

Abstract
Sulfate-assimilating organisms reduce inorganic sulfate for Cys biosynthesis. There are two leading hypotheses for the mechanism of sulfate reduction in higher plants. In one, adenosine 5′-phosphosulfate (APS) (5′-adenylylsulfate) sulfotransferase carries out reductive transfer of sulfate from APS to reduced glutathione. Alternatively, the mechanism may be similar to that in bacteria in which the enzyme, 3′-phosphoadenosine-5′-phosphosulfate (PAPS) reductase, catalyzes thioredoxin (Trx)-dependent reduction of PAPS. Three classes of cDNA were cloned from Arabidopsis thaliana termed APR1, -2, and -3, that functionally complement a cysH, PAPS reductase mutant strain of Escherichia coli. The coding sequence of the APR clones is homologous with PAPS reductases from microorganisms. In addition, a carboxyl-terminal domain is homologous with members of the Trx superfamily. Further genetic analysis showed that the APR clones can functionally complement a mutant strain of E. coli lacking Trx, and an APS kinase, cysC. mutant. These results suggest that the APR enzyme may be a Trx-independent APS reductase. Cell extracts of E. coli expressing APR showed Trx-independent sulfonucleotide reductase activity with a preference for APS over PAPS as a substrate. APR-mediated APS reduction is dependent on dithiothreitol, has a pH optimum of 8.5, is stimulated by high ionic strength, and is sensitive to inactivation by 5′-adenosinemonophosphate (5′-AMP). 2′-AMP, or 3′-phosphoadenosine-5′-phosphate (PAP), a competitive inhibitor of PAPS reductase, do not affect activity. The APR enzymes may be localized in different cellular compartments as evidenced by the presence of an amino-terminal transit peptide for plastid localization in APR1 and APR3 but not APR2. Southern blot analysis confirmed that the APR clones are members of a small gene family, possibly consisting of three members.
The mechanism by which higher plants reduce inorganic sulfate for assimilation into Cys is not fully understood and there are two competing hypotheses. Fig. 1 illustrates the pathways that have been proposed. The first step in sulfate assimilation, for which there is general agreement, is the formation of APS by ATP sulfurylase. Disagreement centers on the mechanism for sulfate reduction. One hypothesis is that APS is reduced through formation of an organic thiosulfate with reduced glutathione (GSH) (1, 2). Since sulfate is thought to be transferred to GSH, the enzyme that catalyzes this reaction was termed APS sulfotransferase. Further reduction to sulfide is believed to be catalyzed by a thiosulfate reductase (3). An alternative hypothesis is that sulfate reduction in plants is similar to that in enteric bacteria and yeast (4). In these organisms, APS is phosphorylated to PAPS by APS kinase. Sulfite is then formed through the action of PAPS reductase, a thioredoxin (Trx)-dependent enzyme (5, 6). Finally, sulfite is reduced to sulfide by sulfite reductase (7).
Figure 1.
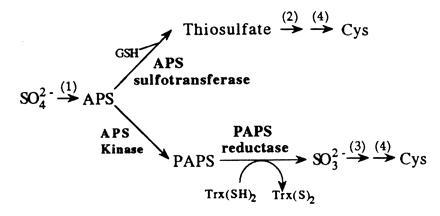
Pathways for sulfate reduction in higher plants. The upper scheme illustrates the adenosine 5′-phosphosulfate (APS) (5′-adenylylsulfate) sulfotransferase pathway and the lower scheme depicts a microorganism-like APS kinase/3′-phosphoadenosine-5′-phosphosulfate (PAPS) reductase pathway. The numbers refer to the following enzymes 1 = ATP sulfurylase, 2 = thiosulfate reductase, 3 = sulfite reductase, 4 = O-acetylserine(thiol)lyase.
Evidence for APS sulfotransferase comes primarily from metabolic studies showing that a cell-free extract of plant chloroplasts is able to form sulfite from APS, but not PAPS (8). Also, Cys-requiring mutants lacking APS sulfotransferase activity have been characterized in the alga Chlorella pyrenoidosa (9). The APS sulfotransferase model for sulfate reduction is bolstered by the finding that this activity is localized in plastids, the primary site for Cys biosynthesis (10). Moreover, sulfotransferase activity is regulated significantly in response to changes in the demand for Cys, as occurs during feeding of reduced sulfur compounds or treatment with heavy metals (11, 12). Despite this evidence, the existence of APS sulfotransferase has been questioned (4, 13). Most perplexing is the observation that plants contain all the enzymes necessary for a bacterial-type sulfate reduction pathway including, APS kinase (14, 15), sulfite reductase (16, 17), and even PAPS reductase (13). Thus, it is argued that, in the absence of definitive evidence for APS sulfotransferase, the idea of this enzyme must be considered tenuous. Indeed, there has been no progress in purifying APS sulfotransferase since its discovery in 1972 (3). Although APS sulfotransferase purification was recently reported from Euglena gracilis (18), the authenticity of the enzyme was subsequently questioned when it was reported that it has properties similar to plant APS kinase, and that plant APS kinase can catalyze an APS sulfotransferase-like activity under in vitro conditions (19).
