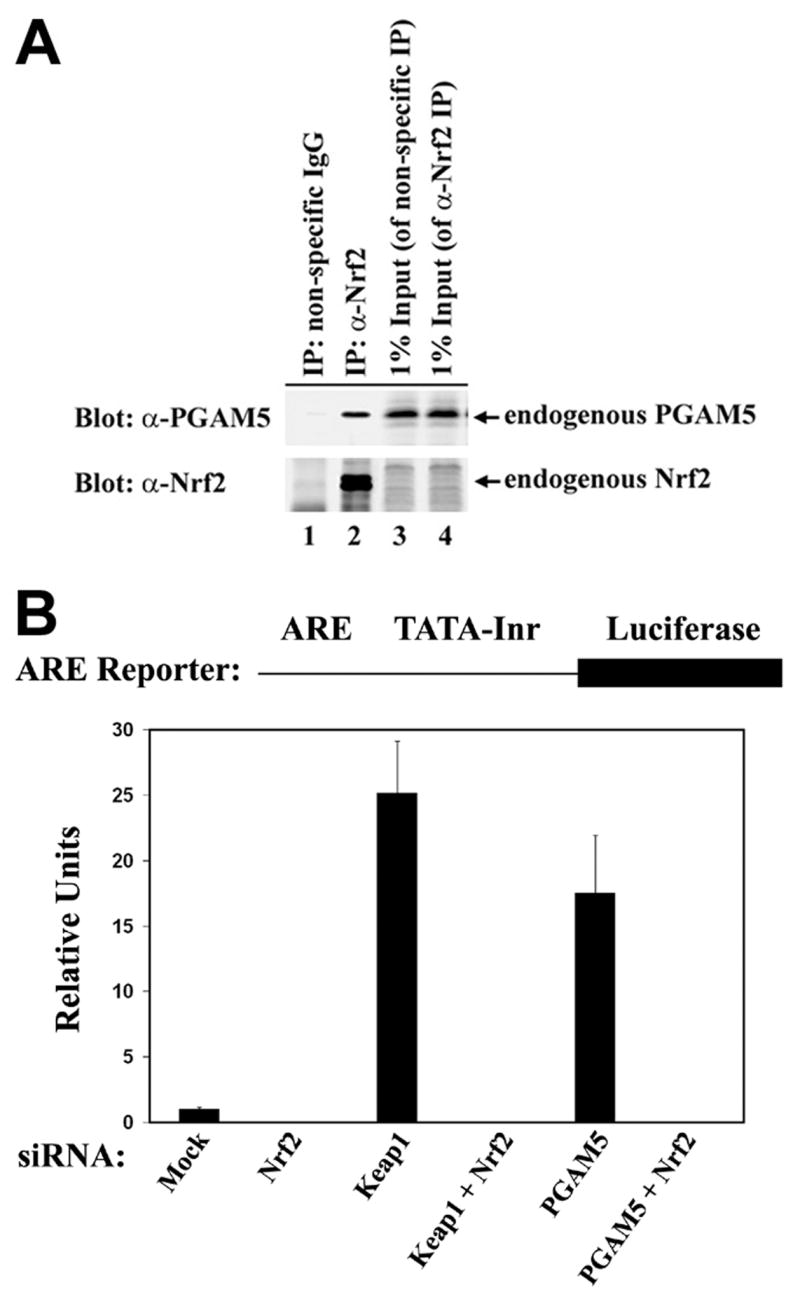
PGAM5 contributes to repression of Nrf2-dependent gene expression. (A) Anti-Nrf2 immunoprecipitates from 9.5 mg of HEK-293T cell lysates (lane 2) were subjected to immunoblot analysis with anti-PGAM5 (top panel) and anti-Nrf2 antibodies (bottom panel).(B) Hela cells were transfected an ARE-dependent firefly luciferase reporter gene construct (0.1 μg) and with siRNA nucleotides (300 nM) against Nrf2, Keap1, or PGAM5 as indicated. A plasmid encoding Renilla luciferase (0.01 μg) was included as a control for transfection efficiency. The data shown are the mean and standard deviation from the mean of three independent experiments.
