

Abstract
The costimulatory molecule OX40 (CD134) is required in many instances for effective T cell-mediated immunity, controlling proliferation and survival of T cells after encountering specific antigen. We previously found that the functional targets of OX40 are survivin and aurora B that regulate proliferation, and Bcl-2 anti-apoptotic family members that regulate survival. However, the intracellular pathways from OX40 that mediate these effects are unclear. Here, we show that OX40 signaling can target the canonical NF–κB (NF–κB1) pathway in peripheral antigen-responding CD4 T cells. Phosphorylation of IκBalpha, nuclear translocation of NF-κB1/p50 and RelA, and NF-κB1 activity, are impaired in OX40-deficient T cells. Retroviral transduction of active IKKβ that constitutively activates NFκB1 rescues the poor expansion and survival of OX40-deficient T cells, directly correlating with increased expression and activity of survivin, aurora B, and Bcl-2 family members. Moreover, active IKKβexpression alone is sufficient to restore the defective expansion and survival of OX40-deficient T cells in vivo when responding to antigen. Thus, OX40 signals regulate T cell number and viability through the NF –κB1 pathway that controls expression and activity of intracellular targets for proliferation and survival.
Keywords: Mouse, T cells, Antigens/peptides, Cell Surface Molecules, Cell activation
Introduction
Costimulatory signals play crucial roles in T cell activation, function, and fate. Since the CD28/B7 interaction was identified as a prominent costimulatory signal for T cells, many other costimulatory molecules have been identified. OX40 (CD134), a tumor necrosis factor receptor (TNFR) family member, which is expressed by activated T lymphocytes, has been shown to play a strong role in regulating proliferation, cytokine production, survival, and memory development of T cells (1). Targeting OX40 with an agonist antibody has shown promise in cancer immunotherapy and prevention of infectious diseases (2–4). In contrast, blocking OX40 signals with an anti-OX40 ligand antibody or an OX40-Ig fusion protein has many potential applications in autoimmune diseases (5–8).
We have previously shown that OX40 can target the PI3K/PKB pathway and that sustained PKB (Akt) signaling driven by OX40 leads to upregulation of several Bcl-2 family members, including Bcl-xL, Bcl-2, and Bfl-1, that control T cell longevity (9, 10). OX40-mediated PKB activation also promotes survivin expression that controls T cell proliferation and expansion (11) in conjunction with a kinase termed aurora B (12). Although the activation of PKB is a critical event for proliferation and survival of T cells, it remains unclear what other downstream pathways contribute to, or are essential for, upregulation of Bcl-2 family members and survivin and aurora B kinase.
One likely candidate responsible for promoting the costimulatory signal regulating proliferation and cell survival of T cells is NF –κB (13). In mammalian cells, the NF –κB family consists of 5 distinct members; c-Rel, p65/RelA, RelB, p50/p105 (NF –κB1), and p52/p100 (NF–κB2), which are able to homodimerize or heterodimerize. Under normal conditions, NF –κB is sequestered in an inactive state by I κB inhibitory molecules in the cytoplasm. Activation of the canonical N –κB (NF –κB1) pathway is initiated by signal-dependent phosphorylation, ubiquitination and subsequent degradation of I κB, which allows cytoplasmic NF –κB complexes, especially NF –κB1-RelA, to stably translocate to the nucleus and activate gene transcription. IκB phosphorylation is catalyzed by the I κB kinase (IKK) complex that contains two homologous catalytic subunits, IKKα and IKKβ, and the regulatory subunit IKKγ (14, 15). Activation of IKKβ subunits is essential for the NF –κB1 pathway in response to all pro-inflammatory stimuli (16–18).
Published data have shown in various ways that the NF –κB family directly or indirectly regulates proliferation, survival, Th1/Th2 differentiation, and tolerance of T cells. A combined IκBα/IκBε deficiency in mice leads to disruption of lymphocyte production and neonatal death. In IκBα/IκBε-deficient fetuses, B cells and T cells die by apoptosis, and IκBα/IκBε−/− reconstituted bone marrow chimeras exhibit a nearly complete absence of T and B cells that is not rescued by cotransfer of wild-type bone marrow. These findings demonstrate that I κBs tightly control NF –κB activity in vivo and that increased NF –κB activity intrinsically impairs lymphocyte survival (19). However, this most likely reflects an activity not directly related to NF –κB activity in T cells. Data from mice expressing IκBα as a transgene in the T lineage, where it was refractory to degradation and NF –κB was constitutively repressed, showed impaired production of peripheral T cells, with those being produced additionally being defective in proliferating and surviving to mitogens or γ-chain cytokines (20, 21). This suggests that activation of NF –κB in T cells might primarily promote proliferation and antagonize apoptosis. In line with this, the anti-apoptotic molecules, Bfl-1/A1 and Bcl-xL, were associated with NF –κB activation in stimulated T cells leading to increased T cell survival (22, 23). Furthermore, mice deficient in NF –κB1 could not develop EAE, associated with defective T cell priming (24). Similarly, also in NF –κB1 knockout mice, antigen-specific CD4 T cell proliferation and IFN-γ production were impaired following infection with the intracellular protozoan parasite Leishmania major (25). In line with a role in Th1 responses, mice transgenic for a su per repressor IκBα, expressed in T cells, were defective in DTH responses and IFN-γ production, but mounted normal Th2 responses (26, 27). Of further interest, tolerant T cells that fail to produce interleukin (IL)-2 have been associated with a predominance of p50-p50 homodimers that bind to the IL-2 promoter κB site, correlating with repressed activity of NF –κB-driven transcription (28).
Collectively, these data suggest that NF –κB1 might be central to many effects of costimulatory receptors that can control clonal expansion and differentiation of T cells to antigen. Several reports have shown that OX40 recruits tumor necrosis factor receptor-associated factors (TRAFs) 2, 3, and 5 to its cytoplasmic tail, and that in transfection systems in a number of mammalian cells, the OX40 interaction with TRAF 2 and 5 can lead to activation of the NF –κB1 pathway (29, 30). However, the ability of OX40 to target NF –κB1 in primary T cells responding to antigen has not been investigated thoroughly, and a direct demonstration that OX40-controlled NF –κB1 activation is relevant for a functional T cell response has not been shown. The studies presented here identify and characterize the NF –κB1 pathway as a principal target of OX40 signals in primary T cells, and that this is intimately involved in regulating both aurora B and survivin activities that sustain T cell expansion, and promoting expression of Bcl-2 anti-apoptotic molecules that control long-term survival.
