

Abstract
Purpose
Novel strategies are needed to prevent the high mortality rates of several types of cancer. These high rates stem from tumor resistance to radiation therapy, which is thought to result from the induction of matrix metalloproteinases and plasminogen activators. In the present study, we show that the modulation of MMP-9 expression, using adenoviral-mediated transfer of the antisense MMP-9 gene (Ad-MMP-9), affects breast cancer sensitivity to radiation.
Experimental Design
In the present study, we used antisense MMP-9 adenoviral construct (Ad-MMP-9) to downregulate the expression of MMP-9 in MDA MB 231 breast cancer cell lines in vitro prior to irradiation and subsequently incubated cells in hypoxic condition. In vivo studies were performed with orthotopic breast tumors and the radiosensitivity evaluated both in vitro and in vivo.
Results
Ad-MMP-9 infection resulted in downregulation of radiation-induced levels of hypoxia-inducible factor 1 alpha (HIF1α) and MMP-9 under hypoxic conditions in MDA MB 231 breast cancer cells. In addition, Ad-MMP-9 in combination with radiation decreased levels of the transcription factors NF-κB and AP 1, both of which contribute to the radioresistance of breast tumors. Finally, the triggering of the Fas-Fas-L apoptotic cascade, which resulted in the cleavage of PARP-1 and caspases 10, 3 and 7, signifies the efficiency of combined treatment of Ad-MMP-9 and radiation. Treatment with Ad-MMP-9 plus radiation completely regressed tumor growth in orthotopic breast cancer model.
Conclusions
In summary, integrating gene therapy (adenovirus-mediated inhibition of MMP-9) with radiotherapy could have a synergistic effect, thereby improving the survival of patients with breast cancer.
Keywords: Ad-MMP-9, Radiation, Orthotopic breast tumor, Hypoxia, Tumor growth
INTRODUCTION
Radiotherapy provides a key management strategy for many epithelial tumor types and forms part of the multidisciplinary approach for breast cancer treatment, and is now of routine value after conservative surgery to reduce locoregional tumor recurrence. However, the significant limitations of radiotherapy make it inefficient as the sole treatment for breast and many other cancers. Some cancer cells are intrinsically resistant to damage by ionizing radiation and treatment can actually induce tumor cell proliferation and re-population, resulting in a diminished response to radiation, resistant growth and poor local control (1). Hypoxia-driven cellular modifications have been shown to contribute to this poor prognostic outlook, giving rise to more aggressive locoregional disease, invasive capacity, and angiogenesis (2).
Intratumoral hypoxia plays a pivotal role in activating the key transcription factor, hypoxia-inducible factor (HIF), which mediates activation of the “survival machinery” in cancer cells. Specifically, HIF1 regulates the expression of numerous genes and controls glycolysis, erythropoiesis, apoptosis and angiogenesis (3). Studies have demonstrated that HIF1α activity promotes tumor growth in vivo (4). Hypoxic tumor regions show increased gene expression caused by hypoxia-induced activation of transcription machinery (5, 6). Several of the gene products induced or upregulated under hypoxic conditions play pivotal roles in the metastatic process, and studies have suggested that hypoxia could promote metastasis in human cancer (5). Hypoxia also activates NF-κB (7), a transcription factor important in the promotion and progression of tumor development and survival (8). As such, inhibition of these pathways represents a potentially important cancer therapeutic.
Cancer cells often develop radioresistance mechanisms related to the DNA repair response. By combining chemotherapy and radiation therapy, radiation efficiency may be strenghtened by inhibiting DNA repair, and overcome resistance to apoptosis. NF-κB is activated by DNA-damaging agents and could be involved in cell cycle arrest and prevention of apoptosis, allowing DNA repair (9). However, sustained NF-κB activation could permit cells with accumulated radiation-induced DNA damage to escape elimination by apoptosis (10). Indeed, high constitutive NF-κB activity prevents cancerous cells from apoptosis (11) resulting in a more aggressive potential seen in prostate cancer (12), malignant melanomas, Hodgkin’s disease, leukemia, breast cancer and cutaneous T-cell lymphoma cancer cell lines (13).
The transcription factor activator protein 1 (AP1) comprises members of the Jun and Fos families, and has been implicated in the regulation of apoptosis and cell proliferation (14). Studies suggest that Jun B and c-Jun may trigger apoptosis and promote proliferation of erythroid cells, respectively (15). Activation of Sp1, another key transcription factor, occurs under hypoxic conditions in various cancer cells including breast carcinoma (16, 17). Thus, agents that enhance apoptosis in irradiated tumor cells could have significant therapeutic benefits.
Degradation of the extracellular matrix plays an important role in tumor metastasis. MMPs have been primarily associated with matrix remodeling, a necessary component of invasion. It has been reported that pre-operative, short course radiotherapy decreases local recurrence rates (e.g., rectal cancer), and combined with optimal surgery, improves patient survival. Although radiotherapy has proven benefits, several reports show an increase in expression and activation of gelatinase MMPs (18). Initially, MMPs were thought to simply break down components of the extracellular matrix, allowing for invasion and metastasis. However, recent studies confirmed that MMP activity involves precise MMP localization on a cell’s invasive front, exposure of key components in the extracellular matrix transform it from a barrier into a scaffold for invasion, and cleavage of free insulin-like growth factors result in cell growth, division and inhibition of apoptosis (19).
Hypoxia-targeted gene therapy presents a number of interesting developments and applications in modern oncology. Attempts are currently being made to overcome the adverse effects and limitations of radiation and exploit resistant hypoxic tumor cells using combination gene therapy and radiotherapy. In this study, we combined radiation with downregulated MMP-9, which reduced hypoxia by using a replication-deficient recombinant adenovirus containing a 528 bp antisense expression segment for human MMP-9 (Ad-MMP-9AS or Ad-MMP-9) (20) to treat breast cancer tumors. Our results indicated that after the combined treatment, transcription factor activity controlling the expression of the oncoproteins were reduced and pro-apoptotic molecules upregulated, resulting in increased apoptosis and tumor suppression.
MATERIALS & METHODS
Cell culture and treatments
MDA MB 231 human breast cancer cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% FBS in a humidified CO2 incubator at 37° C. MDA MB 231 cells were serum-starved for 12 to 18 h and treated with 100 multiplicity of infection (MOI) of the empty vector (Ad-CMV) or antisense MMP-9 adenoviral construct (Ad-MMP-9). Either Ad-MMP-9 or Ad-CMV was added to the cell monolayer (1.0 mL per 60-mm dish or 3 mL per 100-mm dish) and cells were incubated at 37° C for 60 min with brief agitation every 5 min. The necessary amount of culture medium was then added and cells were returned to the incubator. For the radiation treatment, the cells were starved of serum, exposed to 5 gy, and returned to the incubator after the addition of the necessary amount of complete culture medium. For the treatment combination of virus and radiation, the cells were first infected with 100 MOI of Ad MMP-9 or Ad-CMV and 24 h later, the cells were irradiated with 5 gy. After the above-described treatments, the cells were incubated in hypoxic condition for a period of 12 to 16 h. The hypoxic condition was accomplished with the Anaerocult® mini setup (EM Science, Gibbstown, NY).