Aside from the substrate specificity and the absolute requirement of PAPS reductase for Trx (20), APS sulfotransferase and bacterial PAPS reductase have some properties in common, particularly with respect to the reaction they catalyze and the methods used to measure activity. Both enzymes produce sulfite from the appropriate sulfonucleotide in the presence of a thiol-containing reductant such as DTT (6, 21, 22). In the APS sulfotransferase reaction, DTT is thought to function as a sulfate acceptor, although the natural sulfate acceptor has been proposed to be GSH (21, 22). In the PAPS reductase reaction DTT serves as a reductant for Trx (6). While DTT serves as a facile reductant of Trx in vitro, the native reduction system is an NADPH-dependent Trx reductase (20).
It should be noted that it is unnecessary to propose a single mechanism for sulfate reduction in plants, and there may well be two parallel systems consisting of APS sulfotransferase and bacterial-like PAPS reductase pathways. In consideration of the recent successes in defining sulfate assimilation enzymes by functional complementation of microorganism mutants (23), it seemed reasonable to attempt to clone a higher plant PAPS reductase by functional complementation of an Escherichia coli cysH mutant. The cDNAs that were obtained, named APR (APS reductase), revealed that plants contain an APS-preferring reductase that functions independently of Trx. Interestingly, the carboxy terminus of these enzymes is homologous with Trx. This structural feature may explain how APS reductase (APS sulfotransferase) is able to transfer electrons directly from DTT to APS. The results confirm the existence of an APS-dependent sulfate reduction pathway in higher plants.
MATERIALS AND METHODS
General Methods and Specific Materials.
References for preparation of bacteriological media and nucleic acid protocols are Miller (24) and Sambrook et al. (25). Protein was measured using the Bradford (26) assay.
[35S]PAPS was purchased from New England Nuclear. E. coli Trx was from IMCO (Stockholm). All organic compounds were from Sigma.
The E. coli strains used in this study were as follows: JM96 (cysH), JM81A (cysC92, tfr-8), and JM221 (pro-50, lac−, tsx−, galT47, trp-74, his-97, cysD91, argA−, rpsL−, mal−, xyl−, mtl−, ilvA−), all provided by the E. coli Genetic Stock Center (Yale University). A522 (trx, grx::Tn10) was provided by Marjorie Russel and Peter Model (Rockefeller University) (27). DH5α or MV1193 were used for plasmid propagation.
Cloning of APR cDNAs from Arabidopsis thaliana and Complementation Analysis.
A plasmid-based cDNA expression library from A. thaliana, constructed in λYES (28), was used to screen for clones that can complement the Cys requirement of E. coli strain JM96. Plasmid transformation was carried out by electroporation (Bio-Rad Gene Pulser, following the manufacturer’s protocol) using a cuvette with 0.2 cm electrode gap and the following conditions, 25 μF, 200 W, and 2.5 kV. Transformants were selected on Lennox broth medium containing 100 μg/ml Amp, and the colonies were then replica plated onto M-9 minimal medium supplemented with 18 amino acids (excluding Cys and Met), each at a concentration of 25 μg/ml. One million clones were screened and ≈125 growing colonies were obtained. Twenty-five of these were randomly selected for further analysis. The plasmids carried by the positive colonies were isolated and retested for the ability to complement the Cys requirement of JM96. All 25 clones were able to do so. The clones were then mapped with restriction enzymes and found to be of three types. The most abundant type, represented 15 times, was named APR1. At a lower frequency were APR2, represented 9 times, and APR3 was represented once. These plasmids were used for complementation analysis of E. coli strains JM81A, JM221, and A522 as described above for JM96. A522 was tested on M-9 minimal medium lacking all amino acid supplements.
DNA Sequencing.
APR cDNAs were subcloned from the parent vector into pBluescript(SK+) as EcoRI fragments. All clones were completely sequenced on both strands using subclones and plasmid primer sites with an Applied Biosystems model 373 DNA sequencer.
Nucleic Acid Blot Analysis.
Genomic DNA blotting was carried out with DNA isolated from root cultures of A. thaliana exactly as described (29) with a DNA probe labeled by the random primer method (30) using [α-32P]dCTP (3000 Ci/mmol; 1 Ci = 37 GBq).
Enzyme Assays.
Enzyme assays were performed by using soluble cell extracts. E. coli JM96 expressing the APR clones were grown in LB with 2.0 mM isopropyl β-d-thiogalactoside and 100 μg/ml Amp with shaking at 37°C to an absorbance (600 nm) of 1.5. In experiments using E. coli strain A522, cells were grown in M-9 minimal medium containing 40 μg Cys to an A600 of 0.5. The cells were collected by centrifugation, rinsed in Cys-free medium, resuspended in Cys-free medium at the original cell density, and allowed to incubate overnight at 37°C. Cell extracts were prepared at 4°C in buffer containing 50 mM Tris·HCl (pH 8.0) using a bead-mill (Bead Beater or Mini-Bead Beater; Biospec Products, Bartlesville, OK) and 0.1 mm zirconium beads. Cells were disrupted by three 1-min bursts followed by 1-min cooling periods on ice. The extracts were centrifuged at 4°C for 10 min at 12,000 × g, and the supernatant was used for enzyme assay. In some experiments a partially purified preparation of APS2 was tested. A cell extract of JM96 expressing APR2 was fractionated by ammonium sulfate precipitation (70% saturation). The precipitated protein was dissolved in 50 mM Tris·HCl (pH 8.0), dialyzed against this buffer, and then applied to a QSepharose (Pharmacia) ion exchange column. Protein was eluted with a linear gradient of 0 to 250 mM NaCl. Fractions from the elution, centered around 100 mM NaCl, were used for sulfonucleotide reductase assay.
