

Abstract
A unique feature of human alveolar macrophages is their prolonged survival in the face of a stressful environment. We have shown previously that the ERK MAP kinase is constitutively active in these cells and is important in prolonging cell survival. This study examines the role of the ERK pathway in maintaining mitochondrial energy production. The data demonstrate that ATP levels in alveolar macrophages depend on intact mitochondria and optimal functioning of the electron transport chain. Significant levels of MEK and ERK localize to the mitochondria and inhibition of ERK activity induces an early and profound depletion in cellular ATP coincident with a loss of mitochondrial transmembrane potential. The effect of ERK suppression on ATP levels was specific as it did not occur with PI3-kinase/Akt, p38 or JNK suppression. ERK inhibition led to cytosolic release of mitochondrial proteins and caspase activation. Both ERK inhibition and mitochondrial blockers induced loss of plasma membrane permeability and cell death. The cell death induced by ERK inhibition had hallmarks of both apoptotic (caspase activation) and necrotic (ATP loss) cell death. By blocking ERK-inhibition induced reactive oxygen species, caspase activation was prevented, though necrotic pathways continued to induce cell death. This suggests that mitochondrial dysfunction caused by ERK inhibition generates both apoptotic and necrotic cell death-inducing pathways. As a composite, these data demonstrate a novel mitochondrial role for ERK in maintaining mitochondrial membrane potential and ATP production in human alveolar macrophages.
Keywords: Human Macrophages, Lung, Kinases, Apoptosis
INTRODUCTION
Human alveolar macrophages survive for long periods in the lung (1). Survival occurs even in the face of exposure to chemical pollutants, reactive oxygen species, inflammatory mediators and infectious agents (2). This ability to adapt to stress is crucial to their survival. We have shown that human alveolar macrophages have high constitutive activity of two survival pathways, phosphatidylinositol 3 kinase (PI 3-kinase2)/Protein kinase B (Akt) and extracellular signal-regulated kinase (ERK) mitogen-activated protein (MAP) kinase (3, 4). The survival effects of Akt are well described and include inhibition of BH3 only pro-apoptotic proteins (5, 6), stabilization of anti-apoptotic x-linked inhibitor of apoptosis (XIAP) (7), inactivation of protease activity of HtrA2/Omi (8), inactivation of caspase 9 and inactivation of transcription factor, FoxOa (9, 10). The survival mechanisms of ERK activity are less well described and appear to be cell type and system specific. Though ERK has been linked to apoptosis in a few cases (11) in the majority of studies, ERK activity is linked to cell survival (3, 12-16).
ERK is a member of the MAP kinase family of serine/threonine kinases. It is activated by phosphorylation of a tyrosine X threonine motif by the upstream kinases, MAP kinase kinase-1 (MEK1) and 2, and in turn phosphorylates down-stream substrates containing a consensus proline directed motif (PX(S/T)P) (17, 18). Among known ERK substrates are a number of pro-apoptotic proteins that are inhibited by ERK phosphorylation (caspase 9 and BimEL) (19, 20). ERK has also been shown to contribute to cell survival via other mechanisms (13-15, 21-28). We have recently shown that one mechanism of ERK-dependent alveolar macrophage survival is positive regulation of protein translation via an effect on the eukaryotic translation initiation factor, eIF2α (4). It is likely that ERK maintains alveolar macrophage survival via a number of synergistic mechanisms.
ERK is a ubiquitously expressed kinase with more than one hundred and sixty substrates identified to date (29). ERK has been shown to phosphorylate transcription factors, other kinases, phosphatases, cytoskeletal and scaffold proteins, receptors and apoptosis-related proteins (29). These substrates are found all over the cell (nucleus, cytosol and organelles). Some recent papers have localized a subset of ERK to the mitochondria (30-33). The studies demonstrating mitochondrial ERK were performed in neuronal cells and cardiomyocytes. For the cardiomyocytes, Baines et al found that in the mitochondria, ERK complexes with protein kinase C ε (PKCε) and plays a role in protection from ischemia reperfusion (33). In neuronal cells, Alonso et al found that ERK1 and 2 are present in brain mitochondria at the outer membrane/intermembrane space (34). Zhu et al found active ERK associated with mitochondrial proteins (MnSOD for example) and used ultrastructure immuno-gold studies to demonstrate the presence of active ERK in mitochondria from midbrain sections of patients with Parkinson’s disease and diffuse Lewy body disease (35). These studies found ERK associated with outer membrane, intermembrane space and the matrix (31).
Mitochondria perform multiple functions in the cells (36, 37). In addition to generating energy by recycling ADP to ATP via oxidative phosphorylation, mitochondria break down sugars and long chain fatty acids, synthesize steroids and lipids, replicate, transcribe and translate proteins from mitochondrial DNA, produce reactive oxygen species and integrate survival/death signals (36, 37). ATP production relies on the electron transport chain (ETC) and ATP synthase. Movement of electrons down the ETC results in an electrochemical gradient (measured as mitochondrial membrane potential (mitΔψ)). The pumping of protons from the intermembrane space to the matrix through the ATP synthase unit generates ATP from ADP, providing energy for the cell (37). In this study, the role of ERK in mitochondrial ATP production was examined.
The study was performed in human alveolar macrophages obtained from normal volunteers. We found that inhibition of constitutive ERK activity (with dominant negative mutations of the upstream kinase, MEK, cell permeable inhibitory peptides and chemical inhibitors) led to a rapid loss of mitochondrial membrane potential (mitΔψ) and a decrease in ATP levels. These events were an early response to ERK inhibition and were followed at later time points by cytosolic release of mitochondrial proteins, caspase activation, rupture of the plasma membrane and cell death. The apoptotic/necrotic nature of the cell death was at least partially dependent on a requirement for reactive oxygen species for the caspase activation. These data suggest the existence of a previously unknown mitochondria localized pro-survival role for ERK MAP kinase in human alveolar macrophages.
MATERIALS AND METHODS
Materials
Chemicals including Antimycin A (#A8674), NAC (#A8199), and rotenone (#R8875) were obtained from Sigma Chemical Company, St Louis, MO. The MEK inhibitor UO126 (#662005), p38 inhibitor SB203580 (#559389), JNK inhibitor II (SP600125 (#420119)), ERK activation inhibitor peptide I (#328000), oligomycin (#495455) and CCCP (#215911) were obtained from Calbiochem. The dominant negative MEK adenoviral vector (mutated at serine217 and serine 221) was obtained from Cell Biolabs, San Diego, CA. Ethidium homodimer (#E1169) was obtained from Molecular Probes (Eugene, OR). JC-1 was obtained from Guava Technologies. Western blotting reagents include phosphatase inhibitor cocktail (#524625, Calbiochem), complete mini-tab protease inhibitors (#11836170001, Roche Diagnostics, Indianapolis, IN) and ECL (#RPN2106) and ECL Plus (#RPN2132) chemiluminescent detection reagents (Amersham, Piscataway, NJ). Acrylimide (#161-0158), buffers (#161-0798 and #161-0799), PVDF membranes (#162-0174) and Bradford protein assay reagent (#500-0006) were from Bio-Rad, Hercules, CA. Antibodies used in this study were obtained from a variety of sources. Phosphorylation-specific ERK antibody (#9101), Cytochrome c (#4280), cleaved caspase 7 (#9491), cleaved caspase 9 (#9509) and cleaved PARP (#9541) antibodies were obtained from Cell Signaling, Beverly MA. VDAC (#sc-8828), AIF (#sc-5586), a tubulin (#sc-5586) and horseradish peroxidase conjugated antibodies anti-rabbit (#sc-2004), anti-mouse (#sc-2005) and anti-goat (#sc-2020) were all obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Beta actin (A5316) antibody was obtained from Sigma (St. Louis, MO). Culture media used in experiments was serum free RPMI tissue culture media plus Glutamax (#61870-036) from Invitrogen (Piscataway, NJ). Kits utilized in this study included Cell Titer-Glo Luminescent Cell Viability Assay (#G7571) from Promega, Madison, WI and BD Oxygen biosensor System from BD Biosciences, Bedford, MA.
Isolation of human alveolar macrophages
Alveolar macrophages were obtained from normal non-smoking volunteers, as previously described (38). Briefly, normal volunteers with a lifetime non-smoking history, no acute or chronic illness and no current medications, underwent bronchoalveolar lavage. The cell pellet was washed twice in irrigation saline and suspended in complete RPMI medium with Glutamax (Invitrogen) and added gentamycin (80μg/ml)). Cells were cultured in special non-adherent plates from Costar (#3471, #3473) to minimize the effect of adherence on signaling. All experiments were carried out in serum free conditions. Differential cell counts were determined using a Wright-Giemsa stained cytocentrifuge preparation. All cell preparations had between 90-100% alveolar macrophages. This study was approved by the Committee for Investigations Involving Human Subjects at the University of Iowa.
Fibroblast and epithelial cell cultures
Primary human lung fibroblasts (C-12360, Promocell, Heidelberg, Germany), were cultured at 37°C and in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 1% sodium pyruvate, 1% L-glutamine, 40 μg/ml gentamicin and 25 μg/ml fungisone (complete medium). The complete medium was changed every 2 days and the cells were subcultured every 4 to 5 days. The normal fibroblasts were used between the 3rd and 8th passage. For primary tracheobronchial epithelial cells (hTBE’s), all protocols were approved by the University of Iowa Institutional Review Board. Human tracheobronchial epithelial cells were obtained as previously described (39). Epithelial cells were isolated from tracheal and bronchial mucosa by enzymatic dissociation and cultured in Laboratory of carcinogenesis (LHC)-8e medium on plates coated with collagen/albumin for study up to passage 10.
JC-1 stain
JC-1 is a cationic dye that accumulates in mitochondria with intact mitochondrial membrane potential (mitΔψ). Flourescence shifts within the mitochondria from green (~590 nm) to red (~525 nM). For these experiments, alveolar macrophages were seeded on two chamber Titertek microscope slides at one million cells per ml. Experimental groups were incubated with U0126 (20 uM), ERK inhibitory peptide (50 uM) or infected with dnMEK Ad vector (moi of 10 to 100). After various incubation times, live cells were stained with JC-1 (3 uM). The cells with stain were incubated for 30 minutes at 37° and then evaluated by fluorescence microscopy or, in some cases, by confocal microscopy (University of Iowa Microscopy Facility). Photomicrographs were obtained showing orange/red intact mitochondria or diffuse green staining in cells with depolarized mitochondria. For quantitation, alveolar macrophages were seeded in 96 well plates at 50,000 cells per well. After incubation with ERK inhibitors, the cells were stained with JC-1 (30 minutes at 37o) and then fluorescence read in a Tecan Safire Flourescent plate reader. Red aggregates were recorded with an excitation of 535 nM and an emission of 590 nM. Green monomers were red with an excitation of485 nM and an emission of 535 nM. Data is shown as arbitrary aggregate (red) flourescent units (average +/- standard error) of three separate experiments.