Reverse transcription PCR
Total RNA was isolated from cells in all treatment conditions using TRIzol per standard protocol. Total RNA was treated with DNase I (Invitrogen, Carlsbad, CA) to remove contaminating genomic DNA. PCR analysis was performed using the One-step RT-PCR kit (Invitrogen, Carlsbad, CA). GAPDH was used as an internal control. The following primers were used:
| HIF1α | MMP-9 | GAPDH | |
| sense (5′ to 3′) | AGTCTGCAACATGGAAGG | TGGACGATGCCTGCAACGTG | CGGAGTCAACGGATTTGGTCGTAT |
| antisense (5′ to 3′) | CACGACTTGATTTTCTCCC | GTCGTGCGTGTCCAAAGGCA | AGCCTTCTCCATGGTGGTGAAGAC |
The PCR conditions were as follows: 95° C for 5 min, followed by 30 cycles of 95° C for 1 min, annealing temperature set according to the AT and GC content of the primers respectively for 1 min, and 72° C for 1 min. The final extension was at 72° C for 5 min.
Electromobility shift assay (EMSA)
Nuclear extracts were prepared from MDA MB 231 cells that were treated with Ad-MMP-9, radiation or both. Cells were detached with EDTA and resuspended in buffer A (10 mM HEPES [N-2-hydroxyethylenepiperazine-N′-2-ethanesulfonic acid], pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT]) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 5 mM iodoacetamide, 0.1 mM quercetin, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 0.3 mM sodium vanadate) and incubated on ice for 15 min. After homogenization in a Wheaton 0.1-mL homogenizer, the nuclei were collected by centrifugation. The pellet was resuspended in buffer B (20 mM HEPES, pH 7.9, 25% glycerol, 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 0.5 mM DTT) containing protease inhibitors and incubated on ice for 30 min, followed by centrifugation at 13,000g (5 min at 4° C). The supernatant was dialyzed against buffer C (20 mM HEPES, 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 0.5 mM DTT) containing protease inhibitors for 2 h at 4° C, followed by centrifugation at 13,000g (5 min at 4° C). The supernatant proteins were used immediately or aliquoted and stored at −80° C.
Binding reaction was performed for 30 min on ice in a volume of 20 μL, containing 4 μg nuclear protein extracts, 40 ng poly (dI-dC), 4 μL 5X binding buffer (1X binding buffer: 20 mM HEPES, pH 7.9, 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 10% glycerol) with or without 20- to 50-fold excess of cold competitor or of unrelated competitor and a 32P-labeled probe (3× 104 cpm). For supershift EMSA, protein extracts were incubated with 6 μg SP1 monoclonal antibody or isotype control before the addition of the 32P-labeled probe. DNA-protein complexes were separated on 5% polyacrylamide gel in Tris/glycine buffer at 4° C. The following double-stranded oligonucleotides (Santa Cruz Biotechnology, Santa Cruz, CA) were used in this study:
| NF-κB (sc-2505) | AP1 (sc-2501) | |
| 5′ to 3′ | AGT TGA GGG GAC TTT CCC AGG C | CGC TTG ATG ACT CAG CCG GAA |
| 3′ to 5′ | TCA ACT CCC CTG AAA GGG TCC G | GCG AAC TAC TGA GTC GGC CTT |
End-labeled probes were prepared with 40 μCi (1480 MBq) [γ-32P] adenosine triphosphate (ATP) using T4 polynucleotide kinase and were gel-purified on NAP-5 Sephadex G-25 DNA-grade columns.
Western blot analysis
MDA MB 231 cells were treated with Ad-SV, Ad-MMP-9, radiation of 5 gy, or a combination (Ad-MMP-9 plus radiation), and incubated under hypoxic conditions as described earlier. After the incubation period, the cells were washed with ice-cold PBS and lysed in RIPA buffer containing protease inhibitors. Whole cell extracts were subjected to SDS-PAGE and subsequently transferred to a polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA). The membranes were blocked with 7% nonfat dry milk and probed with antibodies for the following molecules: MMP-9, HIF1α, ERK1/2, pERK, NF-κB, p50, p65, c-fos, jun D, AP1, Fas, Fas-L, caspases-10, -3 & -7, and PARP-1. Appropriate antibody conjugate with horseradish peroxidase was used as the secondary antibody. Membranes were developed according to an enhanced chemiluminescence protocol as per the manufacturer’s instructions (Amersham Biosciences, Piscataway, NJ). Nuclear and cytoplasmic fractions for Western blotting were prepared as described elsewhere (21).
Gelatin zymography
Gelatin-substrate gel electrophoresis was performed as described previously (22). MDA MB 231 cells were transfected with Ad-CMV, Ad-MMP-9, radiation of 5 gy, or a combination (Ad-MMP-9 plus radiation), and incubated under hypoxic conditions as described earlier. To collect conditioned media, cells were washed once with serum-free medium and incubated with fresh serum-free medium. After 12 to 14 h, conditioned medium was collected, centrifuged to remove cellular debris, and protein concentrations were determined. Equal amounts of protein were subjected to 0.1% gelatin SDS-PAGE under non-reducing conditions. Gels were washed in 2.5% Triton X-100 and incubated overnight in Tris-CaCl2 buffer. The gels were then stained with 0.2% Coomassie blue for 30 min and destained in 20% methanol and 10% acetic acid. The clear bands represent gelatinase activity.
Clonogenic survival assay
A clonogenic survival assay was used to investigate the sensitivity of MDA MB 231 cells infected with Ad-MMP-9 to radiation therapy as described previously (23). Briefly, the cells were treated as described earlier. The MDA MB 231 cells were trypsinized and plated in 100-mm dishes to assay for their colony-forming ability immediately after irradiation under hypoxic condition. Colonies were counted 10 to 15 days later. Survival curves were plotted using the GraphPad Prism 3.0 software program.