Discussion
Mitochondria are a major source of ROS, particularly when efficient transfer of electrons to oxygen during oxidative phosphorylation is disrupted [8, 9]. Disruption of mitochondrial function and subsequent generation of ROS has been implicated in both cancer and neurodegenerative diseases [2, 66, 67]. For example, mutations in either DJ1 or PINK, both of which are mitochondrial proteins, are responsible for two different monogenic forms of Parkinson’s [2]. Complex I inhibitors such as rotenone or 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPP+) are widely used in animal models of Parkinson’s and oxidative damage to complex 1 has been demonstrated in Parkinson’s [68–70]. Inhibition of complex I has been associated with activation of Nrf2-dependent anti-oxidant genes [12], while loss of DJ1 results in decreased steady-state levels of Nrf2 and reduced Nrf2-dependent gene expression [14]. In this report, we have demonstrated the existence of a ternary complex containing PGAM5, Keap1 and Nrf2 that is localized to mitochondria. We suggest that this ternary complex provides a molecular framework for understanding how nuclear anti-oxidant gene expression is regulated in response to changes in mitochondrial function(s).
Keap1 is generally regarded as a critical molecular sensor for ROS and electrophilic chemicals. However, the molecular and spatial relationships between the endogenous Keap1 protein and subcellular locations of ROS generation are poorly understood. In our experiments, we find that there are two distinct pools of the endogenous Keap1 protein; one pool that is tethered to the outer membrane of mitochondria through a physical association with PGAM5 and another pool that is localized to punctate cytoplasmic dots. In our hands, treatment of HeLa cells with leptomycin B (LMB), an inhibitor of protein export from the nucleus, does not perturb the mitochondrial pool of Keap1, but results in the translocation of the cytoplasmic Keap1-containing dots into the nucleus (data not shown). Our observation that a portion of the endogenous Keap1 protein undergoes LMB-dependent nuclear accumulation is in agreement with several recent reports that ectopically expressed Keap1 and Nrf2 are shifted to the nucleus in LMB-treated cells [39–42]. However, nuclear-cytoplasmic shuttling of Keap1 and Nrf2 may be cell-type dependent, as one recent report indicates that the endogenous Keap1 protein does not undergo nuclear-cytoplasmic shuttling in either mouse embryonic fibroblasts or primary liver cells [37]. In other cell types, the endogenous Keap1 protein has been localized to peripheral focal adhesions and specialized adherens junctions [38, 71]. Nrf2 is activated in many different cell types and in response to a diverse array of cellular stresses [18]. We suggest that the presence of distinct pools of Keap1 at multiple subcellular locations enables activation of Nrf2 in response to different types of cellular stress.
Although the ability of Keap1 to dimerize via its N-terminal BTB domain has been established [56–58], the importance of Keap1 dimerization for binding of Nrf2 and repression of Nrf2-dependent gene expression remains controversial. Structural analyses of complexes between the Kelch domain of Keap1 and several different Nrf2-derived peptides have demonstrated that the two glutamic acid residues within the NxETGE motif of Nrf2 form multiple contacts with positively charged residues in a shallow binding pocket located on the top face of the Kelch domain [26, 27]. The importance of these amino acid contacts for binding of Nrf2 to Keap1 has been confirmed in a detailed mutagenesis study involving more than 30 single and multiple amino acid substitutions in both Keap1 and Nrf2 [26]. Biophysical studies have suggested the existence of a second motif in Nrf2 that also binds to the Kelch domain of Keap1, leading to a model in which the Keap1 dimer binds a single molecule of Nrf2, resulting in a Keap1-Nrf2 complex with a 2:1 stoichiometry [58–60]. However, a Keap1 dimer can bind two different Nrf2 molecules at the same time, giving rise to a Keap1-Nrf2 complex with a 2:2 stoichiometry [26]. Our previous identification of PGAM5 as a Keap1-binding protein revealed the existence of a conserved DxESGE motif in PGAM5 that binds to Keap1 in a manner very similar to that previously described for the NxETGE motif in Nrf2 [46]. In our present experiments, we demonstrate, by both colocalization and co-immunoprecipitation techniques, that Keap1 forms a bridge between PGAM5 and Nrf2 to form a ternary complex. The ability of Keap1 to bridge PGAM5 and Nrf2 is a critical determinant for localization of Nrf2 to mitochondria. It is possible that Keap1 may utilize different binding modes to regulate Nrf2 at other subcellular locations. Several other candidate substrates for Keap1, including prothymosin-α and FAC1 [39, 72], have been identified and these proteins may form similar ternary complexes with Keap1 and Nrf2 that contribute to regulation of Nrf2-dependent gene expression.
