

Abstract
The life cycle of high-risk human papillomaviruses is linked to epithelial differentiation with virion production restricted to highly differentiated suprabasal cells. Two major viral promoters direct high-risk HPV gene expression and their activities are dependent upon differentiation. The early promoter controls initiation of transcripts at sites upstream of the E6 open reading frame and is active in both undifferentiated as well as differentiated cells. The late viral promoter directs transcription from a series of heterogeneous start sites in E7 and is activated upon differentiation. In this study, the state of histones as well as the spectrum of transcription factors bound to the two major HPV 31 viral promoters in undifferentiated and differentiated cells were examined using chromatin immunoprecipitation assays. Our studies indicate that in undifferentiated cells, the chromatin surrounding both promoter regions is in an open, transcriptionally active state as indicated by the presence of dimethylated forms of histone H3 K4 as well as acetylated H3 and acetylated H4. Upon differentiation, there was an increase of four to six fold in the levels of dimethylated H3K4 and acetylated H3 respectively around both promoter regions as well as an increase of approximately nine fold in acetylated H4 at the early promoter. This suggests that nucleosomes of both promoter regions are further activated through histone modifications during differentiation. Chromatin immunoprecipitation assays were also used to examine the binding of transcription factors to the keratinocyte enhancer (KE)/early promoter region in the upstream regulatory region (URR) and late promoter sequences throughout differentiation. Our results suggest a dynamic change in transcription factor binding occurs in both regions upon differentiation; most notably a significant increase in C/EBP-β binding to the KE/early promoter region as well as C/EBP-α binding to the late promoter region upon differentiation. These increases in binding cannot be solely explained by changes in the total cellular levels of these factors following differentiation, but instead reflect increased binding specific to HPV genomes. Finally, transient expression analyses confirmed that the KE/early promoter region of the URR contributes significantly to the activation of late gene expression and this is consistent with regulation through the combinatorial binding of multiple transcription factors
Introduction
Human papillomaviruses (HPV) infect epithelial cells and induce hyperproliferative lesions. A subset of HPV types, referred to as high-risk, infect the mucosal epithelia in the genital track and are the causative agents of most anogenital malignancies (Burd, 2003; zur Hausen, 1996). The productive life cycle of all human papillomavirus types is linked to epithelial differentiation with the synthesis of progeny virions restricted to suprabasal cells (Howley, 1996; Stubenrauch and Laimins, 1999). Papillomaviruses infect cells in the basal layer of stratified epithelia that become exposed through microabrasions (Burd, 2003). Following entry, HPV genomes are established as nuclear episomes at about 50 to 100 copies per cell. Infected basal cells remain in a latent state where they express only low levels of viral genes and stably replicate genomes. The productive replication or amplification of genomes and expression of late genes is restricted to highly differentiated suprabasal cells. Upon differentiation, HPV positive cells re-enter S-phase to induce productive viral replication as well as activate expression of late capsid proteins resulting in virion production. The various phases of the differentiation-dependent HPV life cycle are controlled through the activation of the early and late viral promoters that express distinct spectra of viral genes.
The high-risk human papillomaviruses encode two major promoters whose expression is regulated by differentiation (Figure 1). The early promoter controls expression in undifferentiated cells and directs initiation of viral transcripts at sites upstream of the E6 open reading frame. These polycistronic transcripts encode E6, E7, E1, E2, E4 and E5 and terminate at polyadenylation sites located downstream of the E5 open reading frame (Hummel, Hudson, and Laimins, 1992; Ozbun and Meyers, 1998). The early promoter of HPV 31 is designated p97 and is regulated by transcription factors that bind sequences 5′ of the start sites in the URR. The URR contains early promoter sequences, a keratinocyte-specific enhancer (KE) as well as the origin of viral replication (Apt et al., 1993; Cripe et al., 1987; Gius et al., 1988; Gloss et al., 1987; Kyo, Tam, and Laimins, 1995). The region at the 5′ end of the URR contains an element termed the auxiliary enhancer (AE) that augments the transcriptional activation function of the KE enhancer (Figure 1) (Hubert, Kanaya, and Laimins, 1999; Kanaya, Kyo, and Laimins, 1997). A number of in vitro binding studies have identified cellular transcription factors, including Ap-1, Sp1, YY1, and Oct-1, that associate with sequences in the URR and can activate expression in transient assays (Apt et al., 1996; Butz and Hoppe-Seyler, 1993; del Mar Pena and Laimins, 2001; Hoppe-Seyler and Butz, 1993; Hoppe-Seyler, Butz, and zur Hausen, 1991; Hubert, Kanaya, and Laimins, 1999; Kanaya, Kyo, and Laimins, 1997; Sen, Bromberg-White, and Meyers, 2002; Tan et al., 1994). The viral proteins E1 and E2 have also been shown to bind upstream of the early promoter region and have distinct roles in episome replication and amplification (Chiang et al., 1992; Ustav and Stenlund, 1991). E1 binds to the origin of replication and has an associated helicase activity (Sedman and Stenlund, 1998; zur Hausen, 1996). E2 acts to load E1 unto the viral origin and functions as a regulator of early expression through selective binding to four sites within the URR (Stubenrauch, Lim, and Laimins, 1998; Ustav and Stenlund, 1991).
Figure 1.

Linear depiction of the HPV 31 genome with the early and late regulatory regions expanded.
The second major promoter in HPV 31 directs expression of transcripts encoding the late proteins including E1^E4, E5, L1 and L2. These transcripts are activated upon differentiation and initiate at heterogeneous start sites located around nucleotide 742 in HPV 31 (p742) (Hummel, Hudson, and Laimins, 1992; Ozbun and Meyers, 1998). Previous studies have examined regulation of the late promoter through reporter-driven transient transfection assays using cells that stably maintain HPV episomes and activate expression following differentiation (Bodily and Meyers, 2005; Spink and Laimins, 2005). These studies have implicated sequences within the E6/E7 region as well as in the URR in the regulation of the late promoter. Differentiation alone was found to be sufficient to activate a low level of late transcription and replication was not strictly required. Several minor promoters have also been identified in the E6/E7 regions that are repressed upon differentiation (Ozbun and Meyers, 1998). The identity of the transcription factors that regulate late expression as well as the role that chromatin plays in the regulation of late expression has not been extensively studied.