Animal experiments
MDA MB 231 cells were cultured in complete medium until a 70–80% density was obtained. At this point, cells were trypsinized, washed once with serum-free medium, and counted. Cells were injected bilaterally into the second mammary fat pads of athymic, female, 4- to 6-week-old nu/nu mice (5–6× 106/100 μL serum-free culture medium). Tumor growth was monitored daily. Once the tumor reached approximately 6–8 mm in size, the animals were divided into 5 groups of 5 animals each. Group I received PBS injections, group II received three doses of 5×108 PFU intratumorally on alternate days, group III received three doses of 5×108 PFU intratumorally on alternate days, group IV was irradiated with 2 doses of 5 gy on alternate days, and group V received Ad-MMP-9 and radiation treatments. In the case of the combined treatment, the tumors were first given intratumoral injections of Ad-MMP-9 on alternate days. Then, after the third dosage, the tumors were given 2 doses of 5 gy on alternate days. The regression in the orthotopic tumor growth was followed for up to 8 weeks. Mice were euthanized when the tumor diameter in control mice measured between 1.2 and 1.5 cm were removed and further processed. Additionally, as there was no tumor in animal treated with the Ad-MMP-9 infection in combination with irradiation we included one more group of 5 animals that received combined treatment. The animals with tumor were euthanized after 15 days post combined treatment. Finally, the tumor volume was calculated using the formula V = pi/6 (a×b×c).
Immunohistochemistry
Tumor samples fixed in 10% neutral buffered formalin were embedded in paraffin using automatic embedding equipment, following which 5 μm sections were prepared. Immunohistochemical analysis for MMP-9, HIF1α and caspase 3 was performed on paraffin-embedded breast tumor sections of mice treated with Ad-CMV, Ad-MMP-9, irradiation and Ad-MMP-9 infection in combination with irradiation.
TUNEL assay
TUNEL assay was performed with paraformaldehyde-fixed, paraffin-embedded breast tumor sections as per the manufacturer’s protocol. Briefly, the TUNEL (Terminal deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling) method identifies apoptotic cells in situ by using terminal deoxynucleotidyl transferase (TdT) to transfer biotin-dUTP to the free 3′-OH of cleaved DNA. The biotin-labeled cleavage sites are then visualized by reaction with fluorescein-conjugated avidin (avidin-FITC). The cells were visualized using a fluorescent microscope with appropriate filter sets. DNA fragmentation in these treated tumors is indicative of apoptotic cell population.
RESULTS
Ad-MMP-9 infection inhibits radiation-induced MMP-9 and HIF1α expression at both the mRNA and protein levels in breast cancer cells
We analyzed the effect of Ad-MMP-9 (adenoviral construct of antisense to MMP-9 gene) on the MDA MB 231 cell line, which is the most aggressive breast cancer cell line, for MMP-9 and HIF1α expression at the mRNA level. RT-PCR analysis demonstrated that Ad-MMP-9 infection inhibited MMP-9 and HIF1α at the mRNA level when compared to control or Ad-CMV (empty vector)-infected MDA MB 231 cells under hypoxic conditions (Fig. 1A). In contrast, radiation alone augmented the expression level of these molecules. Treatment with Ad-MMP-9 plus radiation inhibited expression levels more than Ad-MMP-9 infection alone. The level of MMP-9 was reduced by nearly 40% in the Ad-MMP-9-treated cells when compared to mock and Ad-CMV treatments; MMP-9 level was reduced by more than 50% when cells were treated with Ad-MMP-9 plus radiation. Conversely, in the cells that were treated with radiation alone, the level of MMP-9 was 50% higher than in the control cells (Fig. 1A).
Figure 1. Ad-MMP-9 infection inhibits radiation-induced MMP-9 and HIF 1α expression at both the mRNA and protein levels in breast cancer cells.
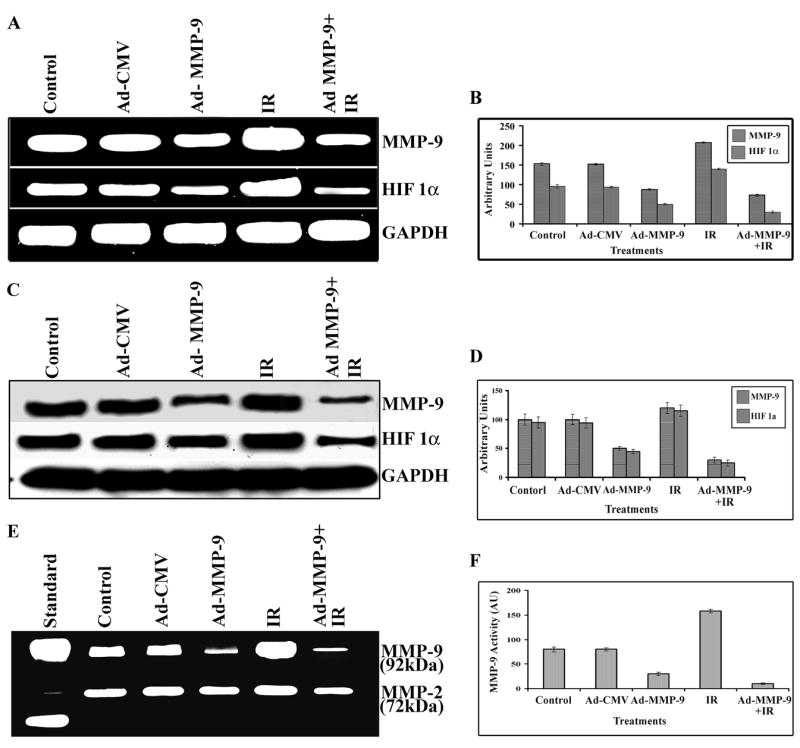
(A) Briefly, MDA MB 231 cells were infected with Ad-CMV (100 MOI), Ad-MMP-9 (100 MOI), Irradiation (IR) (5 gy), or a combination of Ad-MMP-9 (100 MOI) and Irradiation (IR) (5 gy), and incubated under hypoxic conditions as described in Materials & Methods. After 12 to 16 h of incubation, total RNA was extracted using TRIzol reagent, quantitated and RT-PCR was performed for assessment of MMP-9 and HIF1-α levels. Expression of GAPDH was verified for the equal loading of cDNA. (B) Densitometric analysis of MMP-9 and HIF1-α expression at the mRNA level (mean ± S.E.; n=3). (C) After incubation, cell lysates were prepared and used for Western blot analysis to determine the levels of MMP-9 and HIF 1α. GAPDH was also used as a control to confirm equal loading of cell lysates. (D) Densitometric analysis of MMP-9 and HIF1-α expression at the protein level (mean ± S.E.; n=3). (E) MMP-9 activity was analyzed by gelatin zymography using equal amounts of protein from the conditioned medium as described in Materials & Methods. (F) Densitometric analysis of MMP-9 activity (mean ± S.E.; n=4)
Similarly, the expression level of HIF1α at the mRNA level in the irradiated cells was increased by 30% as compared to the control and Ad-CMV-infected cells. Ad-MMP-9 treatment alone inhibited the expression of HIF1α by 25% as compared to the controls; combined treatment of Ad-MMP-9 and radiation resulted in a 60% reduction of HIF1α as compared to the controls. These results support the earlier findings of Moeller et al. (24) that radiation increased the levels of active HIF1 and that the hypoxic condition is responsible for the poor response of the tumors to radiotherapy (Figs. 1A and 1B). Western blot analyses using respective antibodies against MMP-9 and HIF1α support the RT-PCR analysis results, where the level of MMP-9 was reduced nearly 40–45% in the Ad-MMP-9-treated cells as compared to the control and Ad-CMV treatment, and by more than 60–70% when cells were infected with Ad-MMP-9 in combination with radiation. Conversely, in the cells treated with radiation alone, the level of MMP-9 was 30% more than in the control cells (Figs. 1C and 1D).