Whole cell protein isolation
Whole cell protein was obtained by lysing the cells on ice for 20 minutes in 200μl of lysis buffer (0.05M Tris pH 7.4, 0.15M NaCl, 1% NP-40, with added protease and phosphatase inhibitors: 1 protease minitab (Roche Biochemicals)/10 ml and 100ul 100X phosphatase inhibitor cocktail (Calbiochem)/10 ml. The lysates were sonicated for 20 seconds, kept at 4° for 30 minutes, spun at 15,000g for 10 minutes and the supernatant saved. Protein determinations were made using the Bradford Protein assay from Bio-Rad. Cell lysates were stored at −70° until use.
Mitochondria isolation
Following experimental incubations, alveolar macrophages were pelleted in ice cold conditions (all steps of the mitochondrial isolation were performed on ice or in refrigerated centrifuges). Cells were resuspended in cold homogenizing buffer with added protease inhibitors (0.25M sucrose, 0.2 mM EDTA and 10 mM Tris HCl) and homogenized 20-40 times with a cold dounce homogenizer. Unlysed cells and nuclei were removed by centrifuging the sample at 1000 × g (3000 rpm) in a refrigerated table top microfuge. The supernatant containing the mitochondria was centrifuged at 10,000 g (12,000 rpm) for ten minutes at 4° centigrade. The supernatant from this spin was removed and saved as the cytosolic portion. The pellet containing the mitochondria was resuspended in protein lysis buffer (see whole cell protein isolation) and sonicated for ten seconds (mitochondrial fraction). Debris from the mitochondrial fraction was removed with a five minute spin at 12,000 g (14,000 rpm). Protein measurements were performed using the Bradford Protein assay from Bio-Rad.
Western analysis
Western analysis for the presence of particular proteins or for phosphorylated forms of proteins was performed as previously described (40). Briefly, 30 μg of protein was mixed 1:1 with 2x sample buffer (20% glycerol, 4% SDS, 10% β-mercaptoethanol, 0.05% bromophenol blue and 1.25M Tris pH 6.8) and loaded onto a 10% or 12% SDS-PAGE gel and run at 110V for 2 hours. Cell proteins were transferred to PVDF membranes with a Bio-Rad semi-dry transfer system, according to the manufacturer’s instructions. Equal loading of the protein groups on the blots was evaluated using Ponceaus S (Sigma), a staining solution designed for staining total proteins on PVDF membranes. The PVDF was then blocked with 5% milk in TTBS (tris buffered saline with 0.1% Tween 20) for 1 hour, washed, and then incubated with the primary antibody at dilutions of 1:500 to 1:2000 overnight. The blots were washed x4 with TTBS and incubated for 1 hour with horseradish-peroxidase conjugated anti-IgG antibody (1:5000 to 1:20,000). Immunoreactive bands were developed using a chemiluminescent substrate, ECL Plus or ECL (Amersham). An autoradiograph was obtained, with exposure times of 10 seconds to 2 minutes. Protein levels were quantified using a FluorS scanner and Quantity One software for analysis (Bio-Rad). The data were analyzed and statistics performed using Graphpad software. Densitometry is expressed as fold increase (experimental value/control value).
Adenoviral vector infection
To infect alveolar macrophages with an adenoviral vector expressing with GFP or dominant negative MEK, cells were seeded onto 24 well plates (one million cells per well) in RPMI medium with no fetal calf serum. A suspension of vector (AdGFP or Ad dnMEK) (moi of 10) (10 million pfu’s) was mixed with RPMI (167 ul) and Viraductin Adenovirus Transduction Reagent (2 ul) (Cell Biolabs) and allowed to sit at room temperature for 10 minutes. The medium was removed from the alveolar macrophages and replaced with the transduction mixture (169 ul) and incubated at 37° for four hours. Without removing the virus an additional 400 ul of RPMI was added and the culture continued. GFP expression was checked at 16-18 hours and averaged 50-70%.
Cell survival analysis
For analysis of cell survival, alveolar macrophages were cultured in 6 well tissue culture plates with or without pathway inhibitors (ERK, U0126 at 20 uM or ERK activation inhibitor peptide I (50 uM) for the described times. Most cultures were performed in standard RPMI Glutamax media (11 mM glucose levels) with no added serum. Triplicate cultures were performed on all experiments. After the incubation period, the cells were stained with ethidium homodimer (Molecular Probes) at 8 uM and images obtained of both bright-field and fluorescence using a Leica DMRB microscope equipped with a Qimaging RETICA 1300 digital camera and imaging system. After obtaining images, the percentage of EthD-1 positive cells was determined. Quantification was by direct cell count. Two hundred cells were counted from a minimum of four different fields. In some cases, viability was assessed using trypan blue permeability. Cell samples were exposed to 20% trypan blue in media and the percentage of dead cells was calculated. At least 300 cells were counted for each sample from a minimum of six fields. In other cases, plasma membrane integrity was assessed by examining release of lactate dehydrogenase (LDH). This was done using a kit from Promega, Madison, WI (CytoTox-ONE) that measures LDH release via a coupled fluorescent assay. Experiments were performed according to manufacturer’s instructions.
ATP assay
ATP levels were monitored using CellTiter-Glo Luminescent Cell Viability Assay from Promega. Alveolar macrophages were cultured (1 million alveolar macrophages treated with U0126 (20 uM), ERK activation inhibitor peptide I (50 uM) or infected with the dnMEK adenovirus vector) from 6 hours or 24 hours in 96 well plates. ATP was measured by bringing the plate to room temperature and adding CellTiter-Glo directly to each well as directed by the manufacturer. The plate was mixed on an orbital shaker to induce cell lysis and the sample read on a chemiluminescence plate reader (Tecam Safire) (integration time of 1 second). Data is presented as mean +/- standard error of luminescent readings from three separate experiments.
Oxygen consumption
Oxygen consumption was measured using the BD Oxygen Biosensor system. The empty Biosensor plate was initially read at ~485 excitation and ~630 emission in a Tecan Safire II plate reader. Cells were seeded into the Biosensor plate at 50,000 per well. Cells were incubated for zero to 120 minutes in the presence of different ERK inhibitors and mitochondrial inhibitors (U0126 (20 uM), ERK inhibitory peptide (50 uM)), (CCCP (10 uM) and antimycin A (10 uM)). Incubation was in the Tecan Safire plate reader set at 37°. Fluorescent measurements were made every 15 minutes. Oxygen in the media quenches the fluorescent signal. Increased oxygen consumption results in decreased quenching and an increase in arbitrary fluorescent units. The data is shown as arbitrary fluorescent units over the time of the experiment (120 minutes).
GSH/GSSG Measurements
Cells were scraped into cold phosphate-buffered saline and centrifuged at 4 °C for 5 min at 400 × g to obtain cell pellets. The pellets were frozen at −80 °C. Pellets were thawed and homogenized in 50 mM potassium phosphate buffer, pH 7.8, containing 1.34 mM diethylenetriaminepentaacetic acid. Total glutathione content was determined by the method of Spitz (41). Reduced glutathione (GSH) and oxidized glutathione (GSSG) were distinguished by addition of 2 μl of a 1:1 mixture of 2-vinylpyridine and ethanol per 50 μl of sample followed by incubation for 1 h and assay as described previously by Griffith (42). All glutathione determinations were normalized to the protein content of whole homogenates using the method of Lowry et al. (43).
Transmission Electron Microscopy
Samples were fixed overnight with 2.5% glutaraldehyde in 0.1 M cacodylate buffer. Post fixation was carried out for 1 hour at room temperature with a buffered 1% osmium tetroxide solution reduced with 1.5% potassium ferrocyanide. Samples were en bloc stained with 2.5% uranyl acetate. Cells were then rinsed and dehydrated using gradually increasing concentrations of acetone to 100%. Infiltration of Spurr’s epoxy resin and acetone were carried out over several days to 100% resin and cured overnight in a 70°C oven. Sections of 100nm thickness were cut using an Ultracut E ultramicrotome (Reichert-Jung). Grids were then counterstained with 5% uranyl acetate for 12 minutes and Reynold’s lead citrate for 5 minutes. Samples were imaged using a Hitachi H-7000 transmission electron microscope.
Phagocytosis Assay
To evaluate bacterial phagocytosis by alveolar macrophages, cells were cultured in chamber slides (Lab Tek 4 chamber slides) for 2 hours with and without treatments and then exposed to GFP tagged e.coli. at a ratio of 25 bacteria per 1 cell. Cells and bacteria were incubated for a further 30 minutes. Non-phagocytosed cells were washed off by vigorously washing with PBS times 6. Images were obtained using an inverted fluorescent microscope (Zeiss) and then counts of bacteria per cell performed on random fields (fifty cells per group). In some cases, adherent but not phagocytosed bacteria were killed with gentamycin and then remaining bacteria quantified using bacterial plate counts. The data obtained from these studies was not different than those obtained using fluorescent analysis (data not shown).
RESULTS
Alveolar macrophages depend on mitochondria and the electron transport change for ATP production
Studies extending back almost a century have suggested that both macrophages and neutrophils depend on cytosolic glycolysis for the generation of ATP (44-47). This includes macrophages found at sites of inflammation or wound repair that often depend on anaerobic glycolysis for ATP production (44, 45, 48). To determine the source of ATP in human alveolar macrophages, we cultured newly isolated alveolar macrophages with and without a number of inhibitors of mitochondrial ATP production. Oligomycin is an inhibitor of the ATP synthase subunit (F(1)F(0)) (49). Rotenone inhibits complex I of the electron transport chain (leading to generation of reactive oxygen species (ROS)) (50, 51). CCCP is an uncoupler that disperses the proton gradient that drives ATP synthase without interfering directly with the ETC (52). Alveolar macrophages were treated with oligomycin (0.5 uM), rotenone (2.5 uM) and CCCP (10 uM). Combined ATP levels from both intracellular and extracellular sources were measured with a chemiluminescence reagent at various time points. ATP levels rapidly disappeared with all three exposures (Figure 1A). This data demonstrates that ATP levels in alveolar macrophages are maintained via an intact mitochondria and functioning oxidative phosphorylation system (ETC and ATP synthase). In Figure 1B, we show, using transmission electron microscopy, that at baseline alveolar macrophages have multiple mitochondria.