Dimerization of substrate adaptor proteins for cullin-based ubiquitin ligases appears to be a general phenomenon, as several different substrate adaptors for both Cul1 (Cdc4, Fbw7 and Skp2) and Cul3 (Keap1, KLHL9 and KLHL13) form homodimeric or heterodimeric complexes [26, 56–58, 73–77]. Formation of dimeric substrate adaptor complexes may facilitate efficient ubiquitination of a single substrate [74, 76]. An alternative possibility, as suggested by our results, is that the dimeric substrate adaptor may bind two different substrate molecules. As many substrate adaptors have several different substrate proteins, ternary complex formation between a dimeric substrate adaptor and two different substrates may be a general property of cullin-based ubiquitin ligases.
Mitochondria are highly dynamic organelles that are capable of undergoing cycles of fission and fusion as well as intracellular movement along microtubules. In this report, we demonstrate that ectopic expression of the two PGAM5 isoforms results in dramatic alterations in mitochondrial morphology. Specifically, while ectopic expression of PGAM5-L results in perinuclear clustering of mitochondria, ectopic expression of PGAM5-S results in the dispersion of small mitochondria throughout the cell. Although it is possible that ectopic expression of the PGAM5 isoforms interferes with fission/fusion cycles, close examination of the PGAM5-L-induced perinuclear clusters indicates that these clusters are composed of discrete mitochondria rather than giant fused mitochondria. The PGAM5-induced changes in mitochondrial morphology are reminiscent of similar changes caused by ectopic expression of other proteins, such as the atypical GTPase Miro, that have been implicated in mitochondrial movement [78–80]. Mitochondria have critical roles in several aspects of cellular physiology, including ATP generation and regulation of programmed cell death [7, 52]. A better understanding of how PGAM5 regulates mitochondrial morphology may provide novel insights into the functional relationship(s) between mitochondrial fission and fusion, mitochondrial movement and transport, and cellular physiology.
Supplementary Material
Acknowledgments
We thank Carolyn Eberle and Jin Xiaofang for assistance with protein production and antibody purification. We thank Dr. Tom Phillips and the staff at the MU Cytology Core for microscopy assistance and Dr. Marc Johnson, Dr. Shrikesh Sachdev and Jordan Wilkins for helpful discussions. This work was supported by a research grant from NIH (1 RO1 AT003389).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- 2.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006;7:207–19. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 3.Zana M, Janka Z, Kalman J. Oxidative stress: a bridge between Down’s syndrome and Alzheimer’s disease. Neurobiol Aging. 2007;28:648–76. doi: 10.1016/j.neurobiolaging.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol. 2007;292:R18–36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 5.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–47. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 6.Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8:1865–79. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 7.Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–52. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–44. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chem Biol Interact. 2006;163:38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–60. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- 11.Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005;102:244–9. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osburn WO, Wakabayashi N, Misra V, Nilles T, Biswal S, Trush MA, Kensler TW. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch Biochem Biophys. 2006;454:7–15. doi: 10.1016/j.abb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005;280:22925–36. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- 14.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci U S A. 2006;103:15091–6. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics. 2004;18:261–72. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- 16.Ma Q, Battelli L, Hubbs AF. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol. 2006;168:1960–74. doi: 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60:1343–53. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 18.Kensler TW, Wakabayashi N, Biswal S. Cell Survival Responses to Environmental Stresses Via the Keap1-Nrf2-ARE Pathway. Annu Rev Pharmacol Toxicol. 2006 doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 19.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–45. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 20.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhakshinamoorthy S, Jaiswal AK. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2001;20:3906–17. doi: 10.1038/sj.onc.1204506. [DOI] [PubMed] [Google Scholar]