In addition, analysis of MMP-9 activity by gelatin zymography using conditioned medium from the treated cells revealed decreased levels of MMP-9 activity in Ad-MMP-9-infected cells as compared to the controls. MMP-9 activity was even further reduced with the combined treatment. In contrast, we observed an almost 2-fold increase in MMP-9 activity in the irradiated cells when compared to the control and Ad-CMV-treated cells (Figs. 1E and 1F).
Treatment with Ad-MMP-9 and radiation decreases the binding activity of NF-κB and AP1
Ionizing radiation is reported to induce activation of NF-κB and AP-1 (25). Poynter et al. have reported on the phosphorylation and dephosphorylation of members of the extracellular-regulated kinase (ERK) family of the mitogen-activated protein kinase (MAPK) cascade and the events leading to activation of the transcription factor NF-κB. These cascades are critical for the transcriptional upregulation of genes important for cell survival, apoptosis, proliferation, transformation, and inflammation (26). Further, it is known that ionizing radiation augments phosphorylation of ERK (27). Western blot analysis using whole cell extracts of the cells treated with Ad-MMP-9 and Ad-MMP-9 in combination with radiation showed reduced phosphorylation of ERK whereas irradiated cells showed increased phosphorylation when compared to the controls (Fig. 2A). Activation of the binding activities of these transcription factors in tumor cells contributes to MMP-9 transcription and cell invasion (28). Western immunoblot analysis using the nuclear fractions from the treated cells showed reduced translocation of p50 and p65 (subunits of NF-κB) and c-fos and Jun D (subunits of AP1) to the nucleus in the Ad-MMP-9-treated cells, and further reduction in the cells treated with Ad-MMP-9 in combination with radiation as compared to the controls. In contrast, analysis of the cytoplasmic fractions showed reduced signals for the above-said molecules in the Ad-MMP-9-treated cells, and further reduction in the cells treated with Ad-MMP-9 in combination with radiation as compared to the controls (Fig. 2B). However, a slight increase of NF-κB and AP1 translocation to the nucleus was seen in radiated cells alone when compared to the controls. Furthermore, we analyzed the DNA-binding activities of NF-κB and AP1 in the nuclear extracts using NF-κB and AP1-specific oligonucleotide probes (EMSA). In the cells treated with Ad-MMP-9 or Ad-MMP-9 plus radiation, the DNA-protein complex was decreased when compared to the control and irradiated cells. The specificity of the NF-κB-DNA and AP1-DNA complexes was confirmed by supershift assay, using specific antibodies (Fig. 2C).
Figure 2. The combined treatment of Ad-MMP-9 and radiation decreases binding activity of NF-κB and AP1.
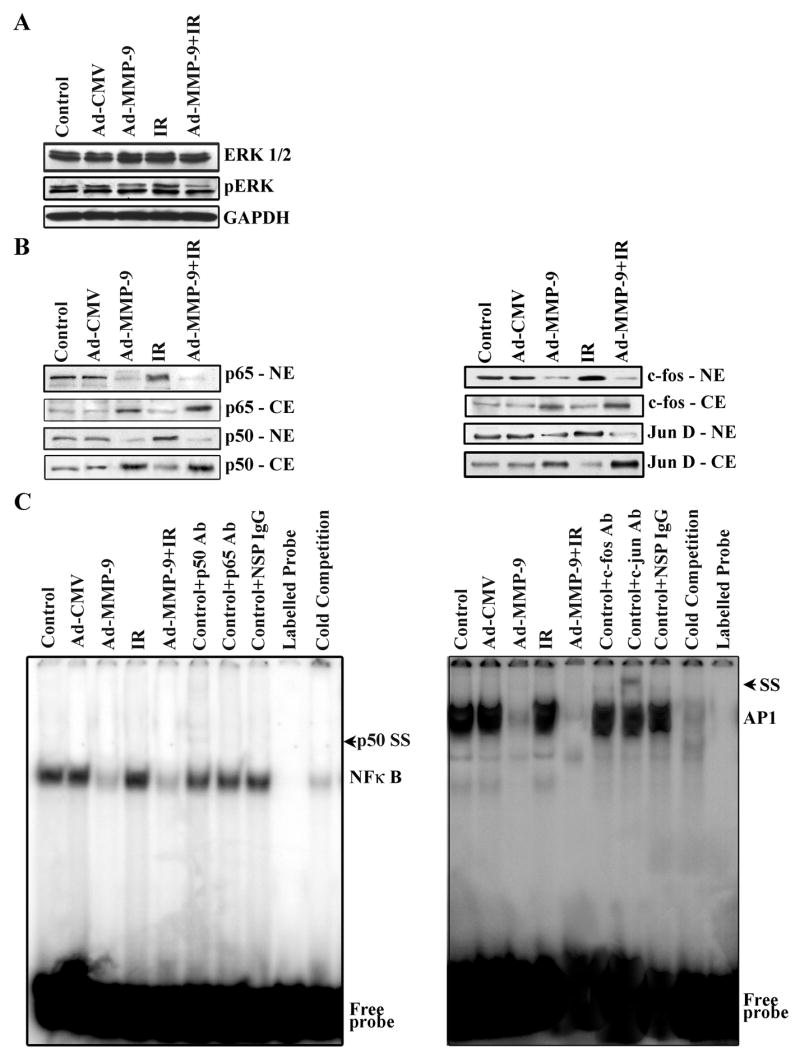
(A) Western blot results for ERK1/2 and pERK. GAPDH was used as a control to confirm equal loading of cell lysates. (B) Western blot analysis of sub-cellular fractions showing the translocation of the p50 and p65 subunits of NF-κB and c-fos and Jun D subunits of AP1. (C) The binding activities of NF-κB and AP1 to DNA were determined using the electromobility shift assay. Oligonucleotide consensus probes for NF-κB and AP1 were end labeled with 32P and used in the shift assay. (IR-Irradiation, SS-Supershift)
Downregulation of MMP-9 and radiation induces apoptosis in breast cancer cells
Because radiation-induced NF-κB activation leads to cell survival, we next examined whether Ad-MMP-9 treatment in combination with radiation could cause apoptosis. Western immunoblot analysis showed the upregulation of Fas in the Ad-MMP-9, radiation and combination treatments. However, Fas-L was upregulated only in the Ad-MMP-9-treated and Ad-MMP-9 plus radiation-treated cells as compared to the controls and radiation alone treatment (Fig. 3A). The densitometric analysis also revealed an increase of Fas (~1.5 folds) and Fas-L (~1.8 folds) expression in the Ad-MMP-9 infected cells in combination with irradiation compared to the irradiation alone. We also looked downstream of this cascade. Western blot analysis revealed the cleavage of PARP-1 and caspases 10, 3 and 7 in Ad-MMP-9, radiation and combination treatments as compared to the control and Ad-CMV-treated cells (Fig. 3B). To confirm these initial observations, we analysed apoptosis by looking for DNA fragmentation (TUNEL) assay. TUNEL-positive, apoptotic MDA MB 231 cells were meagerly present in control or Ad-CMV-infected cells. Ad-MMP-9 infection in combination with irradiation resulted in a distinct increase of TUNEL-positive cells compared to the Ad-MMP-9 alone and irradiation alone treated cells (Figure 3C). Quantitation of TUNEL-positive cells indicated that Ad-MMP-9 infection in combination with irradiation (~85%) had 2 folds more compared to irradiation alone (~40%) (Figure 3D). A Clonogenic survival assay was performed to determine the influence of the downregulation of MMP-9 in breast tumor cell to ionizing radiation. Figure 3E indicates that downregulation of MMP-9 in MDA-MB-231 cells increased the radiosensitivity than uninfected and controls in terms of clonogenic survival. Statistical analysis shows that clonogenic survival in irradiated MDA-MB-231 cells that has reduced MMP-9 activity is significantly reduced compared with irradiated control MDA-MB-231 cells. These data confirm that the downregulation of active MMP-9 increase sensitivity to ionizing radiation.