Sulfonucleotide reductase assays were carried out as described by Berendt et al. (6) with modifications as described in the figure and table legends. This assay measures the formation of [35S]SO32− from sulfonucleotide by its conversion to volatile SO2 after addition of acid. At the end of the incubation, Na2SO3 was added to 40 mM, then H2SO4 was added to 3 M. The uncapped reaction tubes were placed into a scintillation vial containing 1 ml trioctylamine. The vial was capped tightly and incubated overnight at room temp to allow volatilized SO2 to be absorbed by the trioctylamine. The reaction tubes were then removed from the vials and 3 ml scintillation fluid added (Ready-Safe; Beckman). Radioactivity was measured by scintillation counting.
[35S]APS was produced by dephosphorylating [35S]PAPS with P1 nuclease (Sigma; catalog N-8630). Five microliters of [35S]PAPS (57.7 × 103 Bq·nmol−1, 0.45 nmol·μl−1) was added to 90 μl 50 mM Tris·HCl (pH 8.0) containing 7.5 units P1 nuclease. The mixture was incubated 15 min at 30°C. Unlabeled APS was added to achieve the specific radioactivity specified in the text. Conversion of PAPS to APS by P1 nuclease was monitored by high voltage, thin layer electrophoresis (TLE) on cellulose plates at 1000 V for 30 min in 50 mM phosphate buffer (pH 7.5) (31). Following electrophoresis the plates were air dried and autoradiographed. The fate of sulfonucleotides in the reductase assays was similarly monitored. APS kinase activity was measured by directly testing for the production of [35S]PAPS from [35S]APS using the TLE assay, and using a coupled assay with pyruvate kinase and lactate dehydrogenase in which the formation of ADP is linked to reduction of NADH (31).
The functionality of the Trx preparation was measured by using the DTNB reduction assay (32) and the insulin reduction assay (33).
RESULTS
Complementation and Sequence Analysis.
Three classes of cDNA clone were isolated from A. thaliana that can complement E. coli strain JM96, a cysH PAPS reductase mutant. Table 1 lists the major features of the APR clones and their similarity to each other. A search of GenBank with the translation products of the APR clones revealed significant homology with two different enzymes; PAPS reductase from microorganisms, and Trx homologs from a variety of organisms. The highest homology is with E. coli PAPS reductase (26% identical/48% similar, amino acid similarity as defined in ref. 34) and yeast protein disulfide isomerase (20% identical/46% similar). Although the overall identity is rather low the APR enzymes show nearly perfect identity with regions of PAPS reductases and Trx homologs that have been shown to be essential for the catalytic function of these enzymes. A comparison of APR1 with several PAPS reductases and Trx homologs is shown in Fig. 2. It should be noted that all the APR enzymes are identical to APR1 within the conserved domains shown in Fig. 2. Tyr-289 and Cys-322 of APR1 are perfectly conserved with amino acids in PAPS reductases. In the E. coli enzyme, mutation of the corresponding residues results in significant decreases in enzyme Vmax (6). APR1 also shows homology with the conserved active site of all Trx homologs and glutaredoxin, especially at Trp-384, Cys-385, and Cys-388. The Cys residues comprise the vicinal-SH groups in Trx that undergo repeated cycles of oxidation/reduction as the enzyme catalyzes electron transfer (20). In addition, the important 2-amino acid spacing between the Cys residues (35) is maintained in APR1. Most Trx homologs contain a helix-breaker amino acid between the Cys residues and APR1 contains such an amino acid, Pro-386. However, this Pro is positioned next to the first conserved Cys, rather than next to the second conserved Cys, as it is in most Trx homologs. Notable exceptions are the recently identified, cytoplasmically localized forms of Trx from A. thaliana in which Pro is near the first conserved Cys (36) and glutaredoxin which generally contain Pro in this position (37). APR1 differs from all Trx homologs in that Gln at position 389 replaces Lys or Arg. Also, Trx homologs contain conserved residues that are dispersed in the primary structure but are clustered in the native protein to form a flat and hydrophobic region near the active site of the protein. These residues, including Gly-33, Pro-34, Pro-76, Gly-92, and Ala-93 of E. coli Trx, are believed to be involved in binding interactions with other proteins (20). Of these, the APR enzymes show conservation with only Pro-76 of E. coli Trx. Considering that bacterial PAPS reductase accepts electrons for sulfate reduction from Trx it is plausible that the carboxyl terminus of the APR enzymes might function in a manner analogous to Trx. If so, the APR enzymes would be expected to function as a Trx-independent PAPS reductase.
Table 1.
Characteristics of APR clones
| Characteristic | Clone
|
||
|---|---|---|---|
| APR1 | APR2 | APR3 | |
| Size, bp | 1634 | 1450 | 1482 |
| Codons | 455 | 406 | 422 |
| Mass (kDa) | 50.8 | 45.6 | 47.3 |
| pI | 6.6 | 5.7 | 7.6 |
| Amino acid identity | |||
| APR1 | 100 | ||
| APR2 | 79 | 100 | |
| APR3 | 77 | 74 | 100 |
Figure 2.

