

Abstract
Kras is commonly mutated in colon cancers, but mutations in Nras are rare. We have used genetically engineered mice to determine whether and how these related oncogenes regulate homeostasis and tumorigenesis in the colon. Expression of K-RasG12D in the colonic epithelium stimulated hyperproliferation in a Mek-dependent manner. N-RasG12D did not alter the growth properties of the epithelium, but was able to confer resistance to apoptosis. In the context of an Apc-mutant colonic tumor, activation of K-Ras led to defects in terminal differentiation and expansion of putative stem cells within the tumor epithelium. This K-Ras tumor phenotype was associated with attenuated signaling through the MAPK pathway, and human colon cancer cells expressing mutant K-Ras were hypersensitive to inhibition of Raf, but not Mek. These studies demonstrate clear phenotypic differences between mutant Kras and Nras, and suggest that the oncogenic phenotype of mutant K-Ras might be mediated by noncanonical signaling through Ras effector pathways.
Colon cancer affects approximately 5% of the population in the United States and results in almost 60,000 deaths per year (American Cancer Society Cancer Statistics, 2007). Loss of function of the Apc (adenomatous polyposis coli) tumor suppressor gene is thought to initiate neoplastic growth, whereas activating mutations in the Kras oncogene are commonly associated with progression from a benign adenoma to an advanced/dysplastic adenocarcinoma1. Nearly 50% of colon cancers harbor activating mutations in Kras, whereas Nras mutations occur in a smaller percentage (~5%) and seem to arise at a later stage in the development of malignancy1. The basis for this difference remains unclear.
Proteins of the Ras family (K-Ras4A, K-Ras4B, H-Ras and N-Ras) are members of the superfamily of small G proteins. As such, they act as GDP/GTP-regulated switches to convey extracellular signals that influence cell proliferation, remodeling of the actin cytoskeleton, and apoptosis2. Mutations affecting any of these proteins at amino acids 12, 13 or 61 lock the enzyme in the GTP-bound, activated form3. The four Ras proteins are highly homologous to one another, sharing a high degree of identity over the first 90% of the protein. The extreme C terminus of the proteins constitutes the hypervariable region, which diverges radically in primary sequence and undergoes significant post-translational modifications that confer important differences in trafficking and intracellular localization. These differences may underlie the functional identities of the family members.
We have used gene targeting in mice to dissect the functions of mutationally activated Kras and Nras in the colonic epithelium, and these studies have revealed marked functional differences between these two highly related oncogenes. Our results provide novel insight into the Kras mutation bias seen in human colon cancers and show unexpected patterns of signaling from activated K-Ras that may have implications for cancer therapy.
RESULTS
K-RasG12D, but not N-RasG12D, alterss basal homeostasis in the colonic epithelium
A simple hypothesis to explain the mutation data from human colon cancers is that selection for Kras mutations is a reflection of expression patterns of the Ras genes within the colonic epithelium. To explore this hypothesis, we used qRT-PCR to analyze the expression of Kras and Nras in the crypts of mouse and human colon (Fig. 1a and Supplementary Note and Fig. 1a online). In the mouse, Kras was expressed more highly at the top of the crypt than at the bottom of the crypt (Fig. 1a). This was the opposite expression pattern relative to Mcm6 (Fig. 1a), a marker of undifferentiated cells at the bottom of the intestinal epithelial crypt4. Nras was expressed uniformly throughout the crypt in the mouse (Fig. 1a). These results indicate that the selection for Kras mutations in colon cancers cannot be explained simply on the basis of expression patterns of the Ras isoforms within the colonic epithelium.
Figure 1.