Figure 3. Downregulation of MMP-9 and radiation induces apoptosis in breast cancer cells.

(A) Western blot results for Fas and Fas-L. Representation of densitometric analysis of the expression of of Fas and Fas-L (mean ± S.E.; n=4). (B) Western blot results for caspases 10, 3 and 7 show active caspase and PARP cleavage. (C) & (D) TUNEL assay was performed with MDA MB 231 cells with the earlier said treatments as per the manufacturer’s protocol and the densitometric analysis of percentage of TUNEL positive cells are represented (mean ± S.E.; n=3). *, Significant difference between irradiated alone cells and combination treatment of Ad-MMP-9 infection with irradiation (P ≤ 0.05). (E) Clonogenic survival was assessed between 15 d after radiation exposure. Each experiment was done at least four times and triplicates were done for each experiment. Clonogenic survival was assessed between 15 d after radiation exposure. Each experiment was done at least four times and triplicates were done for each experiment. Statistical analysis was done using PRISM and ANOVA. *, Significant difference between irradiated alone cells and combination treatment of Ad-MMP-9 infection with irradiation (P ≤ 0.01). (IR-Irradiation)
MMP-9 and HIF1α mediate the inhibition of tumor growth after treatment with Ad-MMP-9 plus radiation
MDA MB 231 cells were chosen for animal studies because these cells form aggressive primary tumors. We compared the effect of the three treatments (Ad-MMP-9, radiation and a combination) on tumor growth. Tumors that received radiation (2 doses of 5 gy) showed tumor regression of more than 50% when compared to the control and Ad-CMV-treated tumors. Analysis of tumor size revealed that tumors injected with 5×108 PFU of Ad-MMP-9 displayed a more delayed tumor growth than those of control mice. These tumors were suppressed by nearly 40–45% when compared to the controls. However, the combined treatment of Ad-MMP-9 and radiation completely regressed tumor growth in all of the mice by the end of the experiment (after 8 weeks) (Figs. 4A). Further, to show the efficacy of the combined treatment, we also performed another set of experiment where the animals were sacrificed after 15 days of the combined treatment.
Figure 4. MMP-9 and HIF1α mediate the inhibition of tumor growth after treatment with Ad-MMP-9 plus radiation.

(A) Representative tumors from animals treated with Ad-CMV, Ad-MMP-9, Irradiation (IR) and Ad-MMP-9 in combination with IR. Growth delay curve of tumors from animals treated with Ad-CMV, Ad-MMP-9, IR and Ad-MMP-9 in combination with IR. Tumor size was measured using calipers as described in Materials and Methods. Finally the tumor volume was calculated using the formula V = pi/6 (a × b × c). The data represents the mean ± SD (n=5) for each treatment at the said time point. (B) MMP-9 activity was analyzed using gelatin zymography by loading equal amounts of protein from the tissue lysates from the tumors treated with Ad-MMP-9, IR and controls. Additionally, the second panel in the zymogram shows MMP-9 activity in the tumor harvested after 15 days of the combination treatment in comparison to the control. MMP-9 activity is represented densitometrically. (C) H & E staining and immunohistochemical analysis for MMP-9 and HIF1α was performed on paraffin-embedded breast tumor sections of mice treated with Ad-CMV, Ad-MMP-9, IR and Ad-MMP-9 plus radiation using specific antibodies for these molecules. Paraffin-embedded breast tumor sections of mice treated with Ad-CMV, Ad-MMP-9, IR and Ad-MMP-9 plus radiation were immunostained for proteolytically cleaved Caspase-3 active subunits, which exist only when cells undergo apoptosis. TUNEL assay was performed with paraformaldehyde-fixed, paraffin-embedded breast tumor sections as per the manufacturer’s protocol.
Additionally, to show the efficiency of the treatments, we analyzed the activity of MMP-9 in the tumor tissues using zymography. The densitometric analysis showed that the irradiated tumors had a 2 to 3-fold increase in MMP-9 activity as compared to the controls. In contrast, Ad-MMP-9 treatment reduced activity by 2 to 2.5-fold than the control. MMP-9 activity was further reduced by 4 to 5-fold even after only 15 days of treatment with Ad-MMP-9 plus radiation (Fig. 4B). Further, we analyzed the tumor sections from control, Ad-CMV, Ad-MMP-9, irradiation and Ad-MMP-9 plus radiation treatment groups for MMP-9 and HIF1α using immunohistochemistry. We observed more apoptotic cells and necrotic areas in the sections from treated tumors as compared to the control tumors. The sections from control and Ad-CMV-treated tumors revealed a highly aggressive tumor nature and many mitotic dividing cells (Fig. 4C). We observed significant expression of MMP-9 and HIF1α in control, Ad-CMV and radiation-treated tumor sections. However, expression levels were drastically reduced in breast tumor sections of mice treated with Ad-MMP-9 and Ad-MMP-9 plus radiation. Following the in vitro studies for apoptosis, we analyzed the effect of these treatments in vivo. Paraffin-embedded breast tumor sections of mice treated with Ad-CMV, Ad-MMP-9, radiation and Ad-MMP-9 plus radiation were immunostained for proteolytically cleaved active subunits of caspase 3, which exist only when cells undergo apoptosis. The combination-treated tumors showed more active subunits of caspase-3 as compared to the controls and irradiation alone treated tumors, thereby suggesting more apoptosis in the combined treated tumors even at 15 days after treatment (Fig. 4C). In addition, TUNEL assay indicated DNA fragmentation, which is indicative of apoptotic cell population, in Ad-MMP-9-treated and irradiated tumors (Fig. 4C). In tumors treated with Ad-MMP-9 and radiation, we observed a significantly higher presence of DNA fragmentation as compared to the controls. This high level of apoptosis accounts for the 50–60% regression of tumors after treatment with Ad-MMP-9 plus radiation.