Chromatin is constituted from nucleosomes, which consist of octamers of the core histone proteins H2A, H2B, H3, and H4. Nucleosomes bind DNA in 146 base pair intervals and regulate gene expression by controlling the access of transcription factors and transcription machinery to regulatory regions through covalent modifications of the tail regions of histones. Modifications such as acetylation or dimethylation of specific lysine residues decrease the affinity of histones to DNA and so facilitate the binding of transcription factors (Cosma, 2002; Jenuwein and Allis, 2001; Narlikar, Fan, and Kingston, 2002; Schneider et al., 2004; Strahl and Allis, 2000). Conversely, deacetylation induces the tight binding of the histone octamer to DNA, impeding factor binding and thus repressing expression (Narlikar, Fan, and Kingston, 2002). Human papillomavirus genomes have been shown to be bound by nucleosomes in an ordered arrangement around the viral promoters (del Mar Pena and Laimins, 2001; Stunkel and Bernard, 1999; Swindle and Engler, 1998). DNAse hypersensitivity studies performed using cells that maintain HPV 31 episomes indicated that there is a change in chromatin configuration around the late promoter upon differentiation (del Mar Pena and Laimins, 2001). It remains unclear if this opening of chromatin is the result of modifications to histones or increased transcription factor binding.
In the present study we examined the state of histones bound to HPV genomes during the differentiation-dependent productive life cycle through the use of chromatin immunoprecipitation analyses. Our studies indicate that the HPV early and late promoter regions are in an active chromatin state through out the viral life cycle as evidenced by the presence of acetylated H3 and H4 histones, and dimethylation of lysine 4 residues of the H3 histone. Upon differentiation, histones at both early and late promoter regions become further activated as indicated by increased levels of acetylation and dimethylation. In addition, chromatin immunoprecipitation assays identified a series of transcription factors whose binding was significantly enhanced to sites in the URR as well as the late promoter region upon differentiation. Among these factors, C/EBP–β exhibited significantly increased binding to sequences around the KE region in the URR following differentiation and C/EBP-α binding increased on late promoter sequences in E6/E7.
MATERIALS AND METHODS
Cell culture
LKP-1 cells are an immortalized keratinocyte cell line that stably maintain the HPV 31 genome as episomes and CIN 612 9E is an HPV 31 positive cell line derived from a patient biopsy. Both cell lines were maintained in E-medium in the presence of J2 3T3 fibroblast feeders as previously described (Ruesch, Stubenrauch, and Laimins, 1998). Cells were suspended in methylcellulose to induce differentiation as described (Ruesch, Stubenrauch, and Laimins, 1998; Wilson and Laimins, 2005).
Constructs
The HPV31Luc plasmid contains nucleotides 7045–892 of HPV 31 fused in frame in E1 to the firefly luciferase reporter coding sequences in pGL3 (Promega, Madison, WI, USA) and has been previously described (Hubert and Laimins, 2002). Derivatives of HPV31Luc containing deletions 7045–7557, 7045–7816, 7045–125, 7557–7816, and 7816–125 were generated by restriction endonuclease digestion as described previously (Spink and Laimins, 2005).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation assays, (ChIP), were performed using a variation of the Chromatin Immunoprecipitation Assay Kit protocol provided by the manufacturer (Upstate, Lake Placid, NY, USA). Cells grown in monolayer cultures were first treated with 5 ml Versene to remove fibroblast feeders, incubated with 0.05% trypsin-EDTA, resuspended in a final volume of 10 ml in E-media and then crosslinked with addition of formaldehyde to a final concentration of 1% for 10 minutes at room temperature. Cells that had been differentiated in methylcellulose were first washed extensively with phosphate buffered saline pH 7.5 (PBS), resuspended in E-media and then crosslinked as above. A solution of 10X glycine was added to stop the crosslinking reaction, followed by two washes by centrifugation using ice cold PBS. Cells were then resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1) in the presence of Complete Mini protease inhibitors (Roche Diagnostics, NJ, USA) and incubated on ice for ten minutes. The suspension was then sonicated for seven five-second pulses on ice using a microsonicator tip. This sonication procedure resulted in DNA fragments of approximately 400bp or less as determined by agarose gel electrophoresis. Cells were then diluted to a density of one million per ml in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, Upstate) containing Complete Mini protease inhibitors (Roche Diagnostics, Mannheim, Germany). One ml samples of approximately one million cells were then used as for each individual ChIP reaction. The solutions were incubated blocked with 40 μl protein A/salmon sperm agarose slurry (Upstate) for 30 minutes and then the agarose was then removed by centrifugation. The supernatant was then incubated with the desired antibody at 4°C for twelve hours with agitation. 60 μl of the protein A/salmon sperm agarose slurry was then added and samples were incubated for one additional hour at 4°C. The agarose slurry was then washed by rotation and centrifugation with the provided buffers, low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl) high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), LiCl immune complex wash buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris, pH 8.1), and twice with Tris-EDTA pH 8.0 before elution. The eluates were treated with RNAse A and the crosslinking was reversed with 5M NaCl followed by incubation at 65°C overnight. Samples were treated with Proteinase K (Roche Diagnostics) at 45°C for one hour and DNA was extracted with 25:24:1 phenol/chloroform. DNA was then precipitated overnight in ethanol with glycogen (Roche Diagnostics) added as a carrier and then following centrifugation, rinsed with 70% ethanol. The resulting pellets were resuspended in 50 μl Ultra-pure water (Invitrogen, Carlsbad, CA, USA) for further analysis.
Real-Time PCR
Real-time PCR reactions were performed in triplicate using a Taqman based system with primer/probe sets designed by Beacon Designer 5.0 (Premier Biosoft International, Palo Alto, CA, USA). The KE region primer/probe set covers nucleotides 7571–7671 and for the late promoter the primer/probe set covers nucleotides 652–790 nucleotides. Both reactions contained internal sequence-specific probes. All primer/probe sets were found to have comparable efficiencies through a standard curve analysis. Reactions were set up in duplicate with 12.5 μl iQ5 Supermix (BioRad, Hercules, CA, USA), 2.5 μl Primer/Probe mix, 7.5 μl water, and 2.5 μl ChIP DNA in a sealed 96 well optical plate. PCR parameters were: 95°C for 3 min; 95°C for 15 sec, then 65°C for 30 seconds, for 45 cycles. Ct values for the samples were equated to input Ct values to give percent of input values for comparison. The duplicates for each experiment were averaged and standard error values calculated. Fold change was calculated for each antibody used by dividing the average percent of input value for the 24 hour sample by the average percent input value for the 0 hour sample.