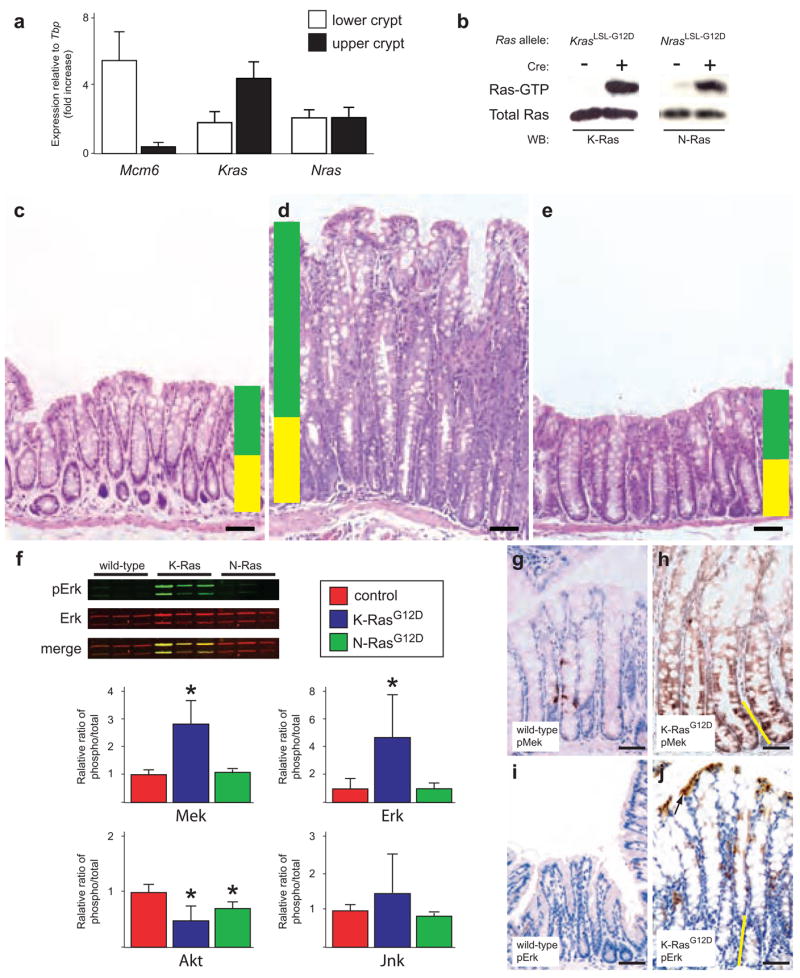
Mutant K-Ras, but not mutant N-Ras, promotes hyperplastic growth in the colonic epithelium. (a) Expression analysis of Kras and Nras in the colonic epithelium. We used Mcm6, which is expressed only in undifferentiated cells at the bottom of the crypt, as a control. Kras is more highly expressed at the top of the crypt, whereas Nras is uniformly expressed throughout the crypt. Expression of Mcm6, Kras and Nras was normalized to the expression of the TATA-box binding protein (Tbp) gene. (b) Biochemical detection of activated K-Ras and N-Ras. Activated (GTP-bound) forms of each protein can be detected in lysates from colonic epithelium, but only in mice carrying the conditional (LSL-G12D) allele and expressing Cre recombinase from the Fabpl promoter. (c) Hematoxylin and eosin staining of normal colonic epithelium. Yellow shading denotes the normal proliferative zone, and green shading marks the zone of differentiation. All animals were killed sacrificed between 4–6 months of age. (d) Hematoxylin and eosin staining of colonic epithelium expressing activated K-RasG12D showing crypt hyperplasia. (e) Hematoxylin and eosin staining of colonic epithelium expressing activated N-RasG12D. (f) Signaling downstream of K-RasG12D and N-RasG12D in vivo. In quantitative protein blots of whole tissue lysates (see example in upper left), mutant K-Ras activates Mek and Erk. Mutant N-Ras, by contrast, does not activate Mek or Erk. Both K-RasG12D and N-RasG12D downregulate phospho-Akt, whereas neither seems to significantly affect phospho-Jnk. *P < 0.05, Wilcoxon rank sum test. The activation state of each molecule is measured as the ratio of phospho-protein to total protein. (g,h) Immunohistochemical detection of phospho-Mek. Few cells in the normal colonic epithelium (g) express phospho-Mek, whereas all cells expressing mutant K-Ras stain positively (h). (i,j) Immunohistochemical detection of phospho-Erk. Phospho-Erk is readily detectable in the differentiated cells at the top of the colonic crypt expressing K-RasG12D (j), but not in the proliferative cells at the bottom of the crypt. In h and j, the height of the proliferative progenitor zone is represented by the yellow line. Scale bar in all panels, 50 μm. Error bars in panels a and f represent standard deviations for each sample class.