Figure 1.
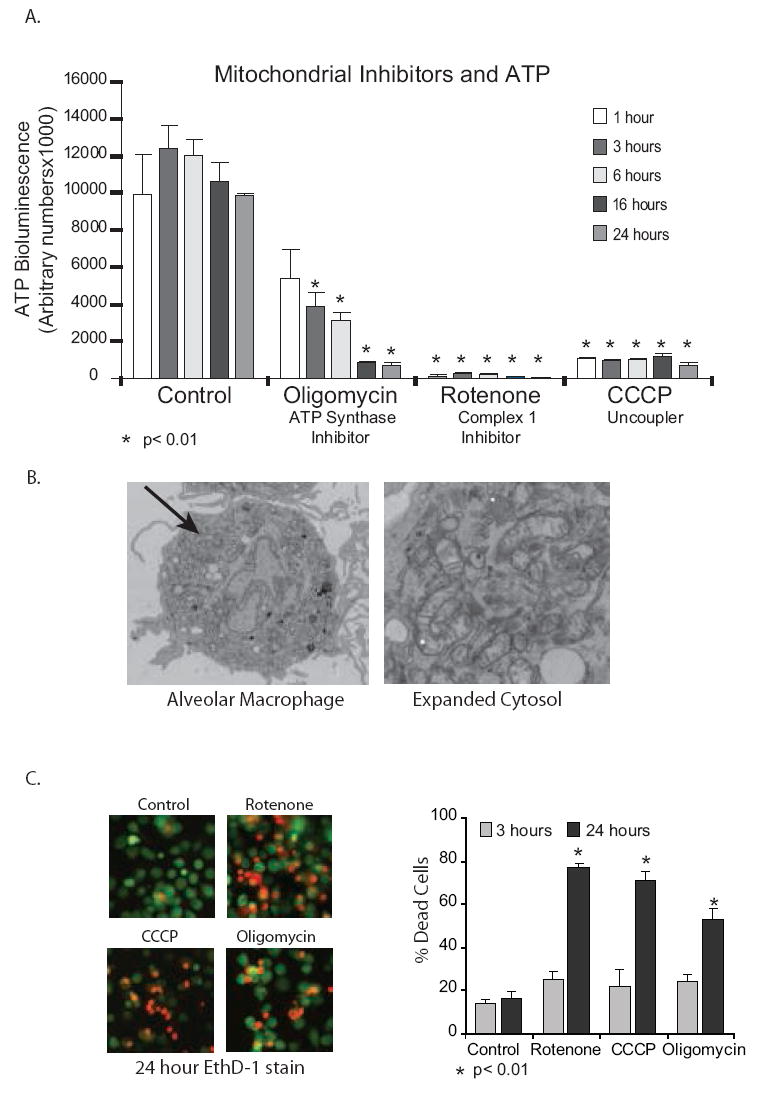
Interfering with mitochondrial function decreases ATP levels and survival of alveolar macrophages. 1A. Human alveolar macrophages (1×106/ml in 96 well tissue culture plates) were cultured with and without inhibitors of the mitochondrial ETC and ATP synthase for time periods between 1 and 24 hours. Cells were exposed to oligomycin (ATP synthase inhibition) (0.5 uM) or rotenone (complex I of the ETC inhibition) (2.5 uM) or CCCP (loss of mitΔψ) (10 uM). At the end of the incubation period, cells were lysed and ATP levels measured using chemiluminescence. Data is presented as arbitrary luminescent units and is a composite of three experiments. Significance at p< 0.01 was determined by analyzing experimental values compared to control values using a non-paired T test. 1B. Human Alveolar macrophages have large numbers of mitochondria. Human alveolar macrophages shortly after harvest with bronchoscopy were fixed overnight with 2.5% glutaraldehyde in 0.1 M cacodylate buffer. They were then processed for transmission electron microscopy (TEM) as described in the Methods section. The image is representative of fifty examined cells and is shown both as an entire cell and as a magnified portion of the cytosol. 1C. Alveolar macrophages were incubated with mitochondrial blockers (oligomycin (ATP synthase inhibition) (0.5 uM) or rotenone (complex I of the ETC inhibition) (2.5 uM) or CCCP (loss of mitΔψ) (10 uM)) for 24 hours. Cell viability was evaluated using ethidium homodimer stain (30 minute stain for plasma membrane permeability followed by fluorescence microscopy. Viability was also assayed using a fluorescent assay for LDH release. At the end of the 24 hour incubation, reagents from Promega’s CytoTox assay were added as per instructions and fluorescent changes monitored. % dead cells were calculated using saponin killed cells as a 100% dead control.
Disruption of ATP production leads to death of alveolar macrophages
Loss of ATP occurs under conditions of stress and if not reversed can lead to cell death. To investigate the effect of blocking the electron transport chain on alveolar macrophage survival, alveolar macrophages were treated with ETC blockers (rotenone (complex 1 inhibitor), CCCP (uncoupler) and oligomycin (ATP synthase inhibition) and cell viability evaluated using ethidium homodimer staining and trypan blue exclusion. Figure 1C demonstrates that loss of mitochondrial derived ATP leads to death of alveolar macrophages.
Alveolar macrophage mitochondria contain members of the ERK MAP kinase family at baseline
A few studies in neuronal cells have found ERK within the mitochondria, both in the intramembrane space and in the matrix. To search for ERK and the upstream kinase MEK in alveolar macrophage mitochondria, we isolated mitochondria from newly harvested alveolar macrophages and used Western analysis of mitochondrial protein. Figure 2A demonstrates partial mitochondrial localization of both MEK and ERK. VDAC and cytochrome C were also examined to control for the efficacy of the mitochondrial protein isolation. We next asked if the ERK present in alveolar macrophage mitochondria was active. Figure 2B shows that in alveolar macrophages from multiple donors (n=3), active ERK (phosphorylated on both threonine 183 and tyrosine 185) is found in proteins from the mitochondrial fraction. Probing the westerns for VDAC (mitochondrial fraction) and α tubulin (cytosolic fraction) was used to control for the protein fractionation. The studies performed in this project rely on a number of ERK inhibitors, a dominant negative adenoviral MEK construct, inhibitory peptides that block the binding of ERK to MEK and U0126, a specific chemical inhibitor of the activity of the upstream kinase MEK. To establish that we were really inhibiting the mitochondrial localized ERK, we treated alveolar macrophages with U0126 for one hour. Figure 2C demonstrates that by one hour of U0126 exposure both cytosolic and mitochondrial localized ERK activity was significantly depressed. As a composite, these data demonstrate that without inflammatory stimuli both ERK and active ERK are localized to the mitochondria and that MEK inhibition (U0126) decreases the mitochondrial-localized ERK activity. The next question was whether ERK activity played a role in production of ATP in alveolar macrophages.
Figure 2.
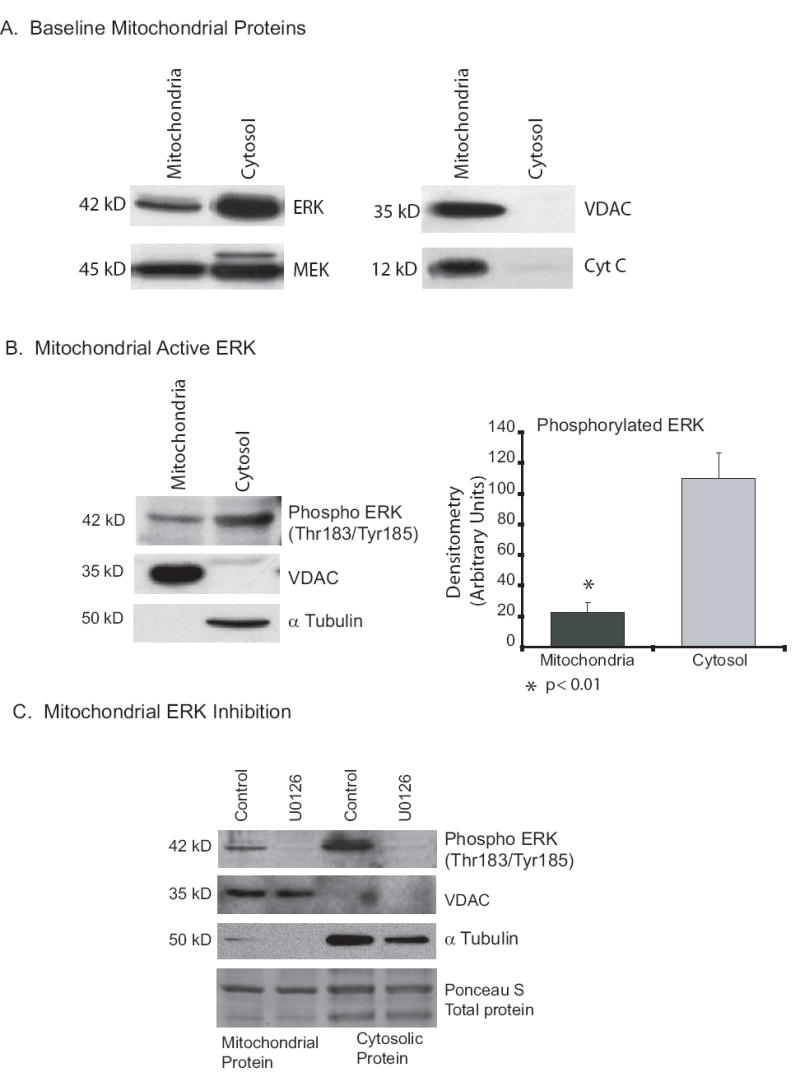
Protein isolations demonstrate mitochondrial localization of both ERK and MEK. 2A. Human alveolar macrophages have ERK and MEK protein in the mitochondria. Human alveolar macrophages collected by bronchoscopy were lysed and mitochondrial and cytosolic fractions isolated. Western analysis was performed for the ERK (ERK 2 is the majority ERK species in human alveolar macrophages) and MEK. Mitochondrial isolation was controlled by staining for the mitochondrial proteins VDAC and cytochrome C. 2B. Active ERK (phosphorylated on Thr185 and Tyr187) is present in alveolar macrophage mitochondria. On the left is Western analysis of mitochondrial and cytosolic proteins for active ERK, VDAC (mitochondria control) and a tubulin (cytosol control). On the right is composite densitometry from three samples. Significance at p< 0.01 was determined using nonpaired t test. The data demonstrates constitutive ERK activity in mitochondria from human alveolar macrophages. 2C. U0126 inhibits active ERK in alveolar macrophage mitochondria. Human alveolar macrophages were placed in culture with and without U0126 (20 uM) for one hour. Following incubation with U0126, mitochondrial proteins were isolated and Western analysis for active ERK performed. The Western blot was probed for active ERK (Phosphorylated on Thr183/Tyr185), VDAC (mitochondrial marker), a tubulin (cytosolic marker) and Ponceau S for total protein levels. This is representative of three separate experiments.