Western blot analyses
Protein lysates were collected using a RIPA cell lysis buffer with protease inhibitors (Roche Diagnostics). Cell lysates were sheared using a 22-gauge needle and spun down at for five minutes at 14,000 rpm (15,000 × g) and the supernatants collected. The protein concentration was quantified by Bradford Assay and protein was boiled in a Laemmli buffer. Western blot analysis was performed as previously described (Longworth and Laimins, 2004) and blots developed using ECL or ECL Plus (Amersham, Piscataway, NJ, USA).
Histone Acid Extraction
Keratinocytes were suspended in 1.5% methylcellulose for up to 48 hour and collected as described previously. Feeders were removed by versene treatment and the remaining keratinocytes were rinsed twice in PBS followed by trypsinization. Cells pellets were resuspended in 250 ul ice-cold lysis buffer (10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl with 0.5 mM DTT, 1.5 mM PMSF, and 200 mM HCl added fresh). Cells were kept on ice for 30 minutes and then the histone-enriched supernatants were collected by centrifugation at 4°C. Samples were precipitated with eight volumes of acetone overnight, centrifuged, and air dried. Pellets were resuspended in deionized water.
Antibodies
Antibodies against unmodified Histone H3, acetyl-Histone H4, acetyl-Histone H3, dimethyl-Histone H3 (Lys4), dimethyl-Histone H3 (Lys9), dimethyl-Histone H3 (Lys27), and Sp-1 were purchased from Upstate. Antibodies against Oct-1, YY1, c-Jun, C/EBP-α, E2F2, and C/EBP-β were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA, USA). An IgG control antibody was used to determine chromatin immunoprecipitation background signal (BD Biosciences, San Jose, CA, USA). An antibody against total histone H3 was purchased from Abcam, Inc. (Cambridge, MA, USA). GAPDH (Acbcam) was used as a protein loading control.
Luciferase Assays
LKP-1 cells were maintained as described previously and split to approximately one million cells per plate the day prior to transfection. Cells were then transiently cotransfected with 2μg of a luciferase construct and 50ng of the Renilla control vector using the Fugene 6 transfection reagent (Roche) according to the manufacturer’s instructions. After 24 hours of incubation, the fibroblast feeders were removed by versene treatment and each plate was divided into a monolayer culture a culture suspended in 1.5% methylcellulose. After 48 hours if incubation, the cells were harvested and lysed for luciferase assays using the Dual Luciferase Kit (Promega). The average percent of wild-type relative luciferase units (RLU) was determined and standard deviation calculated.
RESULTS
HPV 31 genomes are in active chromatin states in both undifferentiated and differentiated keratinocytes
To investigate the factors that regulate differentiation-dependent transcription from the early and late promoters of high-risk HPV 31, we performed chromatin immunoprecipitation assays using cell lines that stably maintain HPV 31 genomes and induce late gene expression following differentiation. We first examined the state of histones around the early and late promoter regions in undifferentiated HPV 31 positive LKP-1 cells as well as following differentiation in methylcellulose for 24 hours. LKP-1 cells stably maintain HPV 31 episomes in undifferentiated cells and active late gene expression as well as genome amplification upon differentiation (Spink and Laimins, 2005). Previous studies indicated that the induction of HPV 31 late viral functions peaks at 24 hours in methylcellulose (Ruesch, Stubenrauch, and Laimins, 1998). Equal numbers of cells that were grown in monolayer culture or suspended in methylcellulose were isolated and factors bound to DNA cross-linked though the addition of formaldehyde. Following cell lysis, the DNA was sonicated into fragments of less than 400 bp on average and DNA protein complexes isolated with antibodies specific for various modified forms of histones.
In order to examine how binding of modified histones to HPV episomes changed during differentiation, we used quantitative PCR in conjunction with the chromatin immunoprecipitation methods. The first primer set used encompassed nucleotides 7571–7642 which corresponds to the KE/early promoter region. The second set of primers was directed at nucleotides 652–767, which corresponded to the late promoter region overlapping E6/E7 coding sequences. These two regions are separated by approximately one kilobase and so our analysis reflects binding to two distinct regions. A standard curve analysis was next performed using the two primer sets and indicated the primer sets bound with equal efficiency (data not shown), permitting direct comparisons to be made of the levels of the factors bound at these two regions of the HPV genome.
A series of chromatin immunoprecipitation assays were then performed for the various forms of modified histones and the levels of bound proteins determined by quantitative PCR. The results shown are representative of three assays and indicated that both regions bound acetylated H3, H4 and dimethylated H3K4 in undifferentiated cells (Figure 2A). This indicates that both regions are in active chromatin states. Interestingly, the late promoter region bound levels of acetylated histones, as well as dimethylated H3K4 that were two to five fold higher than were found associated with the sequences around the KE region. A low level of K9 and K27 methylation was also observed at both promoter regions, which is indicative of repressed chromatin state (Figure 2A). This suggests that a small subset of HPV genomes may be in inactive transcription states.
Figure 2.