DISCUSSION
The goal of this study was to investigate the effect of combining MMP-9 inhibition along with radiation on orthotopic breast tumors. We found that Ad-MMP-9 treatment prior to radiation augmented the effects of radiation and successfully regressed tumors. In recent years, there has been a consensus that hypoxia can influence a broad spectrum of physiological and pathological cellular mechanisms (29). In particular, the combination of hypoxia gene therapy with ionizing radiation represents an exciting and promising approach to overcome and exploit resistant hypoxic tumor cells. Here, we also show that Ad-MMP-9 infection in irradiated cells decreased HIF 1, which is associated with reduction in sensitivity to radiation and causes disease failure after radiation therapy (30). We further show that Ad-MMP-9 infection augmented apoptosis in irradiated cells in vitro and in vivo. In our in vitro studies, we observed increases in HIF1α, both at the mRNA expression and protein levels, in control and irradiated cells. However, in cells treated with Ad-MMP-9 alone and Ad-MMP-9 plus radiation, levels of HIF1α were drastically reduced. Further, the immunohistochemical analysis of tumor tissue sections supported the in vitro results. HIF1 is a key transcription factor that regulates the expression of a variety of genes, which control glycolysis, erythropoiesis, apoptosis and angiogenesis (3). A direct correlation between tumor grade and HIF-1 expression in breast tumors has been shown (31). The role of HIF-1 in solid tumor growth is still not entirely clear, but previous work suggests that this transcription factor is necessary for the growth and angiogenesis of these tumors.
Radiation-induced MMP-9 leads to enhanced tumor growth and metastasis (32). Recent studies have implicated MMPs in multiple roles including tumor growth (33), regulation of apoptosis (34), and angiogenesis (35). Thus, the observed radiation-induced augmentation in MMP-9 activity is not only integral to tumor invasion, but may also aid survival in a relatively hostile setting. Our results show downregulation of MMP-9 in irradiated breast cancer cells decreased MMP activity at both the mRNA and protein levels.
Ionizing radiation acts through the induction of double strand breaks to DNA in order to induce elimination of cancerous cells via apoptosis (36). The efficiency of radiotherapy for cancer treatment is limited by toxic side effects, which impede dose escalation. Moreover, cancer cells often develop radioresistance mechanisms that are related to the DNA repair response. The aim of combining gene therapy and radiation is to strengthen the efficiency of radiation by inhibition of DNA repair, overcoming the clonogenic survival in irradiated cells and in turn apoptotic resistance. Transcription factors like NF-κ B and AP1 are activated by DNA damaging agents and could be involved in cell cycle arrest and prevention of apoptosis in order to allow DNA repair (9, 37). Not only does NF-κB promote survival of cancer cells, but it also contributes to abnormal proliferation and metastasis (25, 38–40). Our Western immunoblot analysis showed a decrease in the translocation of NF-κB subunits (p50 and p65) and AP1 subunits (c-fos and Jun D) in the cells that received the combined treatment of Ad-MMP-9 and radiation. Furthermore, we performed the electrophoretic mobility shift assay (EMSA) to analyze the protein-DNA interaction for NF-κB and AP1. The results indicated that in the case of cells treated with Ad-MMP-9 alone and Ad-MMP-9 combined with radiation, the protein-DNA interaction was reduced as compared to the control and Ad-CMV-treated cells. This observation was supported by the supershift assay using specific antibodies against NF-κB and AP1 respectively (41).
Apoptosis is induced by different stimuli, such as death ligands and chemotherapeutic drugs that lead to the activation of caspases (42). Recently, researchers have shown that of inhibition of NF-κB activity restores sensitivity to Fas-mediated apoptosis (43, 44). Treatment of breast cancer cells with Ad-MMP-9, radiation or both induced caspase-dependent apoptosis, which is associated with the activation of several individual caspases. Our results show that the caspase 10 pathway may be responsible for induction of apoptosis where Fas and Fas-L are involved at the cell membrane. The Fas/Fas ligand (Fas L) death pathway is an important mediator of apoptosis. Deregulation of the Fas pathway is reported to be involved in the immune escape of breast cancer and the resistance to anti-cancer drugs (45). It has been reported that the resistance of leukemic eosinophils to Fas-mediated apoptosis is due to induced NF-κB activation (46). The results of the present study corroborate the findings of these studies; we observed the reduction in NF-κB activation led to increased expression of Fas-L and directed apoptosis via the Fas-Fas-L mediated pathway. This led to the activation of caspase 10, a death effector domain (DED)-containing initiator caspase, which, in turn, cleaves or activates caspases 3 and 7 (effector caspases capable of cleaving PARP-1). Caspase 3 immunofluorescent staining and TUNEL assay revealed the increased apoptosis in the tumors treated with Ad-MMP-9 and radiation as compared to the control and irradiation alone treated tumors. The TUNEL assay using the tissue sections shows synergistic effect when Ad-MMP-9 was given in combination with radiation. Further, the clonogenic survival assay supported the results obtained involving activation of apoptotic cascade.
We suggest a new strategy for improving the radiosensitivity of breast tumors, in treating breast cancer through downregulation of MMP-9 using adenoviral constructs of antisense MMP-9 before radiation (Fig. 5). The tumors’ decreased MMP-9 activity inhibited phosphorylation of ERK, which reduced the transcriptional activity of NF-κB and AP1. This in turn led to increased apoptosis, thereby regressing the tumor. The precise and rapid propagation of this signaling cascade demands strict and flexible regulatory processes that still remain unexplored. The nature of the regulators involved may have therapeutic implications. Our schematic is based on our in vitro and in vivo model data showing an increase in apoptosis and tumor reduction.
Figure 5.