To directly examine the relative phenotypic consequences of mutating the different Ras oncogenes, we used gene targeting to generate mice expressing mutationally activated K-Ras or N-Ras in the colonic epithelium. Mice carrying a Cre-dependent activated allele of Kras (KrasLSL-G12D) have been described previously5. Here we report the first conditional activated allele of Nras (NrasLSL-G12D), where the insertion of a floxed transcriptional stop element renders the mutant allele inactive until Cre recombinase is expressed (Supplementary Fig. 1b). Notably, the mutant alleles of Kras and Nras are expressed from their endogenous genomic loci, allowing us to examine the phenotypic consequences of activating these oncogenes at wild-type levels. To express mutant Ras in the colonic epithelium, KrasLSL-G12D/+ and NrasLSL-G12D/+ animals were crossed with animals carrying the Fapbl4X@-132-Cre transgene (Fabpl-Cre). This transgene directs expression of Cre recombinase to the distal small intestinal and colonic epithelia6. We detected high concentrations levels of activated (GTP-bound) K-Ras or N-Ras in the colonic epithelium of animals carrying the respective conditional alleles, but only in animals also expressing the Fabpl-Cre transgene (Fig. 1b).
Animals expressing K-RasG12D developed widespread hyperplasia throughout the colonic epithelium. This hyperplasia was typified by an extreme lengthening of the crypts and by the development of large, prominent goblet cells when compared with wild-type colon (Fig. 1c,d). We observed a similar phenotype when K-Ras was activated specifically in the adult colonic epithelium (Supplementary Note and Fig. 1c–e). Expression of N-RasG12D had no effect on the histology of the colonic epithelium (Fig. 1e), suggesting that Kras is uniquely capable of modulating basal homeostasis in this tissue. Of note, animals expressing K-RasG12D throughout the colonic epithelium never developed colon cancer, even when aged beyond one year. This observation demonstrates that expression of mutationally activated K-Ras from its endogenous locus is sufficient to promote hyperplasia, but not neoplasia.
K-RasG12D and N-RasG12D show limited downstream signaling in vivo
Activated forms of Ras can signal through a multitude of downstream effector pathways7. We sought to determine which of these are relevant in vivo effectors of K-RasG12D and/or N-RasG12D signaling in the colonic epithelium. We found that K-RasG12D, but not N-RasG12D, could activate both Mek and Erk, two key components of the canonical MAPK pathway (Fig. 1f). K-RasG12D and N-RasG12D both downregulated phospho-Akt, and neither affected the activation state of Jnk. Aside from their effects on MAPK and Akt signaling, we did not detect clear modulation of any other Ras effector pathways (for example, Pdk1, Rac, Rho, Ral or p38) in colons expressing K-RasG12D or N-RasG12D, indicating that the physiological expression of these oncoproteins induces limited downstream signaling in vivo in the colonic epithelium (data not shown).
The colonic epithelium is composed of many different cell types, with the undifferentiated proliferative cells located at the bottom of the crypt and the differentiated postmitotic cells at the top of the crypt (Fig. 1c). To determine whether the phosphorylated forms of Mek and Erk are localized to specific cell types, we analyzed their expression in intact tissues by immunohistochemistry (IHC) (Fig. 1 g–j). Wild-type colons had low or undetectable concentrations of both phospho-Mek and phospho-Erk (Fig. 1g,i). We found that all cells of the colonic epithelium that expressed K-RasG12D had high concentrations of phospho-Mek (Fig. 1h). Notably, even though Erk is the direct downstream target of the Mek kinase, only the differentiated cells at the very top of the crypt expressed high levels of phospho-Erk (Fig. 1j). This observation suggests that the ability of activated Mek to maintain high steady state concentrations of phosphorylated Erk is dependent upon cell type.
K-Ras signals through Mek to promote hyperproliferation
To gain insight into the cellular mechanisms underlying the K-RasG12D phenotype, we analyzed the ability of activated K-Ras and N-Ras to affect the proliferative kinetics of the colonic epithelium. We first measured the number of cells at the bottom of each crypt that express Mcm6, a marker of undifferentiated cells4. Most of these Mcm6-positive cells are the symmetrically dividing transit amplifying cells (TACs)8. The hyperplastic colonic epithelium from Fabpl-Cre;KrasLSL-G12D/+ mice had a larger TAC zone than wild-type controls (23 versus 15 cell positions; Fig. 2a). Fapbl-Cre;KrasLSL-G12D/+ mice also showed a higher mitotic index than wild-type mice, as assessed by staining for phosphorylated histone H3 (PH3) (Fig. 2b). Colons expressing N-RasG12D, by contrast, showed an increase in neither the number of Mcm6-positive nor the number of PH3-positive cells at the bottom of the crypt. Taken together, these data indicate that expression of activated K-Ras, but not N-Ras, can promote hyperplasia in the colonic epithelium by increasing the number of proliferative progenitor cells in the tissue.
Figure 2.