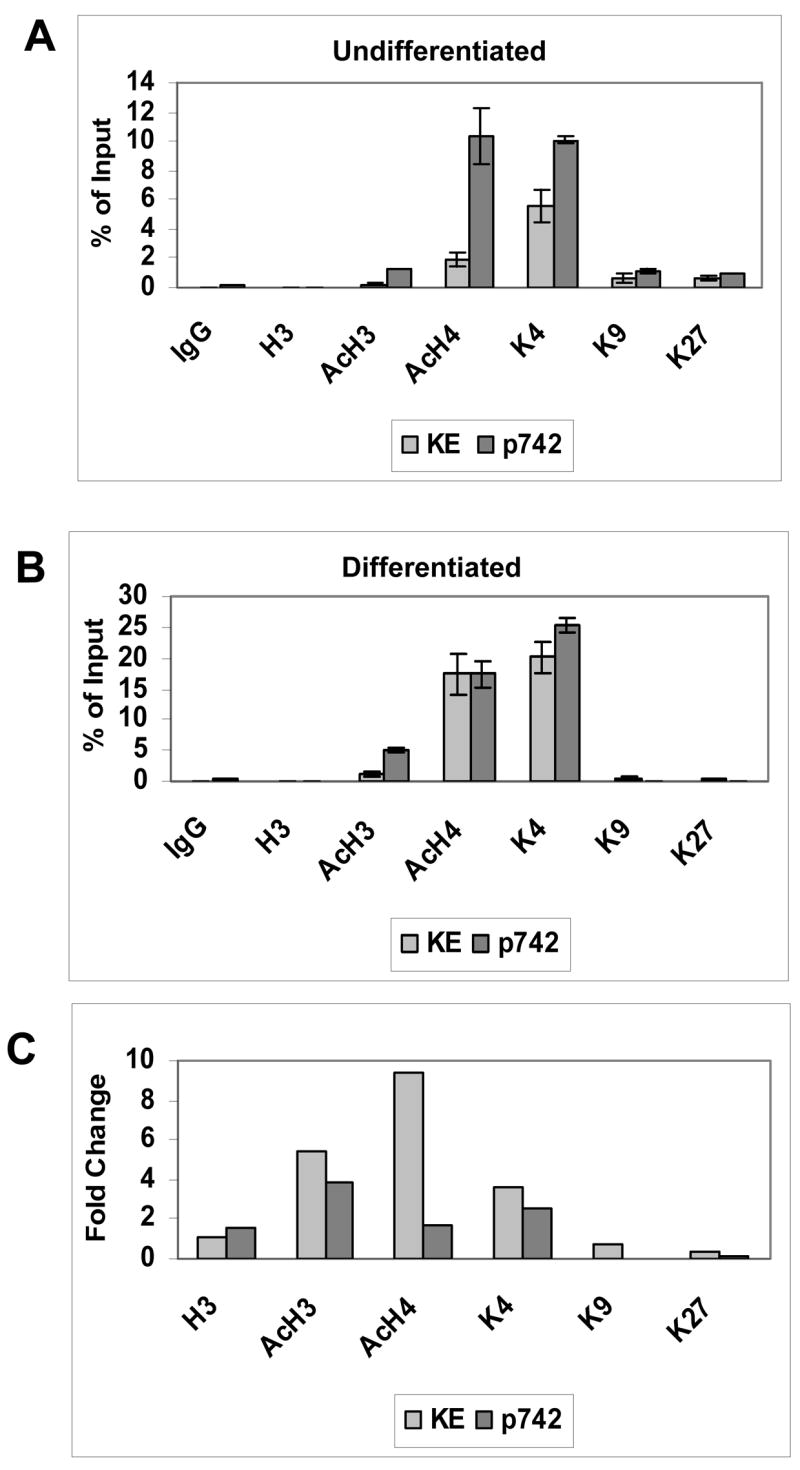
Quantitative PCR analysis of chromatin immunoprecipitation assays of HPV 31 episomes screening for histone modifications. Cells were grown in monolayer (0 hour) or following suspension in methylcellulose for 24 hours. Antibodies used were for IgG control, unmodified H3 (H3), acetylated H3 (AcH3), acetylated H4 (AcH4), dimethylated lysine 4 of H3 (di-MeK4), and dimethylated lysine 9 of H3 (di-MeK9), and dimethylated lysine 27 of H3 (di-MeK27). Following immunoprecipitation with antibodies for specific histone modifications, quantitative real-time PCR amplification was performed. Taqman Primer-probe sets used were specific to the keratinocyte enhancer region (nucleotides 7571–7671), and the later promoter region (652–790), with internal FAMRA/TAMRA labeled sequence-specific probes. Ct (cycle threshold) results are expressed as percent of Input for the Keratinocyte enhancer region and Late promoter region in (A) Undifferentiated cells (B) Differentiating cells. (C) A comparison of the fold change of histone modifications levels from differentiated and undifferentiated cultures for both regions was performed.
It was next important to determine what effect differentiation had on the state of histones bound to HPV genomes. For this analysis we performed chromatin immunoprecipitation assays on cells that had been suspended in methylcellulose for 24 hours followed by quantitative PCR (Figure 2B). Our studies indicated there was no change in the low levels of unmodified histone H3 bound at the KE region while the amounts of acetylated H3 and H4 bound increased by five and nine fold, respectively. Also, levels of dimethylated H3K4 bound increased by 4 four fold in the KE region (Figure 2C). These changes were not the result of increased amounts of viral DNA resulting from amplification, as our analysis is normalized with respect to the levels of HPV 31 genomes in the sample. The levels of acetylated H3, H4 and dimethylated H3K4 also increased two to four fold at the late promoter region following differentiation (Figure 2C). Interestingly, the levels of two repressive forms of histone modifications, dimethylated H3 K9 and K27, were unchanged at the KE region and were found to decrease at the late promoter upon differentiation (Figure 2B). Similar results were obtained in multiple separate assays and in experiments using CIN612 9E cells, another HPV 31 positive cell line that maintains viral DNA as episomes. We conclude that HPV genomes have activated histones bound at both KE and late promoter regions in undifferentiated cells and that these levels increase several fold upon differentiation.
The above analysis indicated that the levels of active histones bound to HPV genomes increased upon differentiation, and we next investigated if these changes were due to alterations in the total levels in cells. To investigate this possibility, total cellular histones were isolated from HFKs and LKP-1 cells by acid extraction and examined by Western analysis (Figure 3). A moderate increase in the levels of acetylated H3 and H4 histones was observed in LKP-1 cells as compared to normal keratinocytes, which is consistent with previous reports that E7 can increase the level of histone acetylation in cells (Zhang et al., 2004). Following differentiation, there was no change in the levels of the modified forms of histones in HPV positive cells while there were minor fluctuations in the amounts present normal keratinocytes (Figure 3). Overall, we conclude that the changes in the levels of acetylated H3, H4 and dimethylated H3K4 bound to HPV genomes upon differentiation cannot be explained by changes in total cellular levels.
Figure 3.
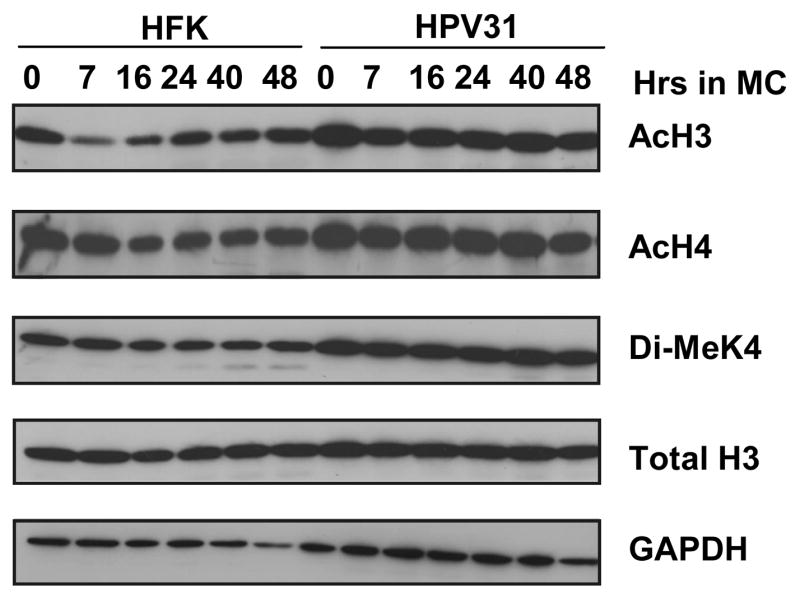
Western blot analysis of acid extracted histones isolated from primary human foreskin keratinocytes (HFKs) and HPV 31 positive LKP-1 cells grown in monolayer (0 hour) or suspended in methylcellulose for 7, 16, 24, 40, and 48 hours. Histones were extracted from cells after lysis by acetone precipitation. Samples were separated on a 15% SDS-PAGE gel and transferred to an Immobilon membrane for western analysis. Antibodies used to probe the membrane were Total H3 (Total H3), acetylated H3 (AcH3), acetylated H4 (AcH4), and dimethylated lysine 4 of H3 (di-MeK4). GAPDH was used as a loading control. MC = cells suspended in 1.5% methylcellulose.