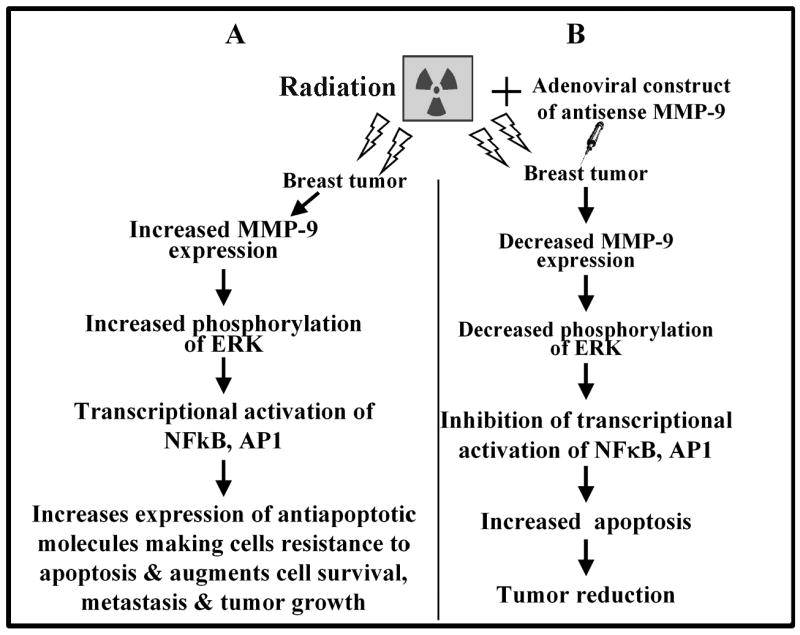
Schematic representations of the effect of irradiation (A) and Ad-MMP-9 in combination with irradiation (B) on breast tumor growth.
In summary, the present study indicates that MMP-9 might be a potential target candidate as an inducer of the HIF-regulated molecular cascade. Downregulating MMP-9 activity augments the effect of radiotherapy by directly or indirectly reducing HIF transcription machinery. Since HIF1α is involved in the pathogenesis of several human diseases (e.g., myocardial and cerebral ischemia, pulmonary hypertension), Ad-MMP-9 may have a future therapeutic role as an HIF1α activity inhibitor.
Acknowledgments
This research was supported by National Cancer Institute Grant CA 75557, CA 92393, CA 95058, CA 116708, N.I.N.D.S. NS47699 and NS57529, and Caterpillar, Inc., OSF Saint Francis, Inc., Peoria, IL (to J.S.R.).
We thank Shellee Abraham for preparing the manuscript, and Diana Meister and Sushma Jasti for reviewing the manuscript. We thank Noorjehan Ali for technical assistance.
Abbreviations used
- MMPs
matrix metalloproteinases
- Ad-MMP-9
adenoviral construct of antisense MMP-9 gene
- Ad-CMV
adenoviral construct of empty vector
- IR
irradiation
- HIF1α
hypoxia-inducible factor 1 alpha
- uPA
urokinase plasminogen activator
- uPAR
urokinase plasminogen activator receptor
- RT-PCR
reverse transcriptase polymerase chain reaction
- TIMP1
tissue inhibitor of metalloproteinase 1
- PARP 1
poly [ADP-ribose] polymerase
- FAS-L
FAS ligand
- EMSA
electromobility shift assay
- TUNEL assay
terminal dUTP nick-end labeling assay
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- PBS
phosphate-buffered saline
- FITC
fluorescein 5-isothiocyanate
Reference List
- 1.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–16. [PubMed] [Google Scholar]
- 2.Chakraborty G, Rangaswami H, Jain S, Kundu GC. Hypoxia regulates cross-talk between Syk and Lck leading to breast cancer progression and angiogenesis. J Biol Chem. 2006;281:11322–31. doi: 10.1074/jbc.M512546200. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8:588–94. doi: 10.1016/s0959-437x(98)80016-6. [DOI] [PubMed] [Google Scholar]
- 4.Unruh A, Ressel A, Mohamed HG, et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene. 2003;22:3213–20. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- 5.Rofstad EK. Microenvironment-induced cancer metastasis. Int J Radiat Biol. 2000;76:589–605. doi: 10.1080/095530000138259. [DOI] [PubMed] [Google Scholar]
- 6.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 7.Cummins EP, Taylor CT. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450:363–71. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 8.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–59. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 9.Russell JS, Raju U, Gumin GJ, et al. Inhibition of radiation-induced nuclear factor-kappaB activation by an anti-Ras single-chain antibody fragment: lack of involvement in radiosensitization. Cancer Res. 2002;62:2318–26. [PubMed] [Google Scholar]
- 10.Jung M, Dritschilo A. NF-kappa B signaling pathway as a target for human tumor radiosensitization. Semin Radiat Oncol. 2001;11:346–51. doi: 10.1053/srao.2001.26034. [DOI] [PubMed] [Google Scholar]
- 11.Smirnov AS, Ruzov AS, Budanov AV, Prokhortchouk AV, Ivanov AV, Prokhortchouk EB. High constitutive level of NF-kappaB is crucial for viability of adenocarcinoma cells. Cell Death Differ. 2001;8:621–30. doi: 10.1038/sj.cdd.4400853. [DOI] [PubMed] [Google Scholar]
- 12.Lindholm PF, Bub J, Kaul S, Shidham VB, Kajdacsy-Balla A. The role of constitutive NF-kappaB activity in PC-3 human prostate cancer cell invasive behavior. Clin Exp Metastasis. 2000;18:471–9. doi: 10.1023/a:1011845725394. [DOI] [PubMed] [Google Scholar]
- 13.Giri DK, Aggarwal BB. Constitutive activation of NF-kappaB causes resistance to apoptosis in human cutaneous T cell lymphoma HuT-78 cells. Autocrine role of tumor necrosis factor and reactive oxygen intermediates. J Biol Chem. 1998;273:14008–14. doi: 10.1074/jbc.273.22.14008. [DOI] [PubMed] [Google Scholar]
- 14.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–6. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs-Helber SM, Wickrema A, Birrer MJ, Sawyer ST. AP1 regulation of proliferation and initiation of apoptosis in erythropoietin-dependent erythroid cells. Mol Cell Biol. 1998;18:3699–707. doi: 10.1128/mcb.18.7.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blasi F. Proteolysis, cell adhesion, chemotaxis, and invasiveness are regulated by the uPA-u-PAR-PAI-1 system. Thromb Haemost. 1999;82:298–304. [PubMed] [Google Scholar]
- 17.Bradbury D, Clarke D, Seedhouse C, Corbett L, Stocks J, Knox A. Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J Biol Chem. 2005;280:29993–30000. doi: 10.1074/jbc.M414530200. [DOI] [PubMed] [Google Scholar]
- 18.Kumar A, Collins H, Van TJ, Scholefield JH, Watson SA. Effect of preoperative radiotherapy on matrilysin gene expression in rectal cancer. Eur J Cancer. 2002;38:505–10. doi: 10.1016/s0959-8049(01)00392-6. [DOI] [PubMed] [Google Scholar]
- 19.Stetler-Stevenson WG, Yu AE. Proteases in invasion: matrix metalloproteinases. Semin Cancer Biol. 2001;11:143–52. doi: 10.1006/scbi.2000.0365. [DOI] [PubMed] [Google Scholar]
- 20.Lakka SS, Rajan M, Gondi CS, et al. Adenovirus-mediated expression of antisense MMP-9 in glioma cells inhibits tumor growth and invasion. Oncogene. 2002;21:8011–9. doi: 10.1038/sj.onc.1205894. [DOI] [PubMed] [Google Scholar]