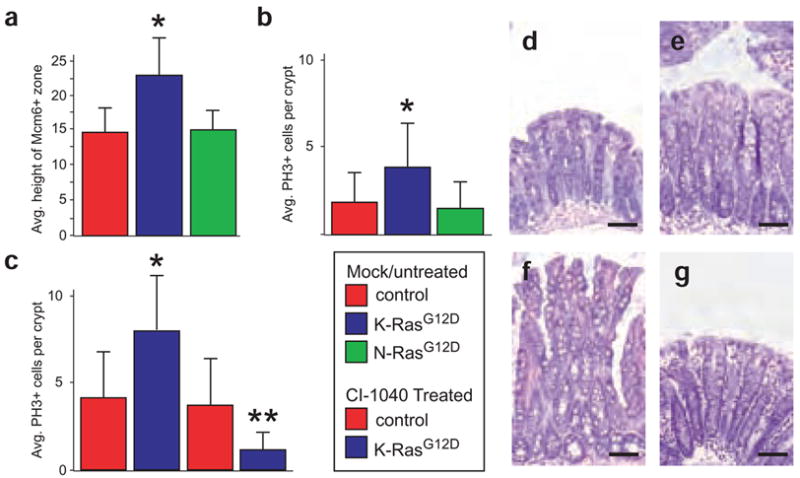
K-RasG12D promotes hyperproliferation through Mek. (a) Quantification of the number of Mcm6-positive cells at the bottom of the crypt shows that mutant K-Ras, but not mutant N-Ras, significantly increases the size of the progenitor cell population. *P < 0.01, Wilcoxon rank sum test. (b) The progenitor cells expressing K-RasG12D are hyperproliferative, as assessed by the number of cells per crypt that are positive for phosphorylated histone-H3, a marker of mitotic cells. * P < 0.01. (c) Inhibition of Mek with CI-1040 suppresses proliferation in colonic epithelium expressing mutant K-Ras. **P < 10−24, Wilcoxon rank sum test. Inhibition of Mek has no effect on proliferation in control colons. (d) Hematoxylin and eosin staining of normal, untreated colonic epithelium. (e) Hematoxylin and eosin staining of wild-type colon in an animal treated with CI-1040 for 24 h. (f) Hematoxylin and eosin staining of colonic epithelium expressing activated K-RasG12D showing crypt hyperplasia. (g) Hematoxylin and eosin staining of K-RasG12D colonic epithelium treated with CI-1040 for 24 h. Note the absence of hyperplasia and the return to somewhat normal histology. Scale bar in all panels, 50 μm. Error bars in panels a-c represent standard deviations for each sample class.
We used in vivo pharmacology to interrogate the K-Ras→Raf→Mek signaling pathway with respect to the hyperproliferative phenotype. CI-1040 (originally known as PD184352) is an orally active inhibitor that is highly selective for Mek9. Our pilot studies indicated that CI-1040 was highly effective in suppressing Mek activity in vivo in the colonic epithelium (Supplementary Note and Fig. 1f,g). We found that CI-1040 treatment did not affect the rate of proliferation in control mice (P = 0.11, Fig. 2c). By contrast, epithelium expressing activated K-Ras was hypersensitive to the effects of Mek inhibition (P < 10−24, Fig. 2c). Not only did inhibition of Mek suppress proliferation in colons expressing K-RasG12D, it also had a considerable effect on tissue morphology. Control animals treated with CI-1040 showed little or no effect on the colonic epithelium (Fig. 2d,e), whereas Fabpl-Cre;KrasLSL-G12D/+ animals showed a return to normal histology within 24 h of treatment (Fig. 2f,g). This pharmacological analysis strongly suggests that K-RasG12D promotes hyperproliferation and hyperplasia in colonic epithelia via a Mek-dependent pathway.
N-Ras, but not K-Ras, regulates apoptosis
In vitro studies have linked N-Ras function to the ability of a cell to respond to apoptotic stimuli10. We sought to determine whether mutant N-Ras could function to suppress apoptosis in the colonic epithelium in vivo. When wild-type mice were exposed to 2.5% dextran sodium sulfate (DSS) in the drinking water, they developed widespread epithelial damage in the colon within 1 week (Fig. 3a–d and Supplementary Note and Fig. 2 online). Animals expressing K-RasG12D were also sensitive to DSS-induced apoptosis (Fig. 3e–h), but those expressing N-RasG12D were almost entirely resistant to this treatment and showed very little effect on the colonic epithelium after 1 week of exposure (Fig. 3i–l). Animals expressing mutant N-Ras were not resistant to irradiation-induced apoptosis in the colonic epithelium, indicating that the effect