ERK inhibition depletes ATP from alveolar macrophages
To study the effect of ERK inhibition on alveolar macrophage ATP levels, alveolar macrophages were cultured in media without serum at a million cells per ml in 96 well tissue culture plates. First, we infected alveolar macrophages with an adenovirus vector expressing dominant negative MEK (Ad dnMEK). MEK is activated by phosphorylation of dual serines (217 and 221). In the dominant negative mutant both serines were mutated to alanine (A217/A221). The control cells were infected with an adenovirus vector expressing green fluorescent protein (eGFP). Ad dnMEK decreased ATP in alveolar macrophages at six hours (Figure 3A). Next we examined two other methods of ERK inhibition at multiple time points. We used a membrane permeable ERK inhibitory peptide (Ste-MEK113;Ste-MPKKKPTPI QLNP-NH2) that binds to ERK and prevents binding to MEK. The ERK inhibitory peptide decreased alveolar macrophage ATP in a time dependent manner (Figure 3B). Of note, by three hours after treatment of cells with the inhibitory peptide, there was a significant decrease in ATP levels. ERK was also inhibited with U0126 and total ATP levels in the cell cultures measured at various time points. Figure 3C demonstrates that inhibition of ERK depletes alveolar macrophage ATP levels in a time dependent manner. As a composite, these data demonstrate that inhibition of ERK rapidly depletes alveolar macrophage ATP in a time dependent manner.
Figure 3.
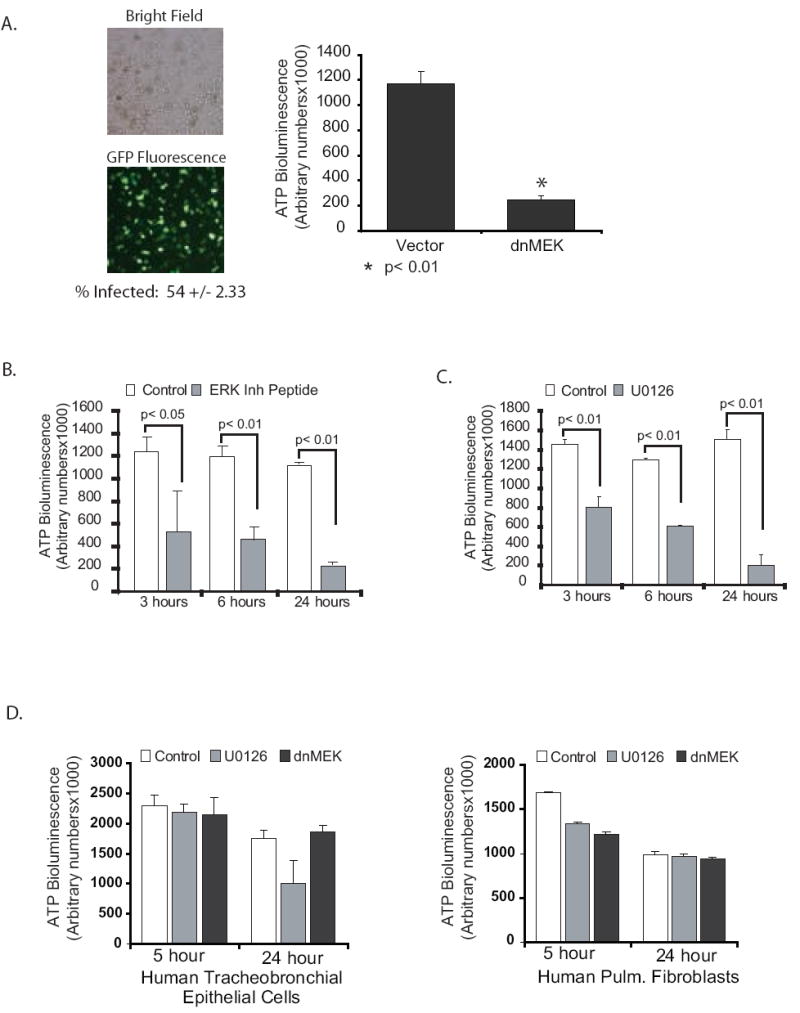
ERK inhibition decreases ATP specifically in alveolar macrophages. 3A. Human alveolar macrophages were cultured (1×106/ml in 96 well tissue culture plates). At time 0 the cells were infected with Ad dnMEK as described in the methods. Control cells were infected with an adenovirus vector expressing GFP. At 6 hours post infection, cells were lysed and total ATP levels analyzed as described. The data represents three separate experiments. Significance was determined using a nonpaired t test. 3B. Human alveolar macrophages were cultured (1×106/ml in 96 well tissue culture plates) with and without a peptide ERK inhibitor (50 uM). At 3, 6 and 24 hours post inhibition, cells were lysed and total ATP levels analyzed as described. The data represents three separate experiments. Significance was determined using nonpaired t tests. 3C. Human alveolar macrophages were cultured (1×106/ml in 96 well tissue culture plates) with and without the MEK inhibitor, U0126 (20 uM). At 3, 6 and 24 hours post inhibition, cells were lysed and total ATP levels analyzed as described. The data represents three separate experiments. Significance was determined using nonpaired t tests. 3D. Primary human tracheobronchial cells and primary human fibroblasts were grown in 96 well plates and then exposed to the ERK/MEK inhibitor, U0126 (20 uM) for various times. ATP was measured as described and is presented as arbitrary bioluminescence numbers. The data represents three experiments.
To determine if the mitochondrial effect of ERK inhibition was unique to alveolar macrophages or also occurred in other cell types, we investigated primary fibroblasts and airway epithelial cells. Both cell types were cultured in 96 well tissue culture plates at 80% confluence. They were treated with and without U0126 or infected with the dominant negative MEK vector as described in the methods. 5 and 24 hours later, ATP levels were measured. Figure 3D demonstrates that there was a minor decrease in ATP levels with the U compound at 24 hours in the hTBE’s, but no change in the ATP levels of the primary lung fibroblasts. This data suggests that the significant ATP depletion with ERK inhibition may be a unique characteristic of alveolar macrophages. A definitive answer would require a more comprehensive survey of other cell types.
ATP depletion is specific to ERK inhibition
Other MAP kinases (p38 and JNK) and the PI 3-kinase effector, Akt, have also been localized to the mitochondria (53-56). To see if the rapid decrease in ATP with inhibition was specific to ERK, alveolar macrophages were treated with inhibitors for JNK (SP600125, 20 uM), p38 (SB203580, 10 uM) and phosphatidylinositol 3-kinase/Akt (LY294002, 20 uM). Figure 4 demonstrates decreased ATP levels after ERK inhibition but not after inhibition of p38, JNK or phosphatidylinositol 3-kinase/Akt.
Figure 4.
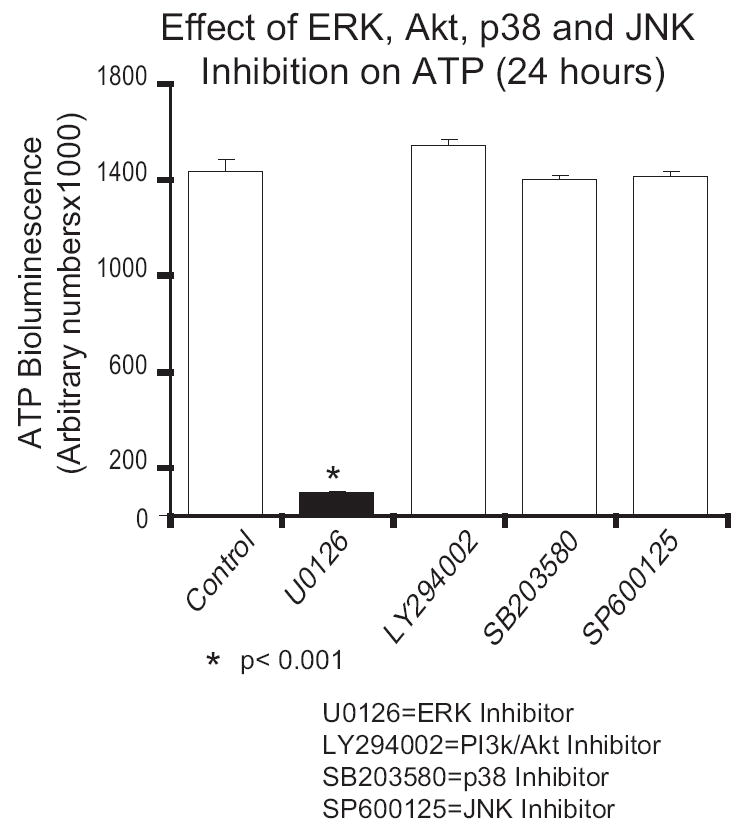
Inhibition of PI 3-kinase and other MAP kinases does not decrease ATP in human alveolar macrophages. Human alveolar macrophages were cultured (1×106/ml in 96 well tissue culture plates) with and without various inhibitors: MEK inhibitor U0126 (20 uM), PI 3-kinase inhibitor LY294002 (20 uM), p38 inhibitor SB203580 (10 uM) and JNK inhibitor SP600125 (20 uM). Cells were cultured for 24 hours and then lysed and ATP levels determined as described. The data represents three separate experiments. Significance was determined using a nonpaired t test.