- 21.Bacqueville D, Deleris P, Mendre C, et al. Characterization of a G protein-activated phosphoinositide 3-kinase in vascular smooth muscle cell nuclei. J Biol Chem. 2001;276:22170–6. doi: 10.1074/jbc.M011572200. [DOI] [PubMed] [Google Scholar]
- 22.Lakka SS, Jasti SL, Kyritsis AP, et al. Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by the extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clin Exp Metastasis. 2000;18:245–52. doi: 10.1023/a:1006724826083. [DOI] [PubMed] [Google Scholar]
- 23.Raju U, Nakata E, Mason KA, Ang KK, Milas L. Flavopiridol, a cyclin-dependent kinase inhibitor, enhances radiosensitivity of ovarian carcinoma cells. Cancer Res. 2003;63:3263–7. [PubMed] [Google Scholar]
- 24.Moeller BJ, Dreher MR, Rabbani ZN, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8:99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 25.Magne N, Toillon RA, Bottero V, et al. NF-kappaB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett. 2006;231:158–68. doi: 10.1016/j.canlet.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 26.Poynter ME, Janssen-Heininger YM, Buder-Hoffmann S, Taatjes DJ, Mossman BT. Measurement of oxidant-induced signal transduction proteins using cell imaging. Free Radic Biol Med. 1999;27:1164–72. doi: 10.1016/s0891-5849(99)00202-6. [DOI] [PubMed] [Google Scholar]
- 27.Wang T, Hu YC, Dong S, et al. Co-activation of ERK, NF-kappaB, and GADD45beta in response to ionizing radiation. J Biol Chem. 2005;280:12593–601. doi: 10.1074/jbc.M410982200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Himelstein BP, Canete-Soler R, Bernhard EJ, Muschel RJ. Induction of fibroblast 92 kDa gelatinase/type IV collagenase expression by direct contact with metastatic tumor cells. J Cell Sci. 1994;107:477–86. doi: 10.1242/jcs.107.2.477. [DOI] [PubMed] [Google Scholar]
- 29.Shih SC, Claffey KP. Hypoxia-mediated regulation of gene expression in mammalian cells. Int J Exp Pathol. 1998;79:347–57. doi: 10.1046/j.1365-2613.1998.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison LB, Chadha M, Hill RJ, Hu K, Shasha D. Impact of tumor hypoxia and anemia on radiation therapy outcomes. Oncologist. 2002;7:492–508. doi: 10.1634/theoncologist.7-6-492. [DOI] [PubMed] [Google Scholar]
- 31.Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006;13:739–49. doi: 10.1677/erc.1.00728. [DOI] [PubMed] [Google Scholar]
- 32.Jadhav U, Mohanam S. Response of neuroblastoma cells to ionizing radiation: modulation of in vitro invasiveness and angiogenesis of human microvascular endothelial cells. Int J Oncol. 2006;29:1525–31. [PMC free article] [PubMed] [Google Scholar]
- 33.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 2000;103:481–90. doi: 10.1016/s0092-8674(00)00139-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–44. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martin DC, Sanchez-Sweatman OH, Ho AT, Inderdeo DS, Tsao MS, Khokha R. Transgenic TIMP-1 inhibits simian virus 40 T antigen-induced hepatocarcinogenesis by impairment of hepatocellular proliferation and tumor angiogenesis. Lab Invest. 1999;79:225–34. [PubMed] [Google Scholar]
- 36.Li L, Story M, Legerski RJ. Cellular responses to ionizing radiation damage. Int J Radiat Oncol Biol Phys. 2001;49:1157–62. doi: 10.1016/s0360-3016(00)01524-8. [DOI] [PubMed] [Google Scholar]
- 37.Takeuchi K, Motoda Y, Ito F. Role of transcription factor activator protein 1 (AP1) in epidermal growth factor-mediated protection against apoptosis induced by a DNA-damaging agent. FEBS J. 2006;273:3743–55. doi: 10.1111/j.1742-4658.2006.05377.x. [DOI] [PubMed] [Google Scholar]
- 38.Aggarwal BB, Shishodia S, Takada Y, et al. Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin Cancer Res. 2005;11:7490–8. doi: 10.1158/1078-0432.CCR-05-1192. [DOI] [PubMed] [Google Scholar]
- 39.Helbig G, Christopherson KW, Bhat-Nakshatri P, et al. NF-kappaB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J Biol Chem. 2003;278:21631–8. doi: 10.1074/jbc.M300609200. [DOI] [PubMed] [Google Scholar]
- 40.Shah N, Thomas T, Shirahata A, Sigal LH, Thomas TJ. Activation of nuclear factor kappaB by polyamines in breast cancer cells. Biochemistry. 1999;38:14763–74. doi: 10.1021/bi991291v. [DOI] [PubMed] [Google Scholar]
- 41.Ho E, Ames BN. Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFkappa B, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line. Proc Natl Acad Sci USA. 2002;99:16770–5. doi: 10.1073/pnas.222679399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–42. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 43.Meli M, D’Alessandro N, Tolomeo M, Rausa L, Notarbartolo M, Dusonchet L. NF-kappaB inhibition restores sensitivity to Fas-mediated apoptosis in lymphoma cell lines. Ann NY Acad Sci. 2003;1010:232–6. doi: 10.1196/annals.1299.041. [DOI] [PubMed] [Google Scholar]
- 44.Shimada K, Nakamura M, Ishida E, Kishi M, Matsuyoshi S, Konishi N. The molecular mechanism of sensitization to Fas-mediated apoptosis by 2-methoxyestradiol in PC3 prostate cancer cells. Mol Carcinog. 2004;39:1–9. doi: 10.1002/mc.10158. [DOI] [PubMed] [Google Scholar]
- 45.Kim R, Tanabe K, Emi M, Uchida Y, Toge T. Death receptor-dependent and -independent pathways in anticancer drug-induced apoptosis of breast cancer cells. Oncol Rep. 2003;10:1925–30. [PubMed] [Google Scholar]
- 46.Qin Y, Camoretti-Mercado B, Blokh L, Long CG, Ko FD, Hamann KJ. Fas resistance of leukemic eosinophils is due to activation of NF-kappa B by Fas ligation. J Immunol. 2002;169:3536–44. doi: 10.4049/jimmunol.169.7.3536. [DOI] [PubMed] [Google Scholar]