- 22.Cullinan SB, Gordan JD, Jin J, Harper JW, Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol. 2004;24:8477–86. doi: 10.1128/MCB.24.19.8477-8486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. 2004;101:2046–51. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol. 2004;24:10941–53. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. Embo J. 2006;25:3605–17. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 28.Lo SC, Hannink M. CAND1-Mediated Substrate Adaptor Recycling Is Required for Efficient Repression of Nrf2 by Keap1. Mol Cell Biol. 2006;26:1235–44. doi: 10.1128/MCB.26.4.1235-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–13. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A. 2005;102:10070–5. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem. 2007;282:2529–37. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 32.He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. J Biol Chem. 2006;281:23620–31. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- 33.Hong F, Freeman ML, Liebler DC. Identification of Sensor Cysteines in Human Keap1 Modified by the Cancer Chemopreventive Agent Sulforaphane. Chem Res Toxicol. 2005;18:1917–1926. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 34.Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong F, Sekhar KR, Freeman ML, Liebler DC. Specific patterns of electrophile adduction trigger keap1 ubiquitination and NRF2 activation. J Biol Chem. 2005;280:31768–75. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 36.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–74. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 37.Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, Itoh K, Yamamoto M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells. 2007;12:1163–78. doi: 10.1111/j.1365-2443.2007.01118.x. [DOI] [PubMed] [Google Scholar]
- 38.Velichkova M, Guttman J, Warren C, Eng L, Kline K, Vogl AW, Hasson T. A human homologue of Drosophila kelch associates with myosin-VIIa in specialized adhesion junctions. Cell Motil Cytoskeleton. 2002;51:147–64. doi: 10.1002/cm.10025. [DOI] [PubMed] [Google Scholar]
- 39.Karapetian RN, Evstafieva AG, Abaeva IS, Chichkova NV, Filonov GS, Rubtsov YP, Sukhacheva EA, Melnikov SV, Schneider U, Wanker EE, Vartapetian AB. Nuclear oncoprotein prothymosin alpha is a partner of Keap1: implications for expression of oxidative stress-protecting genes. Mol Cell Biol. 2005;25:1089–99. doi: 10.1128/MCB.25.3.1089-1099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen T, Sherratt PJ, Nioi P, Yang CS, Pickett CB. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J Biol Chem. 2005;280:32485–92. doi: 10.1074/jbc.M503074200. [DOI] [PubMed] [Google Scholar]
- 41.Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–13. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–49. doi: 10.1128/MCB.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terada LS. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol. 2006;174:615–23. doi: 10.1083/jcb.200605036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–34. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- 45.Jedrzejas MJ. Structure, function, and evolution of phosphoglycerate mutases: comparison with fructose-2,6-bisphosphatase, acid phosphatase, and alkaline phosphatase. Prog Biophys Mol Biol. 2000;73:263–87. doi: 10.1016/s0079-6107(00)00007-9. [DOI] [PubMed] [Google Scholar]
- 46.Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- 47.Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity. 2004;20:37–46. doi: 10.1016/s1074-7613(03)00351-0. [DOI] [PubMed] [Google Scholar]
- 48.Kowanetz K, Crosetto N, Haglund K, Schmidt MH, Heldin CH, Dikic I. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J Biol Chem. 2004;279:32786–95. doi: 10.1074/jbc.M403759200. [DOI] [PubMed] [Google Scholar]
- 49.Davies L, Anderson IP, Turner PC, Shirras AD, Rees HH, Rigden DJ. An unsuspected ecdysteroid/steroid phosphatase activity in the key T-cell regulator, Sts-1: surprising relationship to insect ecdysteroid phosphate phosphatase. Proteins. 2007;67:720–31. doi: 10.1002/prot.21357. [DOI] [PubMed] [Google Scholar]
- 50.Mikhailik A, Ford B, Keller J, Chen Y, Nassar N, Carpino N. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol Cell. 2007;27:486–97. doi: 10.1016/j.molcel.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hammond PW, Alpin J, Rise CE, Wright M, Kreider BL. In vitro selection and characterization of Bcl-X(L)-binding proteins from a mix of tissue-specific mRNA display libraries. J Biol Chem. 2001;276:20898–906. doi: 10.1074/jbc.M011641200. [DOI] [PubMed] [Google Scholar]