ERK inhibition results in a rapid loss of mitochondrial membrane potential (mitΔψ) in human alveolar macrophages
As electrons move down the electron transport chain (ETC), positive hydrogen atoms are pumped from the matrix into the intermitochondrial space. This creates a concentration gradient between the two spaces. It is the flow of protons back into the matrix through the ATP synthase unit that drives ATP production. Our initial ATP depletion data suggested that ERK inhibition affects ATP production as early as one hour post exposure. We next asked if changes in mitΔψ happened within the same time frame. MitΔψ can be measured using the cationic dye, JC-1. In intact mitochondria with a healthy mitΔψ, JC-1 aggregates in the mitochondria and fluoresces red (~590nm). With loss of membrane potential, the dye disaggregates, is seen in the cytosol and fluoresces green (~525). We stained alveolar macrophages after exposure to U0126 for various times (as with the ATP assays, control cells were treated with equal amounts of DMSO). JC-1 staining was assessed using fluorescent microscopy and quantified using a 96 well plate format and fluorescent plate reader. Figure 5A (fluorescent images) shows that in the U0126 treated samples, there was a significant (though not complete) decrease in red staining and increase in green staining by one hour. By six hours most of the red punctate staining was gone and by twenty-four hours it was completely gone. As a positive control for mitochondrial dysfunction, staurosporine was used. Staurosporine rapidly converted punctate red staining to a diffuse green stain (3 hours). The visual data obtained with fluorescent microscopy was confirmed with a 96 well quantitative assay (Figure 5B). These data show that inhibition of ERK causes a rapid decrease in mitΔψ in alveolar macrophages.
Figure 5.
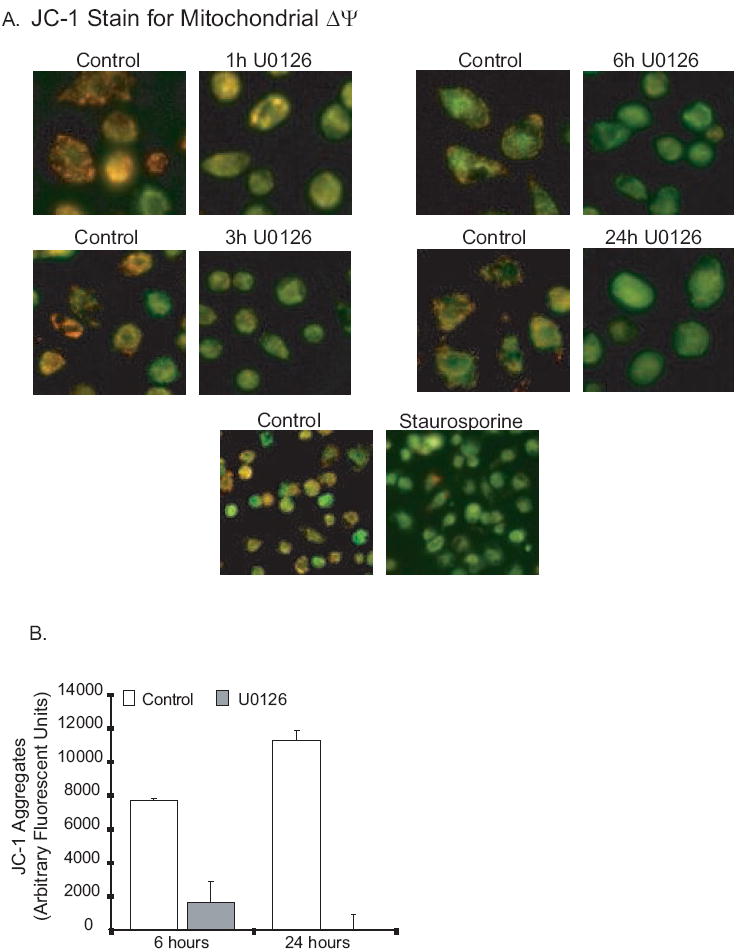
ERK inhibition causes loss of mitochondrial membrane potential (mitΔψ) in human alveolar macrophages. 5A. Human alveolar macrophages were cultured (1×106/ml in 6 well tissue culture plates or in 2 chamber microscope slides) with and without U0126 (20 uM) or staurosporine (1 uM). At the end of the incubation period, the mitochondrial stain JC-1 was added as described in the methods. Cultures were incubated a further 30 minutes and then examined with fluorescence microscopy. Photomicrographs were obtained. Red/orange stain denotes intact mitochondria with no disruption of membrane potential. Green staining denotes loss of mitΔψ. 5B. Identical experiments were performed in 96 well tissue culture plates and red/orange stain quantified using a fluorescence plate reader. The graph represents arbitrary units at an excitation of 535 nM and an emission of 590 nM.
ERK inhibition decreases oxygen consumption by human alveolar macrophages
Alterations in mitΔψ and electron transport chain function can alter rates of oxygen consumption. Uncouplers (that disperse the proton gradient without blocking ETC function) increase rates of oxygen consumption. Blockers of complex I or III in the electron transport chain decrease oxygen consumption by blocking the free flow of electrons through the electron transport chain. We measured oxygen consumption using a BD Oxygen Biosensor System. To examine the effect of ERK inhibition, alveolar macrophages were treated with nothing (control 1), DMSO to match the amount in the U0126 preparation (control 2), the ERK inhibitory peptide, U0126 and the mitochondrial targeted inhibitors, CCCP (uncoupler) and antimycin A (complex III blocker). 11 mM sulfite was used as a positive control and plain media was used as a negative control. The entire plate was pre-blanked and then alveolar macrophages with pre-added inhibitors were seeded into the Biosensor plate. The plate was inserted into a 37° fluorescence plate reader and readings taken every fifteen minutes for two hours. The data is presented as arbitrary fluorescent units. Figure 6 demonstrates that alveolar macrophage oxygen consumption is blocked by both the ERK inhibitory peptide and U0126. This is consistent with a block in the ETC. As expected, the complex III inhibitor, antimycin A blocked oxygen consumption and the uncoupler, CCCP, increased oxygen consumption. This data, combined with the early disruption of mitΔψ suggests that the role of ERK in mitochondrial homeostasis is consistent with disruption of the ETC.
Figure 6.
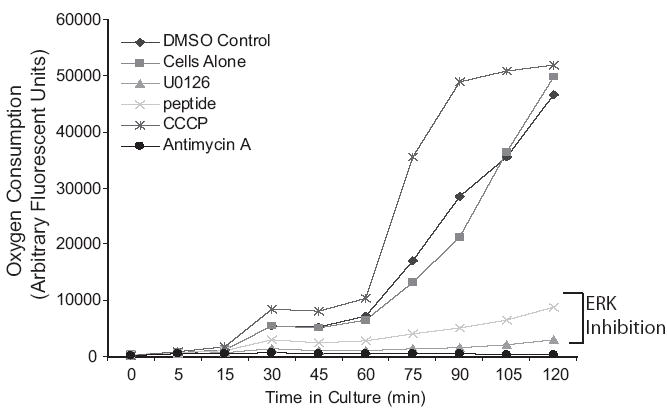
ERK inhibition decreases oxygen consumption in human alveolar macrophages. Human alveolar macrophages were cultured (1×106/ml in 96 well BD Oxygen Biosensor plate) with and without the MEK inhibitor, U0126 (20 uM), an ERK inhibitory peptide (50 uM), CCCP (uncoupler, 10 uM) or antimycin A (complex III inhibitor, 10 uM). Fluorescent readings were taken every 15 minutes. The data is shown as arbitrary fluorescent units. Increased fluorescence denotes increased oxygen consumption (see Methods section).
ERK inhibition induces macrophage death
In order to assess the effect of ERK inhibition on cell viability, a number of markers of cell death were examined. Alveolar macrophages were first examined at 3, 6 and 24 hours after ERK inhibition for disruption of the plasma membrane. Figure 7A demonstrates that cell death, as quantified by a leaky plasma membrane (ethidium homodimer staining) occurs by 24 hours following ERK inhibition. In the next set of experiments, we examined markers of necrosis (AIF release from the mitochondria and disruption of the plasma membrane) and apoptosis (caspases and cytochrome C release from the mitochondria). AIF has been identified as a key player in caspase independent cell death (57). AIF is a biphasic NADH oxidase involved in both mitochondrial respiration and in cell death. Recently Moubarak et al have described AIF as an inducer of programmed necrosis and a part of a novel signaling pathway that distinguishes necrosis from apoptosis (57). Figure 7B demonstrates that there are markers of necrosis induced by ERK inhibition. The Western blot demonstrates loss of AIF from the mitochondria and the electron photomicrograph demonstrates disruption of the plasma membrane after exposure to the ERK inhibitory peptide. We then examined the effect of ERK inhibition on cytochrome c release, caspase activation and PARP inactivation, all of which are linked to apoptosis (Figure 7C). In the same time frame as the loss of mitochondrial contents to the cytosol (twenty-four hours), we found activation of caspases (3, 7 and 9) and cleavage of PARP (Figure 7C). The time frame of both necrosis (AIF release) and apoptosis (caspase activation) markers suggests that ATP loss and loss of mitΔψ precedes opening of the mitochondrial outer membrane pore (MOMP) rather than the other way around.
Figure 7.
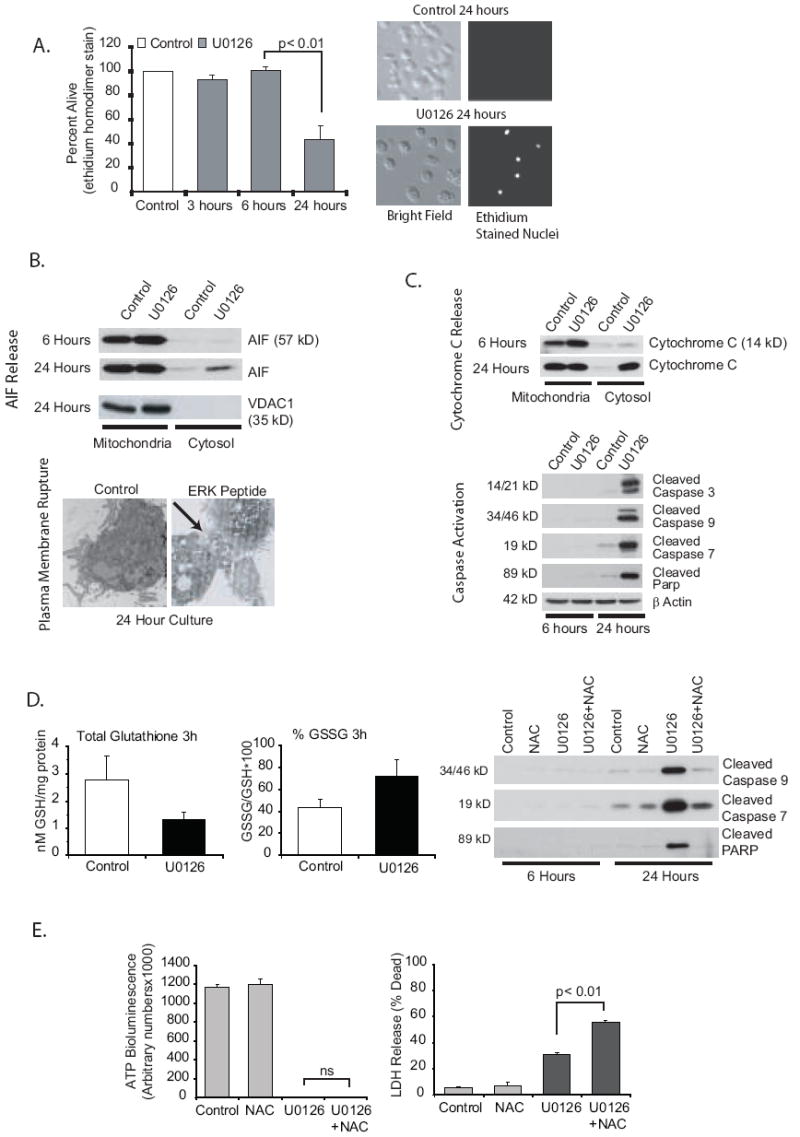
ERK inhibition induces both apoptotic and necrotic pathways. 7A. ERK inhibition leads to alveolar macrophage death. Human alveolar macrophages were cultured (1×106/ml in 6 well tissue culture plates) with or without U0126 (20 uM) for 3, 6 or 24 hours. At the end of each time point, EthD-1 entry was evaluated. Dead cells were counted by examining each cell for red nuclear staining. A total of 300 cells were examined for each data point. The data is graphed as percentage of cells that exclude EthD-1. The graph represents data from three separate experiments. Significance was determined using a nonpaired t test. 7B. ERK inhibition results activation of necrotic pathways. Alveolar macrophages were cultured for 6 or 24 hours (1×106/ml in 6 well tissue culture plates) with or without U0126 (MEK inhibitor, 20 uM). The blots show mitochondrial and cytosolic protein fractions analyzed (Western analysis) for AIF. The 24 hour blot was stained with VDAC as a control for the mitochondrial isolation. Also shown is TEM demonstrating loss of plasma membrane integrity after 24 hours with an ERK inhibitory peptide (50 uM). Cells were fixed overnight with 2.5% glutaraldehyde in 0.1 M cacodylate buffer. They were then processed for TEM as described in the Methods section. 7C. ERK inhibition activates apoptotic pathways. Alveolar macrophages were cultured for 6 or 24 hours (1×106/ml in 6 well tissue culture plates) with or without U0126 (MEK inhibitor, 20 uM). The first blot shows mitochondrial and cytosolic protein fractions analyzed (Western analysis) for cytochrome C. The other blots show total cell lysates stained for cleaved caspase 3, 7 and 9 and cleaved PARP. Equal loading was determined by staining identical blots for β actin. 7D. ERK inhibition increases ROS in alveolar macrophages. Alveolar macrophages were cultured for 3 hours (1×106/ml in 6 well tissue culture plates) with or without U0126 (MEK inhibitor, 20 uM). Frozen cell pellets were used to measure GSH (non-oxidized glutathione) and GSSG (oxidized glutathione) levels. Data is expressed as either total GSH or percentage GSSG (GSSG/GSH × 100). In addition the effect of blocking oxidant increases with NAC (1 mM) on caspase activity was evaluated by Western analysis of whole cell lysates at 6 and 24 hours. 7E. Inhibition of caspase activity does not prevent ERK inhibition-induced loss of ATP and decreased survival. Human alveolar macrophages were cultured (1×105/100 ul in 96 well tissue culture plates) with or without U0126 (20 uM) for 24 hours. At the end of the incubation time ATP levels were measured as described in the methods and LDH release (as described in the methods) was measured as a marker of plasma membrane permeability. NAC did not prevent either the loss of ATP or lass of plasma membrane integrity by ERK inhibition.
To pursue the relative role of the apoptotic pathway in cell death after ERK inhibition, we asked if preventing caspase activation would block the ERK inhibition induced cell death. If that were true it would suggest that this is primarily an apoptotic process that with time results in disruption of the plasma membrane. We found that ERK inhibition increased reactive oxygen species (ROS) in the alveolar macrophages as measured by alterations in glutathione/oxidized glutathione ratios. Non-oxidant stressed cells have a surplus of GSH compared to the oxidized GSSG. A decrease in the ratio is indicative of increased ROS in the cells (58, 59). Treating alveolar macrophages with U0126 for three hours induced a significant decrease in glutathione levels (control: 2.77+/-0.87 compared to U0126: 1.30=+/-0.30) and an increase in the percent of GSSG compared to GSH (Figure 7D). When the increase in ROS was prevented with exogenous N-acetylcysteine (NAC), caspase activation by ERK inhibition was completely prevented (Figure 7D). However, the lack of caspase activity did not protect the cells from ATP loss or plasma membrane rupture (as measured by LDH release) (Figure 7E). We conclude that ERK inhibition induces two cell death pathways, one dependent on ROS and caspase activity and one dependent on ATP depletion and plasma membrane disruption. The balance between the two will be at least partially determined by the anti-oxidant status of alveolar macrophages. It is possible that individual variability in anti-oxidant status among alveolar macrophage donors can alter the balance between apoptotic and necrotic outcomes of ERK inhibition in alveolar macrophages.
ERK inhibition decreases alveolar macrophage phagocytosis of GFP e.coli
To evaluate the role of ERK in the innate immune function of alveolar macrophages, we evaluated the effect of ERK inhibition on phagocytosis. Phagocytosis is a high energy utilizing process that could be impacted by the loss of ATP found with ERK inhibition. Using a GFP tagged e.coli., cells were treated with U0126 for 2 hours (partial ATP depletion without the cell death that comes closer to 24 hours after ERK inhibition) and then exposed to GFP-e.coli. for 30 minutes. After washing off non-phagocytosed cells, fluorescent images were acquired and bacteria/cell counts obtained. The data shown in Figure 8 demonstrates that ERK inhibition decreases bacterial uptake by alveolar macrophages. As well as this newly described link between ERK and mitochondria, the ERK MAP kinase has many other described biological effects. It is possible that the decrease in phagocytosis with ERK inhibition may be only a partial consequence of the loss of ATP. Other ERK dependent processes may also play a role. However, considering the high energy needs of the phagocytic process, we believe that the ERK related mitochondrial dysfunction plays an important role. The phagocytosis data suggests that the role of ERK in mitochondrial homeostasis may play an important part in the immune function of alveolar macrophages.
Figure 8.
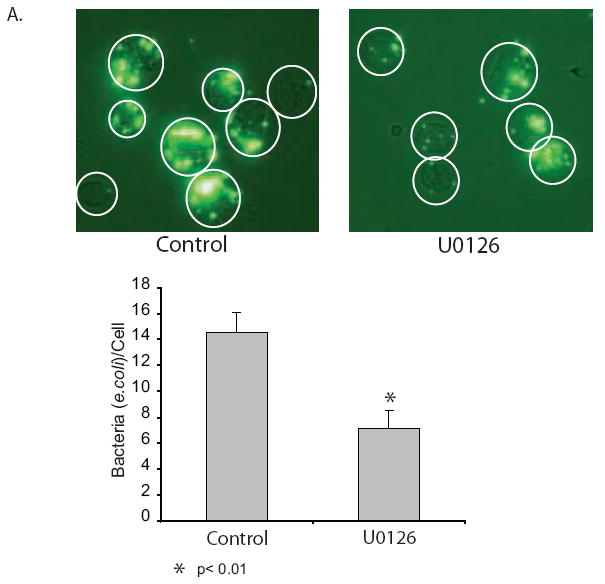
ERK inhibition decreases bacterial phagocytosis by alveolar macrophages. A half million alveolar macrophages/ml were seeded unto chamber slides and allowed to adhere for two hours. The cells were exposed to U0126 (20 uM) or nothing for two hours and then opsonized GFP-e.coli. (25/cells) was added to each well. Following a 30 minute incubation the slides were washed X6 with PBS and adherent or phagocytosed bacteria were evaluated using fluorescent photomicrography or bacteria per cell was counted (50 cells per group).
DISCUSSION
The MAP kinase, ERK, has many described functions. The majority of these functions are linked to increases in ERK activity subsequent to a stimulus (such as lipopolysaccharide or increased oxidants). We have been interested in the role of both stimulated and baselines levels of ERK in alveolar macrophage homeostasis. Earlier studies by our group identified significant levels of baseline ERK activity in human alveolar macrophages (3, 4). This ERK activity was part of a significant pro-survival pathway. In one study, we linked upstream regulation of baseline ERK activity to novel sphingolipid pathways (3). In a second study, we linked cytosolic ERK activity to translation regulation via an effect on eIF2α (regulator of translation initiation) (4). In this study, we identify ERK and its upstream kinase, MEK1/2 as mitochondrial localized proteins in human alveolar macrophages. We found that ATP levels in alveolar macrophages depended on mitochondrial respiration. Inhibition of ERK decreased ATP levels as early as one hour post-inhibition with significant depletion by three hours. The ATP depletion was not due to an effect on cytosolic glycolysis as it could not be reversed by exogenous addition of pyruvate (data not shown). ERK inhibition caused a rapid decrease in mitΔψ and oxygen consumption, suggesting a role for ERK in ETC function.
By twenty-four hours of ERK inhibition, there was substantial caspase activity and loss of plasma membrane integrity. The effects of ERK inhibition on survival, as demonstrated by this study, can be divided into two phases: 1. Early (loss of ATP and mitΔψ) and 2. Late (permeability of the mitochondrial outer membrane, caspase activation and loss of plasma membrane integrity). We found that both necrotic and apoptotic events were induced by ERK inhibition and that blocking caspase activation did not protect the cells from the necrotic component of the response. We conclude that in alveolar macrophages the loss of constitutive ERK activity results in cell death due to a combination of loss of ATP, release of mitochondrial proteins, activation of caspases and loss of plasma membrane integrity (Figure 9).
Figure 9.
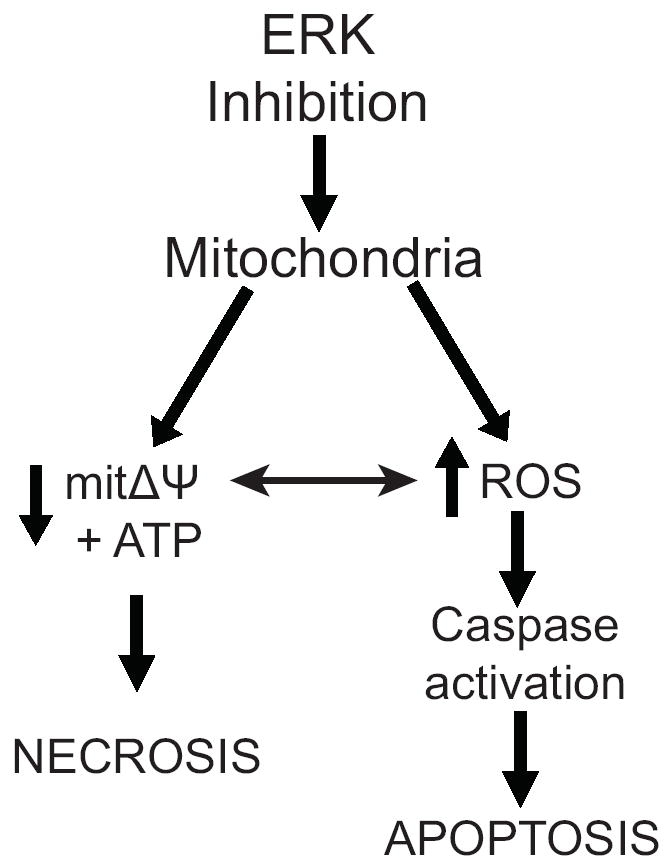
Diagram of the study findings. In human alveolar macrophages a decrease in ERK activity leads to loss of ATP and mitochondrial membrane potential. This results in release of mitochondrial proteins to the cytosol, caspase activation and cell death. Blocking caspase activation by preventing an increase in ROS does not prevent cell death.
Our cultures are carried out in serum free and non-adherent conditions to most closely mimic the alveolar space. We have performed some of these same experiments in adherent conditions and found that the ATP decrease was similar. What is increased by adherence is the inflammation-induced activation of ERK (i.e. by LPS). We don’t know at this time whether the upstream signals that regulate constitutive ERK activity are the same as the upstream signals that activate ERK during an inflammatory response. We are interested in addressing this question.
Reversible phosphorylation as a means of regulating mitochondrial function is an emerging topic of investigation. It is beyond the scope of this paper to identify the mitochondrial site of ERK phosphorylation, however, there are a number of candidates. One known site of regulatory phosphorylation is the pyruvate dehydrogenase complex (PDC) in the mitochondrial matrix. The PDC catalyzes the conversion of pyruvate to acetyl coenzyme A and is made up of a number of subunits. Searching the PDC subunits for potential proline-directed serine or threonines using Scansite (http://scansite.mit.edu/), we found that the E2 subunit has a possible ERK phosphorylation site at threonine 322. Another known site of regulatory phosphorylation is the ATP synthase unit. Two subunits of the F1 catalytic subunit of ATP synthase, delta and beta, also have possible proline-directed sites. ATP synthase delta (ATP5D) has two potential sites, threonine 280 and threonine 290. In addition there is an ERK binding domain in the protein. ATP synthase beta (ATP5B) also contains two potential ERK phosphorylation sites at threonine 641 and 651and a potential ERK docking site. While inhibition of ATP synthase would generate the drop in ATP seen in our data, the block in oxygen consumption with ERK inhibition argues against this as a site of ERK regulation in alveolar macrophages.
It is of note that the only paper to link ERK activity to ATP depletion is a study examining ATP synthase activity. The study by Yung et al examines the effect of ERK inhibition in astrocytes. They found that ERK inhibition induced cell death via a decrease in F1F0 ATP synthase activity. Their data did not find a direct link between ERK and one of the ATP synthase subunits (60) and they concluded only that ERK inhibition might modulate ATP synthase function. These are only a few of many protein possibilities for the mitochondrial ERK target. The human mitochondrial Database (http://bioinfo.nist.gov/hmpf/index.html) lists 1465 mitochondrial proteins, including both nuclear and mitochondrial encoded proteins. We are at this time examining the data base for possible novel ERK substrates that would explain the findings of this study. It is also possible that ERK does not alter ATP production by directly phosphorylating a mitochondrial protein but rather by phosphorylation of a regulatory intermediate. Identification of a relevant substrate is part of an ongoing study.
The release of mitochondrial contents documented here could be a direct response to loss of ATP. We have found that antimycin A (blocking complex III) leads to a rapid release of AIF from the mitochondria (data not shown). However, it is unlikely that the ERK inhibition-linked release of mitochondrial proteins, caspase activation and cell death is totally the result of ATP depletion. It is more likely that the early ATP depletion and loss of mitΔψ synergize with other ERK-dependent changes to effect the subsequent caspase activation and cell death. As stated above, we have shown that ERK inhibition leads to a decrease in translation, which would affect generation of anti-apoptotic proteins like the IAP’s (61). Another possible effector mechanism that could synergize with the loss of ATP is alterations in the Bcl2 protein balance after ERK inhibition. ERK is known to target the pro-apoptotic Bcl-2 protein, BimEL, for proteasome-dependent degradation (20) and phosphorylate Bad, causing it to dissociate from Bclxl (27). ERK inhibition would reverse these events and could certainly contribute to the release of mitochondrial proteins and caspase activation shown at late time points in this study. We are adding mitochondrial dysregulation and ATP depletion subsequent to ERK inhibition as a factor in the death of ERK inhibited alveolar macrophages.
ERK MAP kinase has many functions in the alveolar macrophage. At baseline, active ERK is found in the cytosol, nucleus and, as shown in this study, in the mitochondria. Endogenous regulators of the constitutive ERK activity include redox status of the cells, exposure to exogenous stimuli such as cigarette smoke and age of the alveolar macrophage (i.e. Is it a more monocyte-like cell?). We have shown that hyperoxia causes prolonged activation of the ERK pathway leading to enhanced survival (24, 62). It is not known whether the effect of the prolonged ERK in hyperoxic conditions is due to a protective effect on mitochondria. It is an interesting hypothesis to pursue.
In this paper, we examined a role for baseline ERK activity in mitochondrial homeostasis. As a composite, the data demonstrate that inhibition of ERK leads to disruption of mitΔψ , loss of ATP and eventual death of alveolar macrophages. We have previously shown that baseline ERK activity also regulates protein translation (4). The mitochondrial effects combined with the effects on translation suggest that ERK activity is an important survival pathway in alveolar macrophages. Compared to the short life span of alveolar macrophage precursors (blood monocytes survive approximately twenty-four hours), alveolar macrophages can live for extended periods in the lung (up to one year) (63). Thus, this newly described role for ERK in alveolar macrophage homeostasis may explain, in part, the survival characteristics of these cells.
Footnotes
This manuscript was supported by a VA Merit Review grant, NIH: HL-60316, NIH HL-077431 and HL079901-01A1 to G.W.H., NIH CA-086862 to D.R.S. and RR00059 from the General Clinical Research Centers Program, NCRR, NIH.
The abbreviations used are: Akt, protein kinase B; ERK, extracellular signal-regulated kinase; MAP, mitogen-activated protein; XIAP, x-linked inhibitor of apoptosis; MEK, MAP kinase kinase; eIF2α, eukaryotic initiation factor 2α; PKC ε, protein kinase C epsilon; MnSOD, manganese superoxide dismutase; mitΔψ, mitochondrial membrane potential; CCCP, carbony. Cyanide 4-(trifluoromethoxy) phenylhydrazone; VDAC, voltage-dependent anion channel; JNK, c-jun N-terminal kinase; AIF, apoptosis-inducing factor.
References
- 1.Thomas ED, Ramberg RE, Sale GE, Sparkes RS, Golde DW. Direct evidence for a bone marrow origin of the alveolar macrophage in man. Science (New York, NY) 1976;192:1016–1018. doi: 10.1126/science.775638. [DOI] [PubMed] [Google Scholar]
- 2.Komuro I, Keicho N, Iwamoto A, Akagawa KS. Human alveolar macrophages and granulocyte-macrophage colony-stimulating factor-induced monocyte-derived macrophages are resistant to H2O2 via their high basal and inducible levels of catalase activity. The Journal of biological chemistry. 2001;276:24360–24364. doi: 10.1074/jbc.M102081200. [DOI] [PubMed] [Google Scholar]
- 3.Monick MM, Mallampalli RK, Bradford M, McCoy D, Gross TJ, Flaherty DM, Powers LS, Cameron K, Kelly S, Merrill AH, Jr, Hunninghake GW. Cooperative Prosurvival Activity by ERK and Akt in Human Alveolar Macrophages is Dependent on High Levels of Acid Ceramidase Activity. J Immunol. 2004;173:123–135. doi: 10.4049/jimmunol.173.1.123. [DOI] [PubMed] [Google Scholar]
- 4.Monick MM, Powers LS, Gross TJ, Flaherty DM, Barrett CW, Hunninghake GW. Active ERK Contributes to Protein Translation by Preventing JNK-Dependent Inhibition of Protein Phosphatase 1. J Immunol. 2006;177:1636–1645. doi: 10.4049/jimmunol.177.3.1636. [DOI] [PubMed] [Google Scholar]
- 5.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 6.Kulik G, Weber MJ. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Molecular and cellular biology. 1998;18:6711–6718. doi: 10.1128/mcb.18.11.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashare A, Monick MM, Nymon AB, Morrison JM, Noble M, Powers LS, Yarovinsky TO, Yahr TL, Hunninghake GW. Pseudomonas aeruginosa Delays Kupffer Cell Death via Stabilization of the X-Chromosome-Linked Inhibitor of Apoptosis Protein. J Immunol. 2007;179:505–513. doi: 10.4049/jimmunol.179.1.505. [DOI] [PubMed] [Google Scholar]
- 8.Yang L, Sun M, Sun XM, Cheng GZ, Nicosia SV, Cheng JQ. Akt attenuation of the serine protease activity of HtrA2/Omi through phosphorylation of serine 212. The Journal of biological chemistry. 2007;282:10981–10987. doi: 10.1074/jbc.M700445200. [DOI] [PubMed] [Google Scholar]
- 9.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Molecular and cellular biology. 2000;20:8969–8982. doi: 10.1128/mcb.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zugasti O, Rul W, Roux P, Peyssonnaux C, Eychene A, Franke TF, Fort P, Hibner U. Raf-MEK-Erk cascade in anoikis is controlled by Rac1 and Cdc42 via Akt. Molecular and cellular biology. 2001;21:6706–6717. doi: 10.1128/MCB.21.19.6706-6717.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engedal N, Blomhoff HK. Combined action of ERK and NF kappa B mediates the protective effect of phorbol ester on Fas-induced apoptosis in Jurkat cells. The Journal of biological chemistry. 2003;278:10934–10941. doi: 10.1074/jbc.M211556200. [DOI] [PubMed] [Google Scholar]
- 14.Garcia J, Ye Y, Arranz V, Letourneux C, Pezeron G, Porteu F. IEX-1: a new ERK substrate involved in both ERK survival activity and ERK activation. Embo J. 2002;21:5151–5163. doi: 10.1093/emboj/cdf488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. The Journal of biological chemistry. 2004;279:49420–49429. doi: 10.1074/jbc.M407700200. [DOI] [PubMed] [Google Scholar]
- 16.Luciano F, Jacquel A, Colosetti P, Herrant M, Cagnol S, Pages G, Auberger P. Phosphorylation of Bim-EL by Erk1/2 on serine 69 promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785–6793. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 17.Monick MM, Carter AB, Gudmundsson G, Geist LJ, Hunninghake GW. Changes in PKC isoforms in human alveolar macrophages compared with blood monocytes. Am J Physiol. 1998;275:L389–397. doi: 10.1152/ajplung.1998.275.2.L389. [DOI] [PubMed] [Google Scholar]
- 18.Monick MM, Carter AB, Flaherty DM, Peterson MW, Hunninghake GW. Protein kinase C zeta plays a central role in activation of the p42/44 mitogen-activated protein kinase by endotoxin in alveolar macrophages. J Immunol. 2000;165:4632–4639. doi: 10.4049/jimmunol.165.8.4632. [DOI] [PubMed] [Google Scholar]
- 19.Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nature cell biology. 2003;5:647–654. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- 20.Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nature cell biology. 2003;5:733–740. doi: 10.1038/ncb1026. [DOI] [PubMed] [Google Scholar]
- 21.Phelps M, Phillips A, Darley M, Blaydes JP. MEK-ERK signalling controls Hdm2 oncoprotein expression by regulating hdm2 mRNA export to the cytoplasm. The Journal of biological chemistry. 2005;280:16651–16658. doi: 10.1074/jbc.M412334200. [DOI] [PubMed] [Google Scholar]
- 22.Apati A, Janossy J, Brozik A, Bauer P, Magocsi M. Calcium induces cell survival and proliferation through the activation of MAPK pathway in a human hormone-dependent leukaemia cell line (TF-1) The Journal of biological chemistry. 2003;278:9235–9243. doi: 10.1074/jbc.m205528200. [DOI] [PubMed] [Google Scholar]
- 23.Vial E, Marshall CJ. Elevated ERK-MAP kinase activity protects the FOS family member FRA-1 against proteasomal degradation in colon carcinoma cells. J Cell Sci. 2003;116:4957–4963. doi: 10.1242/jcs.00812. [DOI] [PubMed] [Google Scholar]
- 24.Nyunoya T, Monick MM, Powers LS, Yarovinsky TO, Hunninghake GW. Macrophages survive hyperoxia via prolonged ERK activation due to phosphatase down-regulation. The Journal of biological chemistry. 2005;280:26295–26302. doi: 10.1074/jbc.M500185200. [DOI] [PubMed] [Google Scholar]
- 25.Tamura Y, Simizu S, Osada H. The phosphorylation status and anti-apoptotic activity of Bcl-2 are regulated by ERK and protein phosphatase 2A on the mitochondria. FEBS letters. 2004;569:249–255. doi: 10.1016/j.febslet.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 26.Kurland JF, Voehringer DW, Meyn RE. The MEK/ERK pathway acts upstream of NF kappa B1 (p50) homodimer activity and Bcl-2 expression in a murine B-cell lymphoma cell line. MEK inhibition restores radiation-induced apoptosis. The Journal of biological chemistry. 2003;278:32465–32470. doi: 10.1074/jbc.M212919200. [DOI] [PubMed] [Google Scholar]
- 27.Scheid MP, Schubert KM, Duronio V. Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. The Journal of biological chemistry. 1999;274:31108–31113. doi: 10.1074/jbc.274.43.31108. [DOI] [PubMed] [Google Scholar]
- 28.Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene. 2004;23:5301–5315. doi: 10.1038/sj.onc.1207692. [DOI] [PubMed] [Google Scholar]
- 29.Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24:21–44. doi: 10.1080/02699050500284218. [DOI] [PubMed] [Google Scholar]
- 30.Alonso A, Sasin J, Bottini N, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 31.Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: a matter of life or death. Free radical biology & medicine. 2005;38:2–11. doi: 10.1016/j.freeradbiomed.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Zhang H, Soledad-Conrad V, Zhuang J, Uhal BD. Bleomycin-induced apoptosis of alveolar epithelial cells requires angiotensin synthesis de novo. American journal of physiology. 2003;284:L501–507. doi: 10.1152/ajplung.00273.2002. [DOI] [PubMed] [Google Scholar]
- 33.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. 2002;90:390–397. doi: 10.1161/01.res.0000012702.90501.8d. [DOI] [PubMed] [Google Scholar]
- 34.Alonso M, Melani M, Converso D, Jaitovich A, Paz C, Carreras MC, Medina JH, Poderoso JJ. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J Neurochem. 2004;89:248–256. doi: 10.1111/j.1471-4159.2004.02323.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13:473–481. doi: 10.1111/j.1750-3639.2003.tb00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science (New York, NY) 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 37.McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 38.Monick MM, Carter AB, Hunninghake GW. Human alveolar macrophages are markedly deficient in REF-1 and AP-1 DNA binding activity. The Journal of biological chemistry. 1999;274:18075–18080. doi: 10.1074/jbc.274.25.18075. [DOI] [PubMed] [Google Scholar]
- 39.Joseph TD, Look DC. Specific inhibition of interferon signal transduction pathways by adenoviral infection. The Journal of biological chemistry. 2001;276:47136–47142. doi: 10.1074/jbc.M102030200. [DOI] [PubMed] [Google Scholar]
- 40.Thomas KW, Monick MM, Staber JM, Yarovinsky T, Carter AB, Hunninghake GW. Respiratory Syncytial Virus Inhibits Apoptosis and Induces NF-kappa B Activity through a Phosphatidylinositol 3-Kinase-dependent Pathway. The Journal of biological chemistry. 2002;277:492–501. doi: 10.1074/jbc.M108107200. [DOI] [PubMed] [Google Scholar]
- 41.Lee YJ, Galoforo SS, Berns CM, Chen JC, Davis BH, Sim JE, Corry PM, Spitz DR. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. The Journal of biological chemistry. 1998;273:5294–5299. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 42.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–212. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 43.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. The Journal of biological chemistry. 1951;193:265–275. [PubMed] [Google Scholar]
- 44.Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. The Journal of clinical investigation. 1982;70:550–557. doi: 10.1172/JCI110647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kellett DN. On the mechanism of the anti-inflammatory activity of hexadimethrine bromide. Br J Pharmacol Chemother. 1966;26:351–357. doi: 10.1111/j.1476-5381.1966.tb01914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kempner W. The Nature of Leukemic Blood Cells as Determined by Their Metabolism. The Journal of clinical investigation. 1939;18:291–300. doi: 10.1172/JCI101045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawaguchi T, Veech RL, Uyeda K. Regulation of energy metabolism in macrophages during hypoxia. Roles of fructose 2,6-bisphosphate and ribose 1,5-bisphosphate. The Journal of biological chemistry. 2001;276:28554–28561. doi: 10.1074/jbc.M101396200. [DOI] [PubMed] [Google Scholar]
- 48.Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 49.Galluhn D, Langer T. Reversible assembly of the ATP-binding cassette transporter Mdl1 with the F1F0-ATP synthase in mitochondria. The Journal of biological chemistry. 2004;279:38338–38345. doi: 10.1074/jbc.M405871200. [DOI] [PubMed] [Google Scholar]
- 50.Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science (New York, NY) 2007;316:1345–1348. doi: 10.1126/science.1142984. [DOI] [PubMed] [Google Scholar]
- 51.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. The Journal of biological chemistry. 2003;278:8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 52.Stevens HC, Nichols JW. The Proton Electrochemical Gradient across the Plasma Membrane of Yeast Is Necessary for Phospholipid Flip. The Journal of biological chemistry. 2007;282:17563–17567. doi: 10.1074/jbc.M700454200. [DOI] [PubMed] [Google Scholar]
- 53.Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eminel S, Klettner A, Roemer L, Herdegen T, Waetzig V. JNK2 translocates to the mitochondria and mediates cytochrome c release in PC12 cells in response to 6-hydroxydopamine. The Journal of biological chemistry. 2004;279:55385–55392. doi: 10.1074/jbc.M405858200. [DOI] [PubMed] [Google Scholar]
- 55.Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong JY, Klassen SS, Rabkin SW. Ceramide activates a mitochondrial p38 mitogen-activated protein kinase: a potential mechanism for loss of mitochondrial transmembrane potential and apoptosis. Mol Cell Biochem. 2005;278:39–51. doi: 10.1007/s11010-005-1979-6. [DOI] [PubMed] [Google Scholar]
- 57.Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J, Susin SA. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Molecular and cellular biology. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Georgiou G. How to flip the (redox) switch. Cell. 2002;111:607–610. doi: 10.1016/s0092-8674(02)01165-0. [DOI] [PubMed] [Google Scholar]
- 59.Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal. 2005;7:42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- 60.Yung HW, Wyttenbach A, Tolkovsky AM. Aggravation of necrotic death of glucose-deprived cells by the MEK1 inhibitors U0126 and PD184161 through depletion of ATP. Biochem Pharmacol. 2004;68:351–360. doi: 10.1016/j.bcp.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 61.Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol. 2002;4:439–444. doi: 10.1038/ncb798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Truong SV, Monick MM, Yarovinsky TO, Powers LS, Nyunoya T, Hunninghake GW. Extracellular signal-regulated kinase activation delays hyperoxia-induced epithelial cell death in conditions of Akt downregulation. American journal of respiratory cell and molecular biology. 2004;31:611–618. doi: 10.1165/rcmb.2004-0141OC. [DOI] [PubMed] [Google Scholar]
- 63.Flaherty DM, Monick MM, Hinde SL. Human alveolar macrophages are deficient in PTEN. The role of endogenous oxidants. The Journal of biological chemistry. 2006;281:5058–5064. doi: 10.1074/jbc.M508997200. [DOI] [PubMed] [Google Scholar]