Transcription factor binding to the KE/early promoter and late promoter regions changes upon differentiation
The activation of the late promoter upon differentiation is likely mediated by changes in the levels of cellular transcription factors bound to regulatory sequences. We therefore investigated the binding of transcription factors to the two promoter regions of the HPV 31 genome using quantitative chromatin immunoprecipitation assays. For these analyses, we examined the binding of 14 different transcription factors for which there were putative binding sequences in these two regions. Seven of these factors yielded positive results. Given the degeneracy of binding sites, both regions encoded putative sites for the majority of these factors (Table I). Our results indicate that Sp1, YY1, C/EBP-β and α, E2F2, Oct-1 and c-Jun bound to both the KE/early promoter and late promoter regions in undifferentiated cells (Figure 4A). As discussed, we were able to make comparisons between binding to the two different regions as the efficiency of primer binding to both regions was similar. In undifferentiated cells, the binding of YY1 and Sp1 to the KE/early promoter region was found to be about five fold lower while C/EBP–β about ten fold lower than that seen with the late promoter region (Figure 4A). In addition the binding of C/EBP-α, Oct-1 and c-Jun to both regions was similar in undifferentiated cells while the levels of E2F2 bound to the KE/early promoter were approximately three fold higher than to the late promoter region. Similar results were seen in multiple experiments and results are shown in Figure 4.
Table I.
Putative binding sequences of transcription factors found to bind to HPV genomes. The number of sites found by TransFac analysis is shown.
| Factor | Consensus Sequence | URR/Early Region (Nt 7045–107) | Late Region (Nt 108–850) |
|---|---|---|---|
| Sp1a | G(A/G)GGC(A/G)GGG(A/T) | 22 | 24 |
| YY1b | CCATNT(A/T)N | 22 | 10 |
| C/EBP-ac | NNATT(A/G)CNNAANNN | 7 | 4 |
| C/EBP-Bc | (A/G)N(G/A)T(G/T)NNG(A/C)AA(G/T)NN NN(C/T)AA(T/C)NCGTTN(G/T)N | 6 | 9 |
| E2Fb | TTT(C/G)GCGC (A/C)(G/T)NATTTGCATA(C/T)(C/T) TATGCAAATNNN(A/T)NN(A/T) | 8 | 2 |
| Oct-1c | NNNN(A/T)TATGCAAATNTNNN NNN(A/G)TAATNANNN NNGAAT(G/T)CANNNN TNTATATGNTAATT | 15 | 5 |
| c-Junc | NTGA(C/G)TN(A/C)N(A/T) | 6 | 2 |
60% homology,
70% homology,
80% homology.
Figure 4.

Quantitative PCR analysis of chromatin immunoprecipitation assays for transcription factor binding to regions of HPV 31 episomes grown in monolayer (0 hour) or following suspension in methylcellulose for 24 hours. Antibodies used were for IgG control, Sp1, YY1, C/EBP-β, C/EBP-α, E2F2, Oct-1, and c-Jun. Following immunoprecipitation with antibodies for specific histone modifications, quantitative real-time PCR amplification was performed using Taqman primer-probe sequences specific to the keratinocyte enhancer region (7571–7671), and the later promoter region (652–790) with internal FAMRA/TAMRA labeled sequence-specific probes. Ct results are expressed as percent of Input for the Keratinocyte enhancer region and Late promoter region in (A) Undifferentiated cells (B) Differentiating cells. (C) A comparison of the fold change of levels of transcription factor binding between differentiating and undifferentiating cell cultures for both regions was performed.
Upon differentiation, the binding of YY1, and c-Jun to the KE/early promoter region increased 20 to 50 fold, while the binding of C/EBP-β increased by over 90 fold (Figure 4B and C). In addition, the binding of Oct-1, C/EBP-α, Sp1 and E2F2 exhibited little to no change in binding to this region upon differentiation. A similar analysis was performed for the factors bound to the late promoter region that overlaps the E6/E7 coding region (Figure 4B and C). Upon differentiation, the levels of YY1 and C/EBP-β bound to the late promoter region did not change and this is in contrast to the increased binding of these two factors seen with the KE/early promoter sequences. The most significant increases in the level of binding to the late promoter region were found for C/EBP-α and Sp1 which increased approximately five to six fold while c-Jun increased three fold. A graph showing the fold increases in binding of these factors to these two regions upon differentiation is shown in Figure 4C. Overall our results indicate that there is a dynamic change in the binding of transcription factors to the two HPV 31 promoter regions during the differentiation-dependent life cycle.
As with our analysis of modified histones, it was important to investigate if the increased binding of the factors to HPV genomes corresponded to changes in the total levels of these proteins following differentiation. Total cellular proteins were isolated from HFKs and HPV 31 positive cells induced to differentiate in methylcellulose for various periods of time and examined by Western blot analysis using the antibodies for the factors described above (Figure 5). The levels of Sp1, Oct-1 and YY1 were found to be similar in undifferentiated cells and these levels remained constant or decreased slightly upon differentiation of HFKs and HPV 31 positive cells. C/EBP-β levels were found to be slightly higher in HPV 31 positive cells but did not change appreciably upon differentiation. In contrast, the levels of C/EBP-α were found to be significantly higher in undifferentiated HPV 31 positive cells as compared to HFKs and remained high in differentiated cells. Finally, the levels of c-Jun were found to be similar in both HPV-negative and positive undifferentiated cells (T0). Upon differentiation, the levels of c-Jun decreased in HFKs while they remained at a constant level in LKP-1 cells. Overall these studies suggest that HPV proteins increase the levels of c-Jun, C/EBP-α and C/EBP-β, however these changes alone do not appear to be sufficient to account for the increased binding to viral genomes seen in the ChIP assays.