Materials and Methods
Mice
OT-II and OT-II × OX40-deficient (KO) TCR-transgenic mice, expressing a TCR composed of variable (Vβ5 and Vα2) chains responsive to an ovalbumin (OVA) peptide 323–339, were bred on a BL/6 background; AND or AND × OX40 KO TCR-transgenic mice, expressing Vβ 3 and Vα11 TCR, responsive to a moth cytochrome c (MCC) peptide 88–103, were bred on a B10.BR background. BL/6 and B10.BR mice were purchased from Jackson Laboratory. All experiments were in compliance with the regulations of the La Jolla Institute Animal Care committee in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
Peptides, Chemicals, and Antibodies
OVA 323–339 and MCC 88–103 were synthesized by A&A Laboratories (San Diego). Histone H3 (#14–411) and in vitro kinase assay reagents were from Upstate Inc. Anti-OX40 (OX86) was from a hybridoma obtained from the European cell culture collection (Wiltshire, UK). Anti-CD3 (2C11), anti-IFN-γ (XMG1.2), anti-CD28 (37.51), biotinylated anti-OX40 (OX86), mouse IL-2 and IL-4 were from BD PharMingen. Anti-human/mouse survivin (D-8, sc-17779), Actin (C2, sc-8432), IκBα (C-21, sc-371), p50 (H-119, sc-7178), RelA (A, sc-109) and Lamin B1 (C-20, sc-6216) for Western blot were from Santa Cruz Biotech. Aurora B (#3094) for Western blot and immunoprecipitation, p- IκBα (#9241), Bcl-xL (#2762), Bcl-2 (#2872), Bfl-1 #4622), peroxidase-conjugated anti-rabbit (#7054) or anti-mouse Ig (#7056) for Western blot, were from Cell Signaling Technology (Beverly, MA). All FITC-, PE-, Cyt- conjugated antibodies were from BD Pharmingen. Nuclear and cytoplasmic lysates were prepared by using NE-PER Nuclear and Cytoplasmic Extraction Reagent (Pierce). PKH26-GL was from Sigma.
T Cells and APC
Naïve CD4+ T cells were purified from spleen and lymph nodes by nylon wool depletion, followed by antibody and complement treatment (9). The cells were > 90% CD4+ and > 95% of these cells expressed the appropriate TCR and a naïve phenotype. APC were from spleens of syngeneic non-transgenic mice by depleting T cells. APC were treated with mitomycin c (100 μg/ml) for 30 min at 37°C. In some experiments, a fibroblast DCEK cell line transfected with both I-Ek and OX40L and constitutively expressing B7-1 was also used as an APC (32).
T Cell Cultures
Cultures were in 48-well plates containing 1 ml RPMI 1640 (Invitrogen) with 10% fetal calf serum (Omega Scientific). Naïve CD4 cells were plated at 5 × 105/ml with 2 × 106/ml APCs and various concentrations of antigen. For determining secondary responses, 5 × 105 T cells were recultured with 2 × 106 APCs per ml. For Western blot, live CD4 T cells were isolated from culture with CD4 (L3T4) MicroBeads by Miltenyi Biotec (#130-049–201). For generation of effector T cells for NF-κB1 signaling experiments, CD4 T cells (2 × 106 cells per ml) were stimulated with 5 μml plate-bound anti-CD3, 5 μg/ml soluble anti-CD28, 10 μg/ml anti-IFN-γ, 10 ng/ml IL-2, and 10 ng/ml IL-4. Cells were initially stimulated for 3 days then transferred to flask containing new culture media containing 1 ng/ml IL-2 and expanded for an additional 3 days without anti-CD3 stimulation.
Retroviral Transduction
cDNA for a constitutively active (CA) mutant of IKKβ was subcloned into the murine bicistronic retroviral expression vector pCFG5-IEGZ (a gift from Dr. Thomas Wirth, Ulm University, Germany) (33, 34). Retroviral transduction was performed as before (11). 5 ×105 T cells were stimulated with antigen/APCs. After 2 days, the supernatant was replaced with 1 ml viral supernatant containing 5 μg/ml Polybrene (Sigma), and the cells spun for 1 hr at 32°C and incubated at 32°C for 8 hr. This was repeated the following day. Viral supernatant was removed and replaced with fresh medium, and T cells re-cultured. Expression of GFP was determined by flow cytometry gating on Vβ5+ T cells. GFP-expressing T cells were purified by sorting using a FACS Vantage SE I high-speed cell sorter (BD Immunocytometry Systems, San Jose, CA).
Adoptive Transfer
T cells were cultured with antigen and transduced on day 2/3 with retroviral vectors (11). Cells were recultured for 3 more days. GFP+ CD4 cells were sorted, and 3.5 × 106 injected i.v. into naïve mice. Mice were challenged i.p with 100 μg OVA protein in PBS, or PBS without antigen. Numbers of T cells were calculated based on total cell numbers in spleen and lymph nodes, together with percentages of GFP+Vβ+ cells visualized from FACS.
Cytokine secretion, cell recovery, proliferation
Cytokines were measured by ELISA or intracellular flow staining (9). T cell survival in vitro was determined by trypan blue exclusion. Proliferation was measured in triplicate cultures by incorporation of 3H-thymidine (1 μCi/well; ICN Pharmaceuticals) during the last 12 hr of culture.
Immunopreciptation and Immunoblotting
Live CD4 cells were recovered by Ficoll treatment and positive selection with anti-CD4 microbeads (Miltenyi Biotec Inc). Cells were lysed in ice-cold RIPA Lysis Buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin) for 30 min. Insoluble material was removed and lysates used for Western blotting, or immunoprecipitated overnight with primary antibodies followed by incubation with protein G agarose beads at 4°C for 2 hr. The washed immunoprecipitates were boiled in SDS sample buffer. Protein content was determined by Bio-Rad protein assay kit (Bio-Rad, Hercules, CA). Equal amounts (30–50 μg) were loaded onto 4–12% NuPage Bis-Tris precasting gels (SDS-PAGE), transferred onto PVDF membrane (Invitrogen), and immunoblotted. All blots were developed with the ECL immunodetection system (Amersham Pharmacia Biotech, Piscataway, NJ).
In vitro kinase assay
Kinase activity was measured in immunoprecipitates from live T cell lysates. To assay Aurora B kinase activity, 500 μg of cell lysates were immunoprecipitated with 2 μg anti-Aurora B by Catch and Release v2.0 reversible immunoprecipitation system from Upstate Inc (#17–500A). 10 μl immunoprecipitates were resuspended in 40 μl of kinase reaction buffer [20 μl ADBI assay dilution buffer I (#20–108), 20 μg Histone H3, and 10 μCi of [32P]ATP (3,000 Ci/mmol) in magnesium/ATP cocktail (#20–113) from Upstate Inc], and incubated in a shaking water bath for 10 min at 30°C. 25 μl kinase reactions were transferred on P81 Phosphocellulose Squares (#20–134) from Upstate Inc. Following washes with phosphoric acid and acetone, the phosphocellulose squares were transferred scintillation vials, and phosphorylated Histone H3 was quantitated by using a scintillation counter.