- 52.Setoguchi K, Otera H, Mihara K. Cytosolic factor- and TOM-independent import of C-tail-anchored mitochondrial outer membrane proteins. Embo J. 2006;25:5635–47. doi: 10.1038/sj.emboj.7601438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–11. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guda C, Guda P, Fahy E, Subramaniam S. MITOPRED: a web server for the prediction of mitochondrial proteins. Nucleic Acids Res. 2004;32:W372–4. doi: 10.1093/nar/gkh374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–49. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 56.Chew EH, Poobalasingam T, Hawkey CJ, Hagen T. Characterization of cullin-based E3 ubiquitin ligases in intact mammalian cells--evidence for cullin dimerization. Cell Signal. 2007;19:1071–80. doi: 10.1016/j.cellsig.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 57.Zipper LM, Mulcahy RT. The Keap1 BTB/POZ Dimerization Function Is Required to Sequester Nrf2 in Cytoplasm. J Biol Chem. 2002;277:36544–52. doi: 10.1074/jbc.M206530200. [DOI] [PubMed] [Google Scholar]
- 58.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281:24756–68. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 59.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol. 2006;26:2887–900. doi: 10.1128/MCB.26.8.2887-2900.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol Cell Biol. 2007 doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rashid-Doubell F, Tannetta D, Redman CW, Sargent IL, Boyd CA, Linton EA. Caveolin-1 and lipid rafts in confluent BeWo trophoblasts: evidence for Rock-1 association with caveolin-1. Placenta. 2007;28:139–51. doi: 10.1016/j.placenta.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 62.Bello-Morales R, Fedetz M, Alcina A, Tabares E, Lopez-Guerrero JA. High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J Neurovirol. 2005;11:190–8. doi: 10.1080/13550280590924179. [DOI] [PubMed] [Google Scholar]
- 63.Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, Dawson TM, Dawson VL. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum Mol Genet. 2005;14:71–84. doi: 10.1093/hmg/ddi007. [DOI] [PubMed] [Google Scholar]
- 64.Goetz JG, Nabi IR. Interaction of the smooth endoplasmic reticulum and mitochondria. Biochem Soc Trans. 2006;34:370–3. doi: 10.1042/BST0340370. [DOI] [PubMed] [Google Scholar]
- 65.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 66.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–62. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 67.Zhu X, Smith MA, Perry G, Aliev G. Mitochondrial failures in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2004;19:345–52. doi: 10.1177/153331750401900611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoglinger GU, Oertel WH, Hirsch EC. The rotenone model of parkinsonism--the five years inspection. J Neural Transm Suppl. 2006:269–72. doi: 10.1007/978-3-211-45295-0_41. [DOI] [PubMed] [Google Scholar]
- 69.Keeney PM, Xie J, Capaldi RA, Bennett JP., Jr Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–64. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schmidt WJ, Alam M. Controversies on new animal models of Parkinson’s disease pro and con: the rotenone model of Parkinson’s disease (PD) J Neural Transm Suppl. 2006:273–6. [PubMed] [Google Scholar]
- 71.Velichkova M, Hasson T. Keap1 in adhesion complexes. Cell Motil Cytoskeleton. 2003;56:109–19. doi: 10.1002/cm.10138. [DOI] [PubMed] [Google Scholar]
- 72.Strachan GD, Morgan KL, Otis LL, Caltagarone J, Gittis A, Bowser R, Jordan-Sciutto KL. Fetal Alz-50 clone 1 interacts with the human orthologue of the Kelch-like Ech-associated protein. Biochemistry. 2004;43:12113–22. doi: 10.1021/bi0494166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wimuttisuk W, Singer JD. The Cullin3 ubiquitin ligase functions as a Nedd8-bound heterodimer. Mol Biol Cell. 2007;18:899–909. doi: 10.1091/mbc.E06-06-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W, Koepp DM. Fbw7 isoform interaction contributes to cyclin E proteolysis. Mol Cancer Res. 2006;4:935–43. doi: 10.1158/1541-7786.MCR-06-0253. [DOI] [PubMed] [Google Scholar]
- 75.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 76.Tang X, Orlicky S, Lin Z, Willems A, Neculai D, Ceccarelli D, Mercurio F, Shilton BH, Sicheri F, Tyers M. Suprafacial Orientation of the SCF(Cdc4) Dimer Accommodates Multiple Geometries for Substrate Ubiquitination. Cell. 2007;129:1165–76. doi: 10.1016/j.cell.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 77.Sumara I, Quadroni M, Frei C, Olma MH, Sumara G, Ricci R, Peter M. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell. 2007;12:887–900. doi: 10.1016/j.devcel.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 78.Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- 79.Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–10. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- 80.Nemoto Y, De Camilli P. Recruitment of an alternatively spliced form of synaptojanin 2 to mitochondria by the interaction with the PDZ domain of a mitochondrial outer membrane protein. Embo J. 1999;18:2991–3006. doi: 10.1093/emboj/18.11.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.