Figure 5.
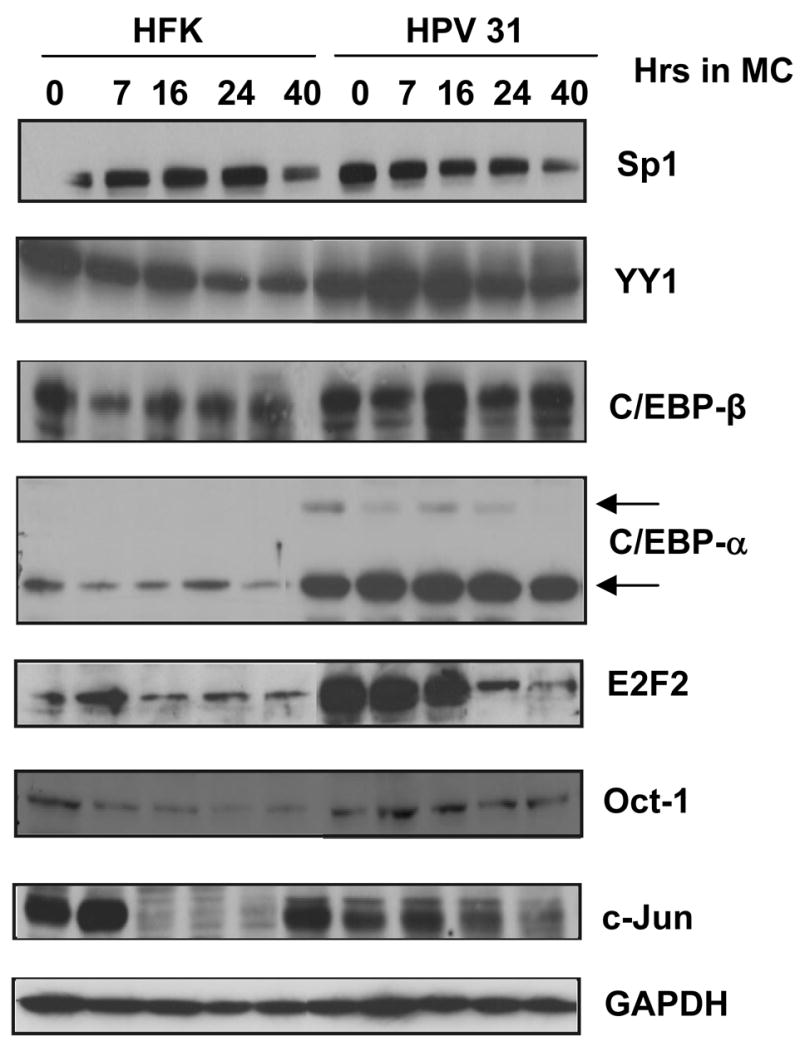
Western blot analysis of transcription factors present in whole cell lysates collected from primary human foreskin keratinocytes (HFKs) and HPV 31 cells grown in monolayer (0 hour) or suspended in methylcellulose for 7, 16, 24, and 40 hours. Cells pellets were resuspended in RIPA lysis buffer and analyzed on SDS-PAGE gels at examined by western analysis. Antibodies against Sp1, YY1, C/EBP-β, C/EBP-α, E2F2, Oct-1, and c-Jun were used to probe the membrane. GAPDH was used as a loading control.
In this analysis only one factor, E2F2, exhibited significant changes in levels in HPV positive cells as a function of the state of differentiation. Consistent with previous reports (Longworth, Wilson, and Laimins, 2005), E2F2 levels were increased in undifferentiated HPV 31 positive cells relative to HFKs and remained high following differentiation for up to 16 hours at which point they rapidly decreased so that at 24 hours the levels were comparable to HFKs (Figure 5). The ChIP analyses shown in Figure 4 were performed 24 hours which correlates with maximal induction of late expression and it was possible we might have missed transient increased binding of E2F2 to HPV genomes if this occurred at an earlier state of differentiation. To address this point, we examined the levels of E2F2 bound to HPV genomes after seven hours of suspension in methylcellulose and found that binding increased five fold to the KE region and twelve fold to the late promoter region and then decreased rapidly at 24 hours to the levels seen in undifferentiated cells (not shown). Since this increase in binding of E2F2 does not directly correlate with the peak of late expression seen at 24 hours, it is unclear how it contributes to late expression or whether it is part of some other viral function.
The KE/early promoter region regulates late gene expression
Our chromatin immunoprecipitation analyses indicated that upon differentiation, the KE/early promoter region binds increased levels of activated histones as well as high levels of transcription factors such as C/EBP-β. This is consistent with this region playing a major role in regulating late gene expression. We therefore investigated the contribution of the KE or AE elements in the activation of late gene expression using transient transfection assays with the HPV 31 Luc late reporter. This plasmid contains sequences from the 5′end of the HPV 31 URR through the E6/E7 open reading frames into E1 fused in frame to luciferase. Previous studies have shown the early promoter p97 contributes minimally to late expressiion in this reporter (Spink and Laimins, 2005). We transfected LKP-1 cells with either the HPV31Luc reporter or constructs in which the KE or AE had been deleted (Figure 6A). HPV 31 Luc exhibited low levels of expression in undifferentiated cells and this is consistent with previous observations showing that the late promoter has a low level of activity in cells (Pray and Laimins, 1995). Transfection of the mutant plasmids resulted in a significant decrease of over 90% in the levels of late gene expression in both monolayer and following differentiation in methylcellulose (Figures 6B and 6C) and supports our previous observations (Spink and Laimins, 2005). Deletion of the region between the end of the KE through the early start sites at around nucleotide 125 (7816–125) had a minimal effect on late gene expression (Figures 6B and 6C). This indicates that the element in the URR that directs late gene expression is localized to the AE/KE regions and may overlap the early keratinocyte enhancer. The magnitude of late expression was significantly reduced upon deletion of the KE/AE elements, however, the remaining sequences retained differentiation-dependent activity (data not shown). This is consistent with previous reports implicating the E6/E7 region as a regulator of differentiation-dependence and suggests this region may act as a promoter element for late expression (Bodily and Meyers, 2005; Spink and Laimins, 2005).
Figure 6.