Conserved domains of APR1. The homology of APR1 amino acid sequence is shown with important amino acids in PAPS reductases (A), or Trx homologs (B). The GenBank accession number, and the amino acid stretch of the sequences are as follows: APR1 [U43412, aa 288–291, 320–327 (A), aa 381–389 (B)], CysH (Y07525, aa 208–211, 237–244), Met16 (J05591, modified version cited in ref 6, aa 213–216, 234–250), PAPS reductase Thiocapsa roseopersicina (Z23169, aa 204–207, 234–240), PAPS reductase Anacystis nidulans (M84476, aa 196–199, 226–232), protein disulfide isomerase (PDI) (P08003, aa 404–412), DsbD (P36655, aa 399–407), Trx (M54881, aa 28–36), TrxH (Z14084, aa 36–44), TrxM (X51462, aa 28–36), TrxF (X14959, aa 112–118), Trx3, 4 and 5 (Z35375-Z353476), glutaredoxin (Grx) E. coli (M13449), S-protein (X81994, aa 202–210).
The availability of a Trx (trxA) mutant of E. coli, A522 (27), afforded the possibility to test this hypothesis genetically. Although A522 is not a Cys auxotroph, it requires Cys for growth because the Trx enzyme is not available for efficient function of PAPS reductase. Fig. 3A shows that expression of APR1 in A522 allows it to grow on minimal medium lacking Cys, just as does a plasmid carrying the E. coli TrxA gene, pPMR18 (27). The λYES vector does not have this effect. APR2 and APR3 behave similarly to APR1 (not shown). This result is consistent with the idea that APR enzymes are able to catalyze sulfate reduction independently of Trx.
Figure 3.
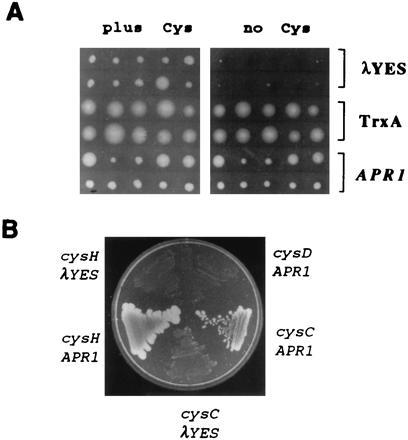
Complementation analysis of APR1. (A) E. coli strain A522 was transformed with λYES, APR1, or a plasmid carrying the E. coli TrxA gene, pPMR18 (27). Transformed colonies were selected on Lennox broth medium containing 100 μg/ml Amp. Ten colonies from each transformation were replica plated onto minimal medium containing 30 μg/ml Cys, and on to minimal medium lacking Cys. (B) APR1 or its parent vector λYES was used to transform three different mutant strains of E. coli, cysH-PAPS reductase (JM96), cysC-APS kinase (JM81A), or cysD-ATP sulfurylase (JM240). A representative colony from each transformation was replica plated onto minimal medium containing 30 μg/ml Cys and onto minimal medium lacking Cys. Only the plate with medium lacking Cys is shown. All the strains grew on the medium in which Cys was provided. The plates were incubated at 30°C for 48 hr prior to photography.
In further complementation tests it was discovered that the APR cDNAs are capable of complementing cysC, an APS kinase mutant of E. coli (Fig. 3B). This strain is unable to produce PAPS, the substrate for PAPS reductase, suggesting that the APR1 enzyme may use APS as a sulfonucleotide substrate. Indeed, APR1 is unable to complement a cysD, ATP sulfurylase E. coli mutant (Fig. 3B) that is unable to produce APS. In total, the sequence information and genetic complementation results indicate that the APR clones may encode novel reductases that use APS as a sulfonucleotide substrate and function independently of Trx. Another possibility for the cysC complementation result is that APR enzymes catalyze both APS phosphorylation and PAPS reduction. However, this is less likely because the APR coding sequences show no homology to any known APS kinase.
Sulfonucleotide Reductase Activity.
The sulfonucleotide specificity and Trx-dependence of the APR1 enzyme was studied in extracts of JM96 using a standard assay for E. coli PAPS reductase (6), and the results are presented in Table 2. Under all conditions tested JM96 showed little to no sulfite formation, confirming the biochemical effect of the cysH mutation. However, JM96 carrying APR1 showed comparatively high rates of sulfite formation, although primarily with APS as a substrate. The activity with APS was 30- to 50-fold greater than with PAPS. Importantly, this activity is dependent on DTT, but is not stimulated by E. coli Trx. The APR-mediated activity is distinct from PAPS reductase activity. An extract of E. coli strain A522, containing a wild-type CysH gene, produced sulfite primarily with PAPS, and this activity was completely dependent on addition of Trx to the assay.
Table 2.
Sulfonucleotide reductase activity of APR1
| Reductant | Specific
activity
|
|||||
|---|---|---|---|---|---|---|
| JM96
|
JM96-APR1
|
A522
|
||||
| PAPS | APS | PAPS | APS | PAPS | APS | |
| No DTT | 0 | 0 | 1 | 5 | 0 | 0 |
| + DTT | 0 | 1 | 7 | 222 | 0 | 0 |
| + DTT + Trx | 0 | 0 | 4 | 226 | 320 | 10 |
Data are given by strain (JM96, JM96–APR1, and A522), and by sulfonucleotide (PAPS and APS). Reaction conditions: 100 μl volume, 50 mM Tris·HCl (pH 8.0), 1 mM EDTA, 25 mM NaF, 0.025 mM [35S]PAPS or APS (57.7 Bq pmol−1), and 0.02 or 0.04 mg cell protein. The following components, when added, were as follows: 5 mM DTT, 7.5 units P1 nuclease, and 0.01 mg Trx. Incubation was for 15 min at 30°C. Each value represents a rate calculated from two measurements with 0.02 and 0.04 mg cell protein. Specific activity = pmoles sulfite min−1·mg−1 protein. Zero activity = less than 1, the minimum, clearly measureable activity is ≈0.1 pmol sulfite formed.
Since the assays were performed using crude cell extracts, and considering the low absolute rate of sulfite formation, it was important to establish the validity of the results. Fig. 4A shows that the rate of sulfite formation is proportional to the amount of protein added to the reaction with both APS and PAPS as substrates. Sulfite formation by the APR1-containing extract with APS as a substrate is linear over time up to 30 min (Fig. 4B). However, with PAPS there is a lag in sulfite production for the first 10 min of incubation and then the reaction is linear up to 30 min (Fig. 4C). This lag period is an indication that PAPS is not used directly by the APR1 enzyme, but must first be converted to APS. Indeed, under the conditions of these assays ≈5% of the PAPS was converted to APS in a 15-min reaction (not shown). Also, APS phosphorylation to PAPS could not be detected in any cell extracts, even those in which APR1 was present. This is likely due to the presence of P1 nuclease in the reaction and/or because there is insufficient ATP in the cell extract to drive APS phosphorylation. Thus the sulfite formed by the APR-containing extract with PAPS is likely due to its slow conversion to APS, and it is unlikely that APR1 is catalyzing APS phosphorylation.
Figure 4.
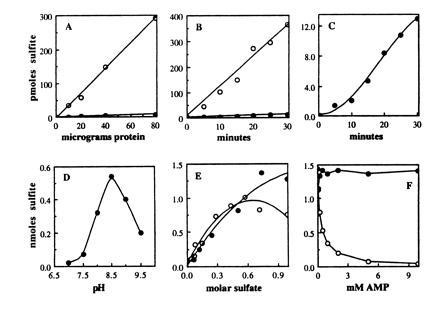
Kinetics of sulfite formation by the APR1 enzyme. (A–C) The open circles reflect reactions with APS as a substrate and the solid circles reflect reactions with PAPS as a substrate. All the abscissas are labeled as in A. (A) Sulfite formed in 15 min at 30°C by varying amounts of protein from an extract of JM96 carrying APR1. (B) Sulfite formation over time in reactions containing 40 μg of protein from an extract of JM96 carrying APR1. (C) The data of graph in C with the scale of the abscissa expanded 30-fold. The sulfonucleotide reductase reaction conditions were as described in Table 2. (D–F) Optimization of APR1-mediated APS reductase activity. All the abscissas are labeled as in D. (D) pH optimum. The reaction mixture contained in a volume of 100 μl: 100 mM Tris·HCl, 500 mM Na2SO4, 1 mM EDTA, 5 mM DTT, 25 μM [35S]APS (≈500 Bq × nmol−1) and 20 μg (2 μl) cell extract. The reaction was incubated for 20 min at 30°C. The pH was adjusted in a 2× mixture of Tris, Na2SO4, and EDTA; then water, DTT, APS, and extract were sequentially added to initiate the reaction. Protein extract was prepared in 50 mM Tris·HCl (pH 8.0) from E. coli strain A522 expressing APR1. (E) Optimal salt concentration. The reaction mixture was as described in D, but with 50 mM Tris·HCl (pH 8.5), 40 μg (4 μl) cell extract from E. coli strain JM96 expressing APR1, and the indicated concentration of Na2SO4 (solid circles) or (NH4)2SO4 (open circles). (F) Inhibition by 5′-AMP. The reaction was as described in E, but with 40 μg (4 μl) cell extract from E. coli strain A522 expressing APR1, and 5′-AMP (open circles) or 2′-AMP (solid circles) at the indicated concentration.
The use of APS and DTT alone for sulfite formation resembles the activity reported for APS sulfotransferase (3, 21, 22). The sulfite formation assay used in Table 2 is optimized for PAPS reductase and differs in some ways from that for APS sulfotransferase. APS sulfotransferase was reported to have a pH optimum of 8.5, it is stimulated by high concentrations of sulfate salts, and is inhibited by the reaction product 5′-adenosinemonophosphate (5′-AMP) (21, 22). Fig. 4D shows that the pH optimum for APR-dependent APS reductase activity is 8.5 and the activity is more than 10-fold greater at pH 8.5 than at 7.0. Sodium sulfate and ammonium sulfate stimulate this activity, with sodium sulfate being the more effective salt (Fig. 4E). Fig. 4F shows that 5′-AMP, but not 2′-AMP, inhibits APS reductase activity. The Ki value of 0.3 mM 5′-AMP is within the same range reported for APS sulfotransferase from a variety of plant sources (38). 3′-Phosphoadenosine-5′-phosphate (PAP), a competitive inhibitor of PAPS reductase (6), also did not affect APS reductase activity at a concentration up to 10 mM (not shown). Using the optimized assay conditions for APS sulfotransferase, the specific activity of the extract of JM96 expressing APR1 (the identical extract used to obtain the results shown in Table 2) was 2.7 nmol/min−1·mg−1 protein, ≈12-fold greater than the activity obtained using the PAPS reductase reaction conditions. All the APR clones gave similar specific activities when assayed using the optimized conditions.
To confirm the major results with the APR enzymes, determined in crude cell extracts as described above, a partially purified preparation of the APR2 enzyme was studied. Ammonium sulfate precipitation and ion exchange chromatography on QSepharose were used to obtain a preparation with an 11.6-fold enrichment of APS reductase specific activity (2.3 nmol/min−1·mg−1 protein in the cell extract compared with 26.6 nmol/min−1·mg−1 protein in the partially purified sample, conditions as in Fig. 3D at pH 8.5). The enzyme in this preparation catalyzed DTT-dependent formation of sulfite from APS. Addition of E. coli Trx had no effect on activity. Sulfite formation using PAPS was still evident, but with a specific activity of 0.03 nmol/min−1·mg−1, 887-fold lower than with APS. In contrast, with the crude cell lysate, sulfite formation from PAPS was 30-fold lower than from APS. This indicates that the purification has partially separated the component necessary for PAPS-dependent activity (probably a 3′-nucleotidase from E. coli) from the APS-dependent activity of the APR2 enzyme. The partially purified preparation was also suitable for directly testing whether APR2 has APS kinase activity. In two different assay systems APR2 showed no ability to form PAPS.
Organelle Localization.
The APR1 enzyme has features of a plastid transit peptide (39). In addition there is a possible transit peptide, protease cleavage site at position 40–43, VHVA (40). The amino termini of APR2 and APR3 are shown in Fig. 5, compared with that of APR1. APR3 is a 5′ truncated clone, but the amino terminus resembles the putative APR1 transit peptide, suggesting that it also is localized in plastids. APR2 encodes the smallest open reading frame. Its amino terminus contains some of the features of an organellar localization sequence, although its short length possibly indicates a mitochondrial or cytoplasmic localization.
Figure 5.

Homology of the amino termini of the APR enzymes. The amino acid alignment of APR1, APR2, and APR3 corresponding to the first 99 amino acids of APR1 is shown. The arrow indicates the position at which homology with E. coli PAPS reductase begins. Periods in the sequences indicate gaps introduced to optimize the alignment. The periods extending from the amino terminus of APR3 indicate that the cDNA is likely truncated at the 5′ end. Identical residues are indicated by a vertical line; conservative substitutions (34) are indicated by a colon (highly conserved) or a period (less conserved).
Analysis of the APR1 Gene Family.
DNA blot analysis was used to examine the size of the APR gene family in A. thaliana. Fig. 6 shows that there are at least three DNA fragments that hybridize with the APR1 cDNA. Identical results were obtained when APR2 or APR3 were used as probes, although the intensity of hybridization to individual bands differed. The multiple smaller bands observed with HindIII-digested DNA can be accounted for by the presence of multiple HindIII sites in all of the APR clones. Overall, the blot results show that there are at least three genes in A. thaliana with significant homology to the APR cDNAs.
Figure 6.
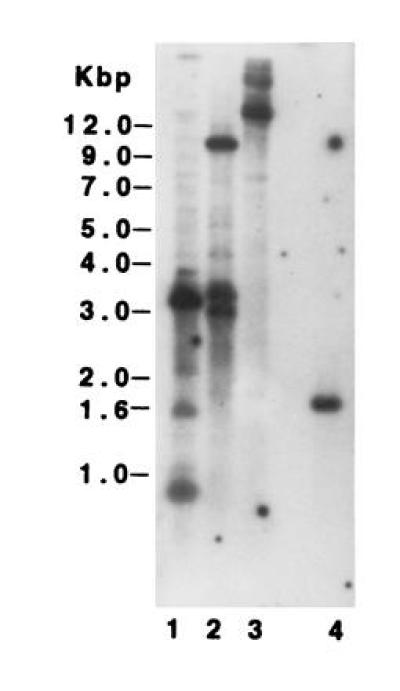
Genomic Southern blot analysis of A. thaliana. DNA was digested with HindIII, lane 1; SstI, lane 2; and XhoI, lane 3. Lane 4 contains 100 pg of the APR1 cDNA. The entire APR1 cDNA was used as the hybridization probe. The position of DNA size markers is shown in kbp.
DISCUSSION
In this report, genetic, sequence, and enzymatic evidence is presented, showing that a gene family exists in the higher plant A. thaliana encoding APS reductase. This result confirms the existence of an APS-dependent sulfate reduction pathway in higher plants. However, our evidence suggests that sulfotransfer is not the catalytic function of the enzyme. First, the homology of APR enzymes with PAPS reductase, especially in the catalytic sites, and the presence of a Trx-like domain, suggests that this enzyme catalyzes sulfonucleotide reduction via a mechanism similar to PAPS reductase. Second, the idea that free sulfite is the product of the APR reaction is supported by the E. coli complementation results. Since E. coli contains only sulfite reductase, an enzyme that is unable to reduce thiosulfate (7), complementation could only occur if free sulfite is produced. The naming of APS sulfotransferase was based on the finding that organic thiosulfate is an in vitro reaction product (3). However, the idea that the enzyme, as originally characterized, transfers sulfate to a thiol was tentative and not based on direct evidence. Indeed, the finding that thiosulfate or free sulfite formation is dependent on the specific thiol compound used in the reaction (41) conflicts with the idea that sulfotransfer is the general mechanism for the enzyme. Based on identical criteria, E. coli PAPS reductase was once thought to be a PAPS sulfotransferase that catalyzes the formation of a thiosulfate with Trx (42, 43). It has now been clearly shown that PAPS reductase accepts electrons from Trx and then uses the electrons for PAPS reduction to free sulfite (5, 6).
The most significant and novel feature of the APR enzymes is the positioning of a Trx-like domain on the same polypeptide as the reductase. This feature provides a potential explanation for the novel property of APS reductase (APS sulfotransferase) to use electrons directly from DTT for sulfate reduction. It is possible that the carboxyl domain, being physically linked to the reductase, serves as an exclusive Trx with the sole function of mediating electron transfer from some donor to the reductase domain. This feature could be related to the unique environment of the chloroplast where Trx serves as the key regulator of entire biosynthetic pathways (44). Since the energy for biosynthesis in the chloroplast is derived from light, a mechanism has evolved in this organelle to activate specific enzymes during illumination when photosynthetic electron transport is active. In the light, Trx reductase transfers electrons from the photosystem I component, ferredoxin to chloroplast Trx. Reduced Trx then alters the redox state of specific enzymes, thereby activating them. Sulfate assimilation in photosynthetic organisms depends primarily on photosynthetically generated reductant. The carboxyl domain of APR enzymes might serve to link APS reduction directly to photosynthetic electron transport, thereby avoiding the competition for Trx, and the diffusion limitation inherent in a system with two separate components. This idea is appealing because chloroplast sulfite reductase is known to be a ferredoxin-dependent enzyme whose activity is linked to photosynthetic electron transport (16, 17). Such a model predicts the existence of a reductase for the APR enzyme. Perhaps this factor could be ferredoxin-dependent Trx. Alternatively, reduced GSH could also serve as an electron source. GSH was previously proposed to serve as the sulfate acceptor for APS sulfotransferase (21, 22).
A feature of most chloroplast enzymes that are subject to redox activation is the presence of vicinal Cys residues. In redox regulated enzymes these Cys residues are the specific targets of Trx (45). Thus the vicinal Cys of APR enzymes might serve as a regulatory domain, although it should be noted that redox regulated enzymes typically do not show homology to Trx, as do the APR enzymes. Moreover, it is significant that not all the APR enzymes may be localized in plastids, yet all contain the Trx-like carboxyl domain.
The APR enzymes are the second example of a substrate reductase containing a Trx-like domain. The pathogen Mycobacterium leprae (46) contains a hybrid Trx reductase–Trx protein in which the Trx domain is located at the carboxyl terminus of the enzyme. In other systems, Trx has been reported as a subunit of phage T7 DNA polymerase (47) and protein disulfide isomerase, a member of the Trx superfamily, is a subunit of prolyl 4-hydroxylase (48). Recently a polypeptide from the higher plant Phalaris coerulescens was reported that contains a Trx-like domain at its carboxyl terminus (49). This protein is thought to be involved in mating compatibility in this species although its exact function is not known.
Acknowledgments
A.S. is a George H. Cook Undergraduate Honors Student at Rutgers University. This project was funded by National Science Foundation Grant IBN-9601145.
Footnotes
Abbreviations: APS, adenosine 5′-phosphosulfate (5′-adenylylsulfate); PAPS, 3′-phosphoadenosine-5′-phosphosulfate; GSH, glutathione; Trx, thioredoxin; PAP, 3′-phosphoadenosine-5′-phosphate.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base (accession nos. U43412U43412, U56921U56921, and U56922U56922).
References
- 1.Schiff J A. In: Encyclopedia of Plant Physiology. Läuchli A, Bieleski R L, editors. 15A. Berlin: Springer; 1983. pp. 401–421. [Google Scholar]
- 2.Schmidt A. Arch Microbiol. 1972;84:77–86. doi: 10.1007/BF00408084. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt A. Arch Microbiol. 1973;93:29–52. [PubMed] [Google Scholar]
- 4.Schwenn J D. Z Naturforsch C. 1994;49:531–539. [Google Scholar]
- 5.Tsang M L-S. In: Thioredoxins: Structure and Function. Gadal P, editor. Paris: Edition Centre National de la Recherches Scientifique; 1983. pp. 21–24. [Google Scholar]
- 6.Berendt U, Haverkamp T, Prior A, Schwenn J D. Eur J Biochem. 1995;233:347–356. doi: 10.1111/j.1432-1033.1995.347_1.x. [DOI] [PubMed] [Google Scholar]
- 7.Kredich N M. In: Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology. Neidhardt F C, Ingram J L, Low K B, Magasanik B, Schaechter M, Umbarger H E, editors. Washington, DC: Am. Soc. Microbiol.; 1987. pp. 419–428. [Google Scholar]
- 8.Goldschmidt E E, Tsang M S-S, Schiff J A. Plant Sci Lett. 1975;4:293–299. [Google Scholar]
- 9.Schmidt A, Abrams W R, Schiff J A. Eur J Biochem. 1974;47:423–434. doi: 10.1111/j.1432-1033.1974.tb03709.x. [DOI] [PubMed] [Google Scholar]
- 10.Brunold C, Suter M. Planta. 1989;179:228–234. doi: 10.1007/BF00393693. [DOI] [PubMed] [Google Scholar]
- 11.Wyss H-R, Brunold C. Planta. 1979;147:37–42. doi: 10.1007/BF00384588. [DOI] [PubMed] [Google Scholar]
- 12.Nussbaum S, Schmutz D, Brunold C. Plant Physiol. 1988;88:1407–1410. doi: 10.1104/pp.88.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwenn J D. Z Naturforsch C. 1989;44:504–508. [Google Scholar]
- 14.Burnell J N, Anderson J W. Biochem J. 1973;134:565–579. doi: 10.1042/bj1340565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain A, Leustek T. Plant Physiol. 1994;105:771–772. doi: 10.1104/pp.105.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aketagawa J, Tamura G. Agric Biol Chem. 1980;44:2371–2378. [Google Scholar]
- 17.Krueger R J, Siegel L M. Biochemistry. 1982;21:2892–2904. doi: 10.1021/bi00541a014. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Schiff J A. Biochem J. 1991;274:355–360. doi: 10.1042/bj2740355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schiffmann S, Schwenn J D. FEBS Lett. 1994;355:229–232. doi: 10.1016/0014-5793(94)01193-1. [DOI] [PubMed] [Google Scholar]
- 20.Holmgren A. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 21.Schmidt A. Planta. 1975;124:267–275. doi: 10.1007/BF00388689. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt A. Planta. 1976;130:257–263. doi: 10.1007/BF00387830. [DOI] [PubMed] [Google Scholar]
- 23.Leustek T. Physiol Plant. 1996;97:411–419. [Google Scholar]
- 24.Miller J H. Experiments in Molecular Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1972. [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 26.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Russel M, Model P, Holmgren A. J Bacteriol. 1990;172:1923–1929. doi: 10.1128/jb.172.4.1923-1929.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elledge S J, Mulligan J T, Ramer S W, Spottswood M, Davis R W. Proc Natl Acad Sci USA. 1991;88:1731–1735. doi: 10.1073/pnas.88.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leustek T, Murillo M, Cervantes M. Plant Physiol. 1994;105:897–902. doi: 10.1104/pp.105.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feinberg A P, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 31.Segel I H, Renosto F, Seubert P A. Methods Enzymol. 1987;143:334–349. doi: 10.1016/0076-6879(87)43061-9. [DOI] [PubMed] [Google Scholar]
- 32.Russel M, Model P. J Biol Chem. 1986;261:14997–15005. [PubMed] [Google Scholar]
- 33.Holmgren A. J Biol Chem. 1979;254:9627–9632. [PubMed] [Google Scholar]
- 34.Gribskoff M, Burgess R R. Nucleic Acids Res. 1986;14:6745–6763. doi: 10.1093/nar/14.16.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gleason F K, Lim C J, Gerami N M, Fuchs J A. Biochemistry. 1990;29:3701–3709. doi: 10.1021/bi00467a016. [DOI] [PubMed] [Google Scholar]
- 36.Rivera-Madrid R, Mestres D, Marinho P, Jacquot J-P, Decottignies P, Miginiac-Maslow M, Meyer Y. Proc Natl Acad Sci USA. 1995;92:5620–5624. doi: 10.1073/pnas.92.12.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holmgren A, Åslund F. Methods Enzymol. 1995;252:283–292. doi: 10.1016/0076-6879(95)52031-7. [DOI] [PubMed] [Google Scholar]
- 38.Schmidt A. Planta. 1975;127:93–95. doi: 10.1007/BF00388867. [DOI] [PubMed] [Google Scholar]
- 39.von Heijne G, Steppuhn J, Herrmann R G. Eur J Biochem. 1989;180:535–545. doi: 10.1111/j.1432-1033.1989.tb14679.x. [DOI] [PubMed] [Google Scholar]
- 40.Gavel Y, von Heijne G. FEBS Lett. 1990;261:455–458. doi: 10.1016/0014-5793(90)80614-o. [DOI] [PubMed] [Google Scholar]
- 41.Tsang M L-S, Schiff J A. Plant Cell Physiol. 1976;17:1209–1220. [Google Scholar]
- 42.Tsang M L-S, Schiff J A. J Bacteriol. 1976;125:923–933. doi: 10.1128/jb.125.3.923-933.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsang M L-S, Schiff J A. J Bacteriol. 1978;134:131–138. doi: 10.1128/jb.134.1.131-138.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buchanan B B, Schürmann P, Decottignies P, Lozano R M. Arch Biochem Biophys. 1994;314:257–260. doi: 10.1006/abbi.1994.1439. [DOI] [PubMed] [Google Scholar]
- 45.Chardot T, Meunier J-C. Biochem J. 1991;278:787–791. doi: 10.1042/bj2780787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wieles B, Van Noort J, Drijfhout J W, Offringa R, Holmgren A, Ottenhoff T H M. J Biol Chem. 1995;270:25604–25606. doi: 10.1074/jbc.270.43.25604. [DOI] [PubMed] [Google Scholar]
- 47.Modrich P, Richardson C C. J Biol Chem. 1975;250:5515–5522. [PubMed] [Google Scholar]
- 48.Pihlajaniemi T, Helaakoski T, Tasanen K, Myllylä R, Huhtala M, Koivu J, Kivirikko K I. EMBO J. 1987;6:643–649. doi: 10.1002/j.1460-2075.1987.tb04803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Nield J, Hayman D, Landrige P. Plant J. 1995;8:133–138. doi: 10.1046/j.1365-313x.1995.08010133.x. [DOI] [PubMed] [Google Scholar]