Nuclear Extraction and Electrophoretic Mobility-Shift Assay (EMSA)
After stimulation, T cells were lysed and nuclei were recovered by using NE-PER Nuclear and Cytoplasmic Extraction Reagent. Nuclear extracts (5 μg protein) were incubated with biotin-labeled double-stranded oligonucleotide probes. The probes specific for activator protein NF–κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′) and EMSA kit were purchased from Panomics (AY1030). Complexes were separated on a 6% non denaturing polyacrylamide gel (Invitrogen, EC62652). The gel was transferred to a nylon membrane (Biodyne B from PALL, P/N 60208) and detected using streptavidin-HRP and a chemiluminescent substrate. The bands were visualized after exposure to X-ray film from KODAK (BioMax MR Film, #870–1302).
Results
OX40 activates NF–κB1 in primary CD4 T cells
Naïve CD4 T cells do not express OX40 but up-regulate it 12 hr or more after Ag/APC or TCR/CD28 engagement (10). To investigate the role of OX40 in activation of the NF–κB1 pathway in physiologically stimulated primary T cells, we pre-activated CD4 T cells for 6 days (Fig. 1a) or 18 hr (Fig. 1b) from WT and OX40 KO TCR transgenic mice. These activated/effector-like T cells, now expressing OX40 in the case of WT T cells, were then stimulated with antigen presented on artificial fibroblast APC bearing B7.1 and OX40L. Nuclear translocation of p50 and RelA was markedly higher in WT T cells than in OX40 KO cells, correlating with reduced phosphorylation of IκBαin the cytoplasm and impaired activation-dependent degradation of IκBα (Fig. 1a). Furthermore, the results demonstrated that maximal p50 and RelA accumulation in the nucleus in response to antigen was dependent on OX40, showing a direct synergy between TCR, CD28, and OX40 signals in targeting the NF-κB1 pathway (Fig. 1b). Similarly, this observation was confirmed in a more physiological antigenic system where WT and OX40 KO CD4 T cells were cultured with antigen and splenic T cell-depleted APC for 2 days. Translocation of NF–κB-binding complexes to the nucleus was significantly decreased in OX40 KO T cells compared to WT T cells (Fig. 1c). Moreover, additional ligation of OX40 in this system with agonist antibody strongly increased NF-κB-binding complexes in WT T cells (Fig. 2a). Lastly, to test the effect of OX40 on the NF–κB1 pathway in the absence of other signals, we preactivated naïve primary CD4 T cells from TCR transgenic mice for 3 days, and then OX40 was triggering by soluble antibody on these T cells in secondary cultures in isolation without TCR or CD28 signals. OX40 stimulation strongly prolonged phosphorylation of IκBα over 4–20 hours (Fig. 2b). In addition, transient upregulation of p50 and RelA in the nucleus was observed, although this activity was obviously not sustained (Fig. 2b). The specificity of the agonist antibody was confirmed in that it had no effect in promoting nuclear accumulation of p50 or RelA in OX40−/− T cells (Fig. 2c). In some cases, a stronger upregulation of p50 and RelA was seen when OX40 was cross-linked by bead coating the agonist antibody, perhaps replicating the degree of cross-linking that might occur with OX40L expressed on an APC (Fig. 2c). These observations clearly indicate that triggering OX40 on primary CD4 T cells activates the NF–κB1 pathway. OX40 largely functions as a classical costimulatory molecule in synergizing with antigen signals and augmenting NF-κB1 activity promoted by the TCR and molecules such as CD28, and under physiological conditions OX40/OX40L interactions contribute to the extent of NF–κB1 activity induced after antigen recognition.
Figure 1.

Defective activation of the NF–κB1 pathway in the absence of OX40. Naïve CD4 T cells from wild-type (WT) or OX40-deficient (OX40 KO) AND TCR transgenic mice were stimulated in vitro with anti-CD3/CD28 (a and c) or APC and peptide (b). (a) After 6 days primary culture, 5 × 105 per ml of effector T cells were restimulated with 1 μM MCC peptide presented on 1 × 106 per ml of DCEK fibroblast cells that expressed I-Ek, B7-1, and OX40L for 1–4 hr. (b) After 18 hr of primary culture with 0.75 μM MCC peptide and T cell-depleted APC, purified T cells were re-stimulated with 1 μM antigen and DCEK/B7/OX40L cells for 24 hr. Protein expression of IκB, p-IκB, Rel A and p50, as well as β-actin and Lamin B, in total or cytosol and nuclear lysates were determined by western blotting. Densitometer quantitation was relative to the first data set in each case (indicated by a value of 1). (c) After 6 days of primary culture, WT or OX40 KO T cells were restimulated with T-depleted APC and 0.5 μM peptide. 48 hr later, live T cells were isolated and NF–κB activity was determined by EMSA from nuclear extracts. All data are representative of at least two independent experiments.
Figure 2.

OX40 signaling sustains phosphorylation of IκBα and activates NF-κB1. (a) Naïve CD4 T cells from wild-type (WT) or OX40-deficient (OX40 KO) AND TCR transgenic mice were stimulated in vitro with anti-CD3/CD28. After 6 days of primary culture, T cells were restimulated with T-depleted APC and 0.5 μM peptide in the presence or absence of 10 μg/ml agonist anti-OX40 or rat IgG. 48 hr later, live T cells were isolated and NF–κB activity was determined by EMSA from nuclear extracts. (b) WT T cells were stimulated for 3 days with anti-CD3/CD28, and then extensively washed to remove excess agonistic antibodies and cytokines. Three million cells per ml of T cells were recultured with 200 μg/ml agonist anti-OX40 or rat IgG for the indicated periods. Protein expression of IκB, p-IκB, Rel A and p50, as well as β-actin and Lamin B, in total or cytosol and nuclear lysates were determined by western blotting. Densitometer quantitation was relative to the first data set in each case (indicated by a value of 1). (c) Naïve CD4 T cells from WT or OX40 KO AND TCR transgenic mice were stimulated in vitro with anti-CD3/CD28. After 24 hr primary culture, T cells were restimulated with control Ig or anti-OX40 coated beads for 24 hr, and protein expression of Rel A and p50 and Lamin B determined in nuclear lysates. Densitometer quantitation was relative to the first data set in each case (indicated by a value of 1). All data are representative of at least two independent experiments.
Active IKKβ Restores passive Proliferation and Survival in OX40-deficient T Cells in vitro
To determine if weak NF–κB1 activity contributes to the defective proliferation and survival of OX40 KO T cells (11), we retrovirally transduced antigen-stimulated T cells after 2 days with a GFP-IRES vector containing constitutively active IKKβ (CA-IKKβ), which bears two serine to glutamic acid mutations in the activation loop allowing direct targeting and activation of NF–κB1 (33). After transduction, T cells were passively recultured in the absence of further antigen stimulation and their proliferation assessed by thymidine incorporation after 1 (day 4) and 5 days (day 8). Previously, we found that the inability to express OX40 resulted in strongly reduced passive division over time, as well as poor survival (9, 11). IKKβ-reconstitution in OX40 KO T cells led to high amounts of expression of IKKβ (Fig. 3a), and this restored passive proliferation at late times, as measured by thymidine incorporation, to a level similar to that of WT T cells (Fig. 3b). No effect on T cell division was observed when dilution of the dye PKH26 was used as a monitor of this activity (Fig. 3c), whereas analyses of the percent of dead/dying cells showed that CA-IKKβ reversed the excessive death seen in OX40 KO T cells (Fig. 3d). In line with this, enumerating the recovery of live T cells through monitoring GFP expression showed that expression of active IKKβ allowed OX40 KO T cells to persist similarly to WT T cells through to day 8. Moreover, active IKKβ enhanced the ability of OX40 KO T cells to survive for a longer time in vitro, comparable to WT T cells, after day 8 (Fig. 3e).
Figure 3.

Retroviral transduction of OX40 KO T cells with active IKKβ reverses defective passive proliferation and survival in vitro. Naïve CD4 T cells from WT or OX40 KO OT-II TCR transgenic mice were stimulated with peptide/APCs, and transduced on days 2/3 with retroviral vectors expressing GFP, or GFP with CA-IKKβ and then recultured without any further stimulation. In some cases, T cells were also labeled with the dye PKH26. (a) On day 6 of primary culture, GFP+ T cells were sorted and analyzed for IKKβ and β-actin. (b) Primary passive proliferation on day 4 and day 8 measured in unseparated cultures by pulsing with tritiated thymidine for 20 hr. Data are mean cpm ± SD from triplicate cultures and are representative of three experiments (* P < 0.05, Student’s unpaired t-test). (c) Primary passive cell division of GFP+ T cells on day 6 based on dilution of PKH26. Data are representative of three experiments. (d) Quantitation of percent dead or dying cells based on FSC/SSC gating within the GFP+ T cell fraction on day 6. Data are representative of three experiments. (* P < 0.05, Student’s unpaired t-test). (e) GFP+Vβ5+ T cell recovery normalized to take into account differences in initial transduction efficiency between cultures. Numbers of GFP+ cells present on day 4 were assigned a value of 100%, and numbers surviving on day 8 and day 12 were used to calculate the percentage recovery relative to day 4. Data represent the mean ± s.d. percentage change from three separate experiments (* P < 0.05, ** P < 0.01, Student’s unpaired t-test).
Active IKKβ Reverses Defective Recall Responses of OX40 KO T Cells in vitro
Our previous data have additionally shown that recall responses of already primed CD4 T cells are also impaired without OX40 signals (10, 11). Effector T cells expressing active IKKβ from primary naïve cultures were therefore sorted based on GFP expression, and equal numbers restimulated with antigen. OX40 KO cells transduced with active IKKβ displayed enhanced proliferation essentially restoring the response to WT levels (Fig. 4a), and furthermore the defective recovery of OX40 KO T cells over time was also reversed (Fig. 4b). Again, no difference in the rate of cell division was observed through analysis of dilution of PKH26 (Fig. 4c), but CA-IKKβ suppressed the enhanced accumulation of dead/dying OX40 KO T cells (Fig. 4d). Active IKKβ did not promote greater IL-2 or IL-4 production in OX40 KO T cells when assessed early after antigen restimulation, either at 16h by intracellular staining (Fig. 4e) or at 40h by ELISA assay of supernatants (Fig. 4f). At 144h post stimulation, increased levels of both IL-2 and IL-4 were evident in T cells transduced with active IKKβ, particularly in WT cells (Fig. 4f). Thus, the NF-κB1 pathway can strongly regulate sustained production of cytokines, especially IL-2. However, as the control vector-transduced OX40 KO T cells were not substantially impaired in producing IL-2 or IL-4 compared to control WT T cells, and as short and long-term production of IL-2 and IL-4 in the various cultures did not correlate with cell expansion/survival over time (Fig. 4b), it is unlikely that enhanced T cell proliferation and survival induced by activation of this pathway was mediated solely by a feedback loop through these growth-promoting cytokines.
Figure 4.

Active IKKβ restores the proliferation and survival of OX40 KO T cells in secondary responses in vitro. Naïve CD4 cells from WT or OX40 KO OT-II TCR transgenic mice were stimulated with peptide and APCs. On day 2/3, T cells were transduced with retroviral vectors expressing GFP, or GFP with CA-IKKβ. On day 6 of primary culture, GFP+ CD4 T cells were sorted, and restimulated with APCs and peptide. In some cases, T cells were also labeled with the dye PKH26. (a) Recall proliferation on day 1 to day 7 measured by pulsing with tritiated thymidine for the last 20 hr. Data are mean cpm ± SD from triplicate cultures and are representative of three experiments (* P<0.05, ** P<0.01, Student’s unpaired t-test). (b) Recall survival, based on recovery of GFP+Vβ5+ T cells over time. Cell numbers present on day 0 were assigned a value of 100%, and cell numbers surviving on day 3 to day 12 were used to calculate the percentage recovery. Data represent the mean ± s.d. percentage change from three separate experiments (* P < 0.05, Student’s unpaired t-test). ). (c) Recall cell division of GFP+ T cells on day 4 based on dilution of PKH26. Data are representative of three experiments. (d) Quantitation of percent dead or dying cells based on FSC/SSC gating within the GFP+ T cell fraction on day 4. Data are representative of three experiments. (* P < 0.05, Student’s unpaired t-test). (e–f) Recall IL-2 and IL-4 production by intracellular staining at 16 h (e) and by ELISA at 40 h and 144 h (f, left and right respectively). Data are representative of three experiments (e) or means ± s.d. from three experiments (f) (* P < 0.05, Student’s unpaired t-test)
Active IKKβ Restores Defective Expression of Aurora B, Survivin, and Bcl-2 anti-apoptotic molecules in the absence of OX40
OX40 signals promote T cell expansion by sustaining aurora B and survivin expression (11, 12), while controlling T cell long-term survival by regulating Bcl-2 family proteins (9, 10). To determine whether active IKKβ altered expression of aurora B, survivin and Bcl-2 family members, GFP-expressing T cells were sorted after retroviral transduction with active IKKβ restimulated with antigen, and then CD4 cell lysates examined by western blot. OX40 KO T cells with control vector had reduced amounts of these proteins as previously observed. This was essentially fully reversed in OX40 KO T cells expressing active IKKβ (Fig. 5a). Furthermore, active IKKβ completely rescued aurora B kinase activity of OX40 KO T cells as measured by an in vitro kinase assay after restimulation with antigen (Fig. 5b).
Figure 5.
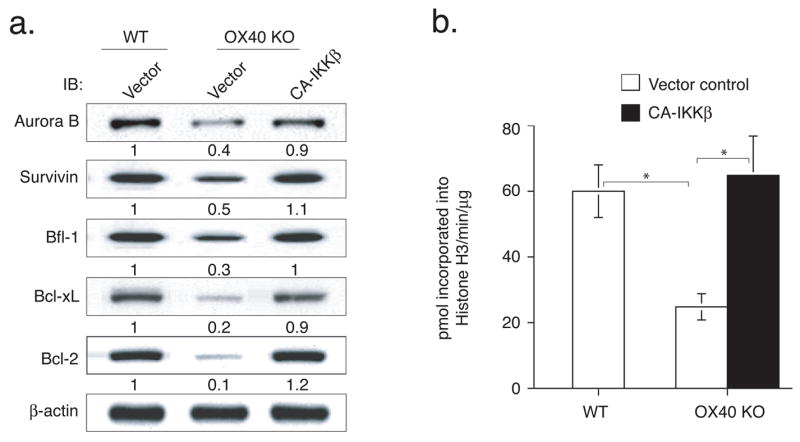
Active IKKβ regulates the expression of Aurora B, Survivin, Bfl-1, Bcl-xL and Bcl-2 and promotes Aurora B kinase activity in OX40 KO T cells. Naïve CD4 cells from WT or OX40 KO OT-II TCR transgenic mice were stimulated with peptide and APCs. On day 2/3, T cells were transduced with retroviral vectors expressing GFP, or GFP with CA-IKKβ. On day 6 of primary culture, GFP+ CD4 T cells were sorted and restimulated with APCs and peptide. (a) On day 2 of recall culture, GFP+ T cells were isolated and analyzed for Aurora B, Survivin, Bfl-1, Bcl-xL and Bcl-2. Protein amounts were determined by densitometry and are shown relative to expression in wild-type T cells transduced with control vector (taken as 1). (b) On day 3, Aurora B kinase activity was assessed with histone H3 as a substrate. Data are representative of four experiments (* P < 0.05, Student’s unpaired t-test).
Active IKKβ Reverses Defective Expansion and Survivial of OX40 KO T Cells in vivo
Lastly, to show that defective activation of the NF–κB1 pathway can account for much of the inability of OX40 KO T cells to respond well in vivo in a truly physiological setting, GFP-sorted OT-II T cells were adoptively transferred into syngeneic recipients. These mice were subsequently challenged with OVA protein. OX40 KO T cells expanded less than WT T cells over 3 days in lymph nodes or spleen, and this defect was rescued by active IKKβ ( Figs. 6a and 6b), supporting the in vitro results (Figs. 3–5). More impressively, the effect of targeting the NF–κB1 pathway was long lasting, with enhanced numbers of antigen-specific T cells not only present 7 days after antigen challenge through the peak of response, but also after 14 days and 20 days when the secondary in vivo response was over and contraction of T cell populations had occurred in all recipients (Fig 7). At each time point examined, active IKKβ fully restored the defective accumulation of OX40 KO T cells to WT levels. Active IKKβ did not result in any enhanced elevation in OX40 KO T cell number over that seen in WT T cells, suggesting that the IKKβ construct was replicating physiological antigen- and OX40-activated IKKβ. Overall, these data strongly support the conclusion that OX40/OX40L interactions sustain T cell expansion through targeting the canonical NF–κB1 pathway.
Figure 6.

Active IKKβ promotes initial CD4 T cell expansion in vivo. Naïve CD4 T cells from WT or OX40 KO OT-II TCR transgenic mice were stimulated with APCs/peptide. On day 2/3, T cells were transduced with retroviral vectors expressing GFP, or GFP with CA-IKKβ. On day 6 of primary culture, GFP+ CD4 T cells were sorted and adoptively transferred into naive wild-type recipient mice that were subsequently challenged i.p. with whole OVA protein in PBS (filled bars) or with PBS alone (open bars). (a) On day 3, percent GFP+CD44+ T cells were analyzed by flow cytometry, after gating on live CD4 T cells in the spleen. Only OVA challenged mice shown. Results are representative of three experiments. (b) Actual numbers of GFP+CD44+ T cells in pooled lymph nodes (top) and spleen (bottom) on day 3. Data are mean number of GFP+CD44+ T cells ± s.d from four individual mice (* P < 0.05, ** P < 0.01, Student’s unpaired t-test).
Figure 7.

Active IKKβ restores the ability of OX40 KO T cells to accumulate and survive over time in vivo. Naïve CD4 T cells from WT or OX40 KO OT-II TCR transgenic mice were stimulated with APCs/peptide. On day 2/3, T cells were transduced with retroviral vectors expressing GFP, or GFP with CA-IKKβ. On day 6 of primary culture, GFP+ CD4 T cells were sorted and adoptively transferred into naive wild-type recipient mice that were subsequently challenged i.p. with whole OVA protein in PBS (filled bars) or PBS alone (open bars). On days 7, 14, and 20, GFP+Vβ5+CD4+ T cells were enumerated from pooled lymph nodes and spleen. Data are mean number of GFP+Vβ5+CD4+ ± s.d from four individual mice and representative of two experiments (* P < 0.05, ** P < 0.01, Student’s unpaired t-test).
Discussion
NF–κB pathways regulate genes involved in inflammatory and immune responses as well as in some aspects of cell growth, survival and differentiation (36–38). In antigen-responding T cells, OX40 functions as a critical driver of proliferation and survival by promoting continued and maximal expression of anti-apoptotic Bcl-2 family members, and an inhibitor of apoptosis family member, survivin (9–11). In this paper, we now show that OX40-m ediated activation of NF–κB1 is a key event proceeding expression of specific genes responsible for proliferation and survival in T cells.
We previously reported that PKB activation from OX40 is required for upregulation and maintenance of the cell division and survival factors, Bcl-2, Bcl-xL, Bfl-1, and survivin (9–11). Triggering OX40 on primary antigen-specific CD4 T cells supports prolonged activation of the PI3K-PKB pathway, which in turn contributes to the high rate of clonal expansion of recently activated T cells and to increased generation of high frequencies of effector T cells in the later phases of T cell differentiation. As now shown here, both by expression analyses and retroviral reconstitution experiments, the cellular and molecular functions regulated by the OX40-PκB axis are quite similar if not identical to those targeted by the OX40-NF–κB1 axis. Using a gene-complementation approach, we found that active PKB restored the defective proliferation and survival of OX40 KO CD4 T cells, with a concomitant effect on promoting expression of Bcl-2, Bcl-xL, Bfl-1, and survivin (9). In the current study, an active IKKβ, which can facilitate activation of the canonical NF–κB1 pathway, led to a similar effect in OX40 KO T cells. This suggests that OX40 controlled PKB and IKKβ synergistically maintain T cell division and survival over time, and hence coordinately regulate the extent of clonal expansion of primary effector and memory effector T cells.
This conclusion correlates with reports suggesting that PKB and NF–κB1 pathways cooperatively regulate downstream signaling events and cellular fates (39), and that there is a level of synergy between PKB and NF–κB1 in the regulation of T cell homeostasis in vivo (40–42). Transgenic expression of active PKB in thymocytes and mature T cells enhanced their viability and resistance to apoptotic stimuli, accompanied by enhanced activation of the NF–κB1 pathway (41, 43). Furthermore, transgenic expression of a dominant negative form of IκBα or mutation of NF–κB1/p50 rendered PKB-transgenic T cells susceptible to Fas-mediated cell death, again suggesting that NF–κB1 controls at least some of the functional proinflammatory activity of PI3K-PKB signaling and that PKB-NF–κB1 cooperation controls some de novo gene transcription responsible for T cell survival (37, 44).
One possible explanation for this is that PKB directly associates and controls activity of the IKK complex. Several reports show activation-induced association between IKKα/β and PKB (17, 45, 46), and that PKB can directly phosphorylate IKKα at threonine 23 (17, 46). In addition, kinase dead forms of IKKα and IKKβ have been shown to be capable of blocking PKB-mediated NF–κB activation (47, 48). These data suggest a direct regulatory function of PKB for the IKK complex. Alternatively, it has been reported that PKB can activate CARMA1 and Bcl10, two of the important upstream regulators of the IKK complex (49–51). Thus PKB has a great potential, directly or indirectly, to regulate activity of the IKK complex which is essential for the initiation of NF–κB1 signaling events. Our data provide further indirect evidence for close cooperation between PKB (9) and IKKβ (the present study) in that active versions of these molecules produce the same overall effect when introduced into OX40-deficient CD4 cells, allowing induction of survivin, aurora B, and Bcl-2 anti-apoptotic molecules.
Our finding that activation of NF–κB1 in primary T cells leads to upregulation of anti-apoptotic molecules in the Bcl-2 family is in line with prior data also showing that blocking NF–κB in T cells suppressed expression of Bcl-xL (23), and that CD40-induced NF–κB activation in B cells can promote Bcl-xL and Bfl-1 expression (52). The demonstration that Bfl-1 and Bcl-xL can be direct transcriptional targets of NF–κB supports these conclusions (53, 54). To our knowledge, there is no prior published data linking NF–κB activation to the upregulation of aurora B expression or to aurora B kinase activity. However, given the notion that PI3K/PKB cooperate with IKKβ this correlates with our recent data showing that aurora B is strongly regulated in T cells by PI3K (12). Furthermore, somewhat indirect data using inhibitors of NF–κB have shown suppression of survivin expression in various non-T cell types (55, 56), as well as activation of the survivin promoter by HTLV-1 Tax through an NF–κB dependent mechanism (57). This also correlates with our observation that PKB activation targets survivin (11), again supporting a cooperative action between PKB and IKKβ. Survivin can regulate the kinase activity of aurora B in T cells, leading to cooperation with mTOR and cell cycle progression (12), but whether the genes for survivin and aurora B kinase are both direct targets of NF–κB1 transcriptional activity needs to be addressed in the future.
In conclusion, we have provided complementary evidence in primary T cells to that gained in prior studies of OX40 signaling in transformed cells that the NF–κB1 pathway plays a strong and arguably dominant role in the costimulatory activity of OX40. Moreover, we additionally provide data that indicates that IKKβ/NF–κB1 is central to the ability of OX40 to control proliferation, survival, and overall clonal expansion of T cells when responding to antigen. Our data which demonstrate that restoring IKKβ activity in OX40 KO T cells can result in high expression of Bcl-xL, Bcl-2, Bfl-1, survivin, and aurora kinase B further reinforce the notion that NF–κB1 is a central mediator of T cell longevity. Moreover, the similar phenotypes produced by targeting PKB and IKKβ in primary T cells suggest a great deal of overlap in the requirement for these molecules when costimulatory receptors such as OX40 are engaged. However, more fundamental research is needed to fully understand how OX40 initiates downstream signaling events that recruit specific signaling transducers, and whether cooperative signaling from other costimulatory molecules, or antagonistic signaling from coinhibitory molecules, modulates the use and dominance of one or both molecules, or more significantly might reveal separate activities of NF–κB1 that contribute to cosignaling that are not co-regulated by PKB.
Acknowledgments
We thank Dr. Thomas Wirth (Department of Physiological Chemistry, University of Ulm, Germany) for active IKKβ cDNA, and X. Tang and Y. Adams for technical assistance.
The work was supported by grants from the National Institutes of Health (AI50498 and AI49453 to M.C.) and the Universitywide AIDS Research Program (J.S.). This is manuscript #911 from the La Jolla Institute for Allergy and Immunology.
The abbreviations used are
- NF–κB1
nuclear factor of kappa light polypeptide gene enhancer in B-cells 1
- MCC
moth cytochrome c
- PKB
protein kinase B/Akt
- OVA
ovalbumin
- PI3K
phosphoinositide Kinase-3
- IKK
IκB kinase
- TNFR
TNF receptor
- TRAF
TNFR-associated factor
- GFP
green fluorescent protein
- IRES
internal ribosome entry site
References
- 1.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 2.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 3.Song A, Song J, Tang X, Croft M. Cooperation between CD4 and CD8 T cells for anti-tumor activity is enhanced by OX40 signals. Eur J Immunol. 2007;37:1224–1232. doi: 10.1002/eji.200636957. [DOI] [PubMed] [Google Scholar]
- 4.Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J Immunol. 2005;175:3534–3541. doi: 10.4049/jimmunol.175.6.3534. [DOI] [PubMed] [Google Scholar]
- 5.Chitnis T, Najafian N, Abdallah KA, Dong V, Yagita H, Sayegh MH, Khoury SJ. CD28-independent induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;107:575–583. doi: 10.1172/JCI11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malmstrom V, Shipton D, Singh B, Al-Shamkhani A, Puklavec MJ, Barclay AN, Powrie F. CD134L expression on dendritic cells in the mesenteric lymph nodes drives colitis in T cell-restored SCID mice. J Immunol. 2001;166:6972–6981. doi: 10.4049/jimmunol.166.11.6972. [DOI] [PubMed] [Google Scholar]
- 7.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, Croft M. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J Exp Med. 2003;198:315–324. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vu MD, Amanullah F, Li Y, Demirci G, Sayegh MH, Li XC. Different costimulatory and growth factor requirements for CD4+ and CD8+ T cell-mediated rejection. J Immunol. 2004;173:214–221. doi: 10.4049/jimmunol.173.1.214. [DOI] [PubMed] [Google Scholar]
- 9.Song J, Salek-Ardakani S, Rogers PR, Cheng M, Van Parijs L, Croft M. The costimulation-regulated duration of PKB activation controls T cell longevity. Nat Immunol. 2004;5:150–158. doi: 10.1038/ni1030. [DOI] [PubMed] [Google Scholar]
- 10.Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity. 2001;15:445–455. doi: 10.1016/s1074-7613(01)00191-1. [DOI] [PubMed] [Google Scholar]
- 11.Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity. 2005;22:621–631. doi: 10.1016/j.immuni.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 12.Song J, Salek-Ardakani S, So T, Croft M. The kinases aurora B and mTOR regulate the G1-S cell cycle progression of T lymphocytes. Nat Immunol. 2007;8:64–73. doi: 10.1038/ni1413. [DOI] [PubMed] [Google Scholar]
- 13.Ruland J, Mak TW. Transducing signals from antigen receptors to nuclear factor kappaB. Immunol Rev. 2003;193:93–100. doi: 10.1034/j.1600-065x.2003.00049.x. [DOI] [PubMed] [Google Scholar]
- 14.Chariot A, Leonardi A, Muller J, Bonif M, Brown K, Siebenlist U. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem. 2002;277:37029–37036. doi: 10.1074/jbc.M205069200. [DOI] [PubMed] [Google Scholar]
- 15.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 16.Harhaj NS, Sun SC, Harhaj EW. Activation of NF-kappa B by the human T cell leukemia virus type I Tax oncoprotein is associated with ubiquitin-dependent relocalization of I kappa B kinase. J Biol Chem. 2007;282:4185–4192. doi: 10.1074/jbc.M611031200. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka H, Fujita N, Tsuruo T. 3-Phosphoinositide-dependent protein kinase-1-mediated IkappaB kinase beta (IkkB) phosphorylation activates NF-kappaB signaling. J Biol Chem. 2005;280:40965–40973. doi: 10.1074/jbc.M506235200. [DOI] [PubMed] [Google Scholar]
- 18.Li ZW, Omori SA, Labuda T, Karin M, Rickert RC. IKK beta is required for peripheral B cell survival and proliferation. J Immunol. 2003;170:4630–4637. doi: 10.4049/jimmunol.170.9.4630. [DOI] [PubMed] [Google Scholar]
- 19.Goudeau B, Huetz F, Samson S, Di Santo JP, Cumano A, Beg A, Israel A, Memet S. IkappaBalpha/IkappaBepsilon deficiency reveals that a critical NF-kappaB dosage is required for lymphocyte survival. Proc Natl Acad Sci U S A. 2003;100:15800–15805. doi: 10.1073/pnas.2535880100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mora A, Youn J, Keegan A, Boothby M. NF-kappa B/Rel participation in the lymphokine-dependent proliferation of T lymphoid cells. J Immunol. 2001;166:2218–2227. doi: 10.4049/jimmunol.166.4.2218. [DOI] [PubMed] [Google Scholar]
- 22.Edelstein LC, Lagos L, Simmons M, Tirumalai H, Gelinas C. NF-kappa B-dependent assembly of an enhanceosome-like complex on the promoter region of apoptosis inhibitor Bfl-1/A1. Mol Cell Biol. 2003;23:2749–2761. doi: 10.1128/MCB.23.8.2749-2761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mora AL, Corn RA, Stanic AK, Goenka S, Aronica M, Stanley S, Ballard DW, Joyce S, Boothby M. Antiapoptotic function of NF-kappaB in T lymphocytes is influenced by their differentiation status: roles of Fas, c-FLIP, and Bcl-xL. Cell Death Differ. 2003;10:1032–1044. doi: 10.1038/sj.cdd.4401257. [DOI] [PubMed] [Google Scholar]
- 24.Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y. Experimental autoimmune encephalomyelitis in NF-kappa B-deficient mice:roles of NF-kappa B in the activation and differentiation of autoreactive T cells. J Immunol. 1999;163:2937–2943. [PubMed] [Google Scholar]
- 25.Artis D, Speirs K, Joyce K, Goldschmidt M, Caamano J, Hunter CA, Scott P. NF-kappa B1 is required for optimal CD4+ Th1 cell development and resistance to Leishmania major. J Immunol. 2003;170:1995–2003. doi: 10.4049/jimmunol.170.4.1995. [DOI] [PubMed] [Google Scholar]
- 26.Aronica MA, Mora AL, Mitchell DB, Finn PW, Johnson JE, Sheller JR, Boothby MR. Preferential role for NF-kappa B/Rel signaling in the type 1 but not type 2 T cell-dependent immune response in vivo. J Immunol. 1999;163:5116–5124. [PubMed] [Google Scholar]
- 27.Corn RA, Aronica MA, Zhang F, Tong Y, Stanley SA, Kim SR, Stephenson L, Enerson B, McCarthy S, Mora A, Boothby M. T cell-intrinsic requirement for NF-kappa B induction in postdifferentiation IFN-gamma production and clonal expansion in a Th1 response. J Immunol. 2003;171:1816–1824. doi: 10.4049/jimmunol.171.4.1816. [DOI] [PubMed] [Google Scholar]
- 28.Grundstrom S, Anderson P, Scheipers P, Sundstedt A. Bcl-3 and NFkappaB p50-p50 homodimers act as transcriptional repressors in tolerant CD4+ T cells. J Biol Chem. 2004;279:8460–8468. doi: 10.1074/jbc.M312398200. [DOI] [PubMed] [Google Scholar]
- 29.Arch RH, Thompson CB. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol Cell Biol. 1998;18:558–565. doi: 10.1128/mcb.18.1.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem. 1998;273:5808–5814. doi: 10.1074/jbc.273.10.5808. [DOI] [PubMed] [Google Scholar]
- 31.So T, Song J, Sugie K, Altman A, Croft M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc Natl Acad Sci U S A. 2006;103:3740–3745. doi: 10.1073/pnas.0600205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 33.Denk A, Goebeler M, Schmid S, Berberich I, Ritz O, Lindemann D, Ludwig S, Wirth T. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J Biol Chem. 2001;276:28451–28458. doi: 10.1074/jbc.M102698200. [DOI] [PubMed] [Google Scholar]
- 34.Herrmann O, Baumann B, de Lorenzi R, Muhammad S, Zhang W, Kleesiek J, Malfertheiner M, Kohrmann M, Potrovita I, Maegele I, Beyer C, Burke JR, Hasan MT, Bujard H, Wirth T, Pasparakis M, Schwaninger M. IKK mediates ischemia-induced neuronal death. Nat Med. 2005;11:1322–1329. doi: 10.1038/nm1323. [DOI] [PubMed] [Google Scholar]
- 35.Sugie K, Jeon MS, Grey HM. Activation of naive CD4 T cells by anti-CD3 reveals an important role for Fyn in Lck-mediated signaling. Proc Natl Acad Sci U S A. 2004;101:14859–14864. doi: 10.1073/pnas.0406168101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabatingan MS, Schmidt MR, Sen R, Woodland RT. Naive B lymphocytes undergo homeostatic proliferation in response to B cell deficit. J Immunol. 2002;169:6795–6805. doi: 10.4049/jimmunol.169.12.6795. [DOI] [PubMed] [Google Scholar]
- 37.Gelman AE, LaRosa DF, Zhang J, Walsh PT, Choi Y, Sunyer JO, Turka LA. The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity. 2006;25:783–793. doi: 10.1016/j.immuni.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapira S, Harb OS, Caamano J, Hunter CA. The NF-kappaB signaling pathway: immune evasion and immunoregulation during toxoplasmosis. Int J Parasitol. 2004;34:393–400. doi: 10.1016/j.ijpara.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki H, Matsuda S, Terauchi Y, Fujiwara M, Ohteki T, Asano T, Behrens TW, Kouro T, Takatsu K, Kadowaki T, Koyasu S. PI3K and Btk differentially regulate B cell antigen receptor-mediated signal transduction. Nat Immunol. 2003;4:280–286. doi: 10.1038/ni890. [DOI] [PubMed] [Google Scholar]
- 40.Jones RG, Elford AR, Parsons MJ, Wu L, Krawczyk CM, Yeh WC, Hakem R, Rottapel R, Woodgett JR, Ohashi PS. CD28-dependent activation of protein kinase B/Akt blocks Fas-mediated apoptosis by preventing death-inducing signaling complex assembly. J Exp Med. 2002;196:335–348. doi: 10.1084/jem.20020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones RG, Saibil SD, Pun JM, Elford AR, Bonnard M, Pellegrini M, Arya S, Parsons ME, Krawczyk CM, Gerondakis S, Yeh WC, Woodgett JR, Boothby MR, Ohashi PS. NF-kappaB couples protein kinase B/Akt signaling to distinct survival pathways and the regulation of lymphocyte homeostasis in vivo. J Immunol. 2005;175:3790–3799. doi: 10.4049/jimmunol.175.6.3790. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, Yoshida R, Wakeham A, Higuchi T, Fukumoto M, Tsubata T, Ohashi PS, Koyasu S, Penninger JM, Nakano T, Mak TW. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- 43.Jones RG, Parsons M, Bonnard M, Chan VS, Yeh WC, Woodgett JR, Ohashi PS. Protein kinase B regulates T lymphocyte survival, nuclear factor kappaB activation, and Bcl-X(L) levels in vivo. J Exp Med. 2000;191:1721–1734. doi: 10.1084/jem.191.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gustin JA, Korgaonkar CK, Pincheira R, Li Q, Donner DB. Akt regulates basal and induced processing of NF-kappaB2 (p100) to p52. J Biol Chem. 2006;281:16473–16481. doi: 10.1074/jbc.M507373200. [DOI] [PubMed] [Google Scholar]
- 45.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 46.Vandermoere F, El Yazidi-Belkoura I, Demont Y, Slomianny C, Antol J, Lemoine J, Hondermarck H. Proteomics exploration reveals that actin is a signaling target of the kinase Akt. Mol Cell Proteomics. 2007;6:114–124. doi: 10.1074/mcp.M600335-MCP200. [DOI] [PubMed] [Google Scholar]
- 47.Agarwal A, Das K, Lerner N, Sathe S, Cicek M, Casey G, Sizemore N. The AKT/I kappa B kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-kappa B and beta-catenin. Oncogene. 2005;24:1021–1031. doi: 10.1038/sj.onc.1208296. [DOI] [PubMed] [Google Scholar]
- 48.Kane LP, V, Shapiro S, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 49.Narayan P, Holt B, Tosti R, Kane LP. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol Cell Biol. 2006;26:2327–2336. doi: 10.1128/MCB.26.6.2327-2336.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klemm S, Zimmermann S, Peschel C, Mak TW, Ruland J. Bcl10 and Malt1 control lysophosphatidic acid-induced NF-kappaB activation and cytokine production. Proc Natl Acad Sci U S A. 2007;104:134–138. doi: 10.1073/pnas.0608388103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yeh PY, Kuo SH, Yeh KH, Chuang SE, Hsu CH, Chang WC, Lin HI, Gao M, Cheng AL. A pathway for tumor necrosis factor-alpha-induced Bcl10 nuclear translocation. Bcl10 is up-regulated by NF-kappaB and phosphorylated by Akt1 and then complexes with Bcl3 to enter the nucleus. J Biol Chem. 2006;281:167–175. doi: 10.1074/jbc.M511014200. [DOI] [PubMed] [Google Scholar]
- 52.Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc Natl Acad Sci U S A. 1999;96:9136–9141. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zong WX, Edelstein LC, Chen C, Bash J, Gelinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999;13:382–387. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, Munshi N, Treon SP, Anderson KC. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99:4079–4086. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 56.Shishodia S, Aggarwal BB. Guggulsterone inhibits NF-kappaB and IkappaBalpha kinase activation, suppresses expression of anti-apoptotic gene products, and enhances apoptosis. J Biol Chem. 2004;279:47148–47158. doi: 10.1074/jbc.M408093200. [DOI] [PubMed] [Google Scholar]
- 57.Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M, Hatano M, Tokuhisa T, Mori N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. International journal of cancer. 2005;115:967–974. doi: 10.1002/ijc.20954. [DOI] [PubMed] [Google Scholar]