Transient expression analysis of the contribution of the upstream regulatory region to late gene expression. (A) The reporter construct contained the entire URR from nucleotide 7045 through the N terminus of E1 (nucleotide 892) fused to the firefly luciferase gene, as previously described (Hubert and Laimins, 2002). Deletions of the auxiliary enhancer (7045–7515), keratinocyte enhancer (7515–7816), the combined mutation (7045–7816), the 3′ region through the early promoter (7816–125), as well as the entire URR (7045–125) were generated using restriction endonuclease digestion as described previously (Spink and Laimins, 2005). HPV 31 positive LKP1 cells were co-transfected with a luciferase reporter construct as well as the Renilla control vector and incubated for 24 hours. The culture was then split into monolayer and methylcellulose cultures. After differentiation, cells were harvested for luciferase assays. Cells were harvested 48 hours later and luciferase assays were completed. Results shown are averages from a representative experiment completed in triplicate with standard deviation. (B) Levels of relative luciferase units (RLU) for the mutant constructs were compared to wild-type in monolayer culture. (C) Levels of relative luciferase units (RLU) for the mutant constructs as compared to wild-type cells suspended in methylcellulose.
CONCLUSIONS
In this study we examined the state of histones and the spectrum of transcription factors bound to two regions of the HPV 31 genome corresponding to the early and late promoters. Our results demonstrate that both promoter regions are in active states in undifferentiated cells as indicated by the binding of acetylated H3, acetylated H4, and dimethylated H3K4. It had been previously hypothesized that the early promoter/KE enhancer region was in active state while the late promoter region in late promoter region was is a repressed state in undifferentiated cells (del Mar Pena and Laimins, 2001). Our studies show this not to be the case and demonstrate that both regions bind activated forms of histones. An active chromatin state for the late promoter in undifferentiated cells is consistent with the observations that low levels of late gene expression can be detected in monolayer cultures of cells that maintain either HPV 31 or 16 episomes (Koffa et al., 2000; Pray and Laimins, 1995). Upon differentiation, we observed an increase in the levels of acetylated histones bound to the KE/early promoter as well as the late promoter region in E6/E7. This is consistent with the KE/early promoter region playing an active role in regulating late expression. The KE/early promoter region includes the origin of replication and the maintenance of an active chromatin state upon differentiation may also be required for productive replication. While both the KE and late promoter regions bound acetylated histones indicate of active chromatin, we also observed low level binding of K9 and K27 dimethylated histones, which correlates with a repressed state. It is possible that a subset of HPV episomes are in transcriptionally inactive states while the remainder are in active states, but our studies do not allow us to separate populations of genomes.
The activation of gene expression is also regulated by changes in the levels of transcription factors bound to promoter sequences. Our studies indicate that binding of several transcription factors to the KE/early promoter region increased significantly upon differentiation. The binding of C/EBP-β increased over 90 fold, while the binding of c-Jun was enhanced by 50 fold and that of YY1 by 20 fold. This increase in binding supports the idea that the KE/early promoter region is a key regulator of the late promoter and that this is mediated through the binding of a combination of cellular transcription factors. It is possible that increased binding to this region also acts to augment genome amplification upon differentiation. In previous studies, we determined that transcription factor binding to URR sequences could contribute to the replication of viral genomes (Hubert, Kanaya, and Laimins, 1999) and this may also occur during amplification.
Analysis of transcription factor binding to the late promoter sequences in the E6/E7 coding region demonstrated that the spectrum of factors is distinct from those that bind the KE/early promoter. We observed a five fold increase in binding of C/EBP-α along with a six fold enhancement in Sp1 and a three fold change in c-Jun binding. No change in the levels of YY1, C/EBP-β, or Oct-1 to late promoter sequences was observed. Our studies are consistent with a previous analysis of transcription factors bound to HPV 16 episomes in vivo following differentiation (Carson and Khan, 2006). These authors looked at a several factors by chromatin immunoprecipitation and found binding of C/EBP-α to increase seven fold while YY1 increased two fold to HPV 16 genomes but did not examine other factors addressed in our study. In addition, their analysis did not discriminate binding between the early and late promoter regions.
Our studies also demonstrated that E2F2 binding to both the early and late promoters increased upon differentiation but that it preceded full activation of late promoter activity. Maximal activation of late expression occurs around 24 hours after suspension in methylcellulose at which time the amounts of E2F2 bound had decreased. It is possible that E2F2 binding acts to help recruit other factors to the HPV genome but itself is not required for late promoter function or that it is important for other activities such as productive replication. None of the other factors we examined by ChIP exhibited such significant changes in expression levels as a function of differentiation. Further investigation of the role of E2F2 in viral pathogenesis will require the identification and mutation of the actual E2F2 binding sites in HPV genomes to see how these act in late viral functions.
The chromatin immunoprecipitation assays indicate that the KE region may play an active role in regulating late expression. Further support for this hypothesis is provided by transient reporter assays, which indicate that deletion of the KE/AE elements alone reduced late expression by over 90% and is consistent with previous reporter studies (Bodily and Meyers, 2005; Spink and Laimins, 2005). In addition, deletion of the sequences from the end of the KE to the start site for early transcription (7816–125) had minimal effect on late gene expression. These sequences are important for early promoter function and this observation is consistent with the early promoter not contributing significantly to the activity in the late HPV 31 reporter. Overall, our studies indicate that sequences within the URR that are responsible for high levels of late gene expression. The element within the URR that regulates late expression appears to overlap the early KE enhancer and itself exhibits some similarities to an enhancer.
One factor we found whose binding to the URR upon differentiation correlated with activation of late expression was C/EBP-β. C/EBP-β and C/EBP-α are CCAAT enhancer binding proteins of the bZIP family of transcription factors. These factors have been suggested to have a role in keratinocyte differentiation, in part through regulation of the K10 promoter (Maytin et al., 1999) and are expressed in the epidermis (Jin, Yang, and Auborn, 1998; Maytin and Habener, 1998). C/EBP-β has been previously suggested to be both an activator and a repressor of HPV gene expression (Kukimoto, Takeuchi, and Kanda, 2006; Kyo et al., 1993; Wang et al., 1996; Zhao et al., 1999). The HPV 31 URR contains six potential binding sites for C/EBP-β and nine additional sites are present in the E6/E7 region at 80% homology to consensus binding sites (Table I). Similar sequences are found in other HPV types. Kukimoto et al reported that overexpression of C/EBP-β in transient assays could induce low-level activation of an HPV 16 late reporter in undifferentiated cells (Kukimoto, Takeuchi, and Kanda, 2006), while Sen et al. suggested that C/EBP-β may play a role in regulating HPV 31 early gene expression (Sen, Bromberg-White, and Meyers, 2002). It is thus possible that C/EBP-β has two roles in differentiating cells: the repression of the early promoter and the activation of the late promoter region, but this has yet to be demonstrated. In our studies we observed significantly enhanced binding of C/EBP-β and YY1 to the KE/early promoter region, while there were only modest changes in the total cellular levels of these factors. We believe this may be due cooperative effects mediated through the binding of multiple transcription factors to the same region. For example, both E2 and YY1 have been reported to form complexes with C/EBP-β and this may contribute to the enhanced binding we observed in our analyses.
Our studies also provide insights into previous hypersensitivity experiments examining cells with HPV episomes. Pena et al. (del Mar Pena and Laimins, 2001) used DNAse hypersensitivity studies to demonstrate that a change in chromatin occurs around the late promoter region following differentiation. This increased sensitivity could be the result of changes in the state of histones in the late promoter region or altered binding of transcription factors. Our current studies provide support for the latter hypothesis. In conclusion, we have found that the entire HPV genome is in an open, active chromatin conformation throughout the viral life cycle. Furthermore, transcription factor binding to both KE/early promoter and late promoter regions was found to increase upon differentiation. This included significant increases in the binding of C/EBP-β as well as C/EBP-α, YY1, and c-Jun. It is likely that combinations of these factors act to regulate late gene expression and this is an active area for future investigation.
Acknowledgments
We thank Ann Roman, Cary Moody, Katie Spink, Walter Hubert, and members of the Laimins lab for helpful advice as well as reagents. This study was supported by a grant from the NCI to LAL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bodily JM, Meyers C. Genetic analysis of the human papillomavirus type 31 differentiation-dependent late promoter. J Virol. 2005;79(6):3309–21. doi: 10.1128/JVI.79.6.3309-3321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd EM. Human papillomavirus and cervical cancer. Clin Microbiol Rev. 2003;16(1):1–17. doi: 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson A, Khan SA. Characterization of transcription factor binding to human papillomavirus type 16 DNA during cellular differentiation. J Virol. 2006;80(9):4356–62. doi: 10.1128/JVI.80.9.4356-4362.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CM, Ustav M, Stenlund A, Ho TF, Broker TR, Chow LT. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc Natl Acad Sci U S A. 1992;89(13):5799–803. doi: 10.1073/pnas.89.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Mar Pena LM, Laimins LA. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J Virol. 2001;75(20):10005–13. doi: 10.1128/JVI.75.20.10005-10013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howley PM. Papillomaviridae: the viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fundamental Virology. Vol. 3. Lippincott-Raven Publishers; Philidelphia, PA: 1996. pp. 947–978. [Google Scholar]
- Hubert WG, Kanaya T, Laimins LA. DNA replication of human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5′ of the minimal origin. J Virol. 1999;73(3):1835–45. doi: 10.1128/jvi.73.3.1835-1845.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert WG, Laimins LA. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of E1 and E2 expression. J Virol. 2002;76(5):2263–73. doi: 10.1128/jvi.76.5.2263-2273.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Hudson JB, Laimins LA. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol. 1992;66(10):6070–80. doi: 10.1128/jvi.66.10.6070-6080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Yang GY, Auborn K. Differences in C/EBPs in normal tissue and papillomas of the larynx. Cell Prolif. 1998;31(3–4):127–38. doi: 10.1046/j.1365-2184.1998.00115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya T, Kyo S, Laimins LA. The 5′ region of the human papillomavirus type 31 upstream regulatory region acts as an enhancer which augments viral early expression through the action of YY1. Virology. 1997;237(1):159–69. doi: 10.1006/viro.1997.8771. [DOI] [PubMed] [Google Scholar]
- Kukimoto I, Takeuchi T, Kanda T. CCAAT/enhancer binding protein beta binds to and activates the P670 promoter of human papillomavirus type 16. Virology. 2006;346(1):98–107. doi: 10.1016/j.virol.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol. 2004;78(7):3533–41. doi: 10.1128/JVI.78.7.3533-3541.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. Embo J. 2005;24(10):1821–30. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maytin EV, Habener JF. Transcription factors C/EBP alpha, C/EBP beta, and CHOP (Gadd153) expressed during the differentiation program of keratinocytes in vitro and in vivo. J Invest Dermatol. 1998;110(3):238–46. doi: 10.1046/j.1523-1747.1998.00123.x. [DOI] [PubMed] [Google Scholar]
- Maytin EV, Lin JC, Krishnamurthy R, Batchvarova N, Ron D, Mitchell PJ, Habener JF. Keratin 10 gene expression during differentiation of mouse epidermis requires transcription factors C/EBP and AP-2. Dev Biol. 1999;216(1):164–81. doi: 10.1006/dbio.1999.9460. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108(4):475–87. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Ozbun MA, Meyers C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J Virol. 1998;72(4):2715–22. doi: 10.1128/jvi.72.4.2715-2722.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pray TR, Laimins LA. Differentiation-dependent expression of E1--E4 proteins in cell lines maintaining episomes of human papillomavirus type 31b. Virology. 1995;206(1):679–85. doi: 10.1016/s0042-6822(95)80088-3. [DOI] [PubMed] [Google Scholar]
- Ruesch MN, Stubenrauch F, Laimins LA. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J Virol. 1998;72(6):5016–24. doi: 10.1128/jvi.72.6.5016-5024.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedman J, Stenlund A. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J Virol. 1998;72(8):6893–7. doi: 10.1128/jvi.72.8.6893-6897.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen E, Bromberg-White JL, Meyers C. Genetic analysis of cis regulatory elements within the 5′ region of the human papillomavirus type 31 upstream regulatory region during different stages of the viral life cycle. J Virol. 2002;76(10):4798–809. doi: 10.1128/JVI.76.10.4798-4809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spink KM, Laimins LA. Induction of the human papillomavirus type 31 late promoter requires differentiation but not DNA amplification. J Virol. 2005;79(8):4918–26. doi: 10.1128/JVI.79.8.4918-4926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubenrauch F, Laimins LA. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol. 1999;9(6):379–86. doi: 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- Stubenrauch F, Lim HB, Laimins LA. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J Virol. 1998;72(2):1071–7. doi: 10.1128/jvi.72.2.1071-1077.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. Embo J. 1991;10(2):449–57. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Laimins LA. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol Med. 2005;119:157–69. doi: 10.1385/1-59259-982-6:157. [DOI] [PubMed] [Google Scholar]
- Zhang B, Laribee RN, Klemsz MJ, Roman A. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology. 2004;329(1):189–98. doi: 10.1016/j.virol.2004.08.009. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochim Biophys Acta. 1996;1288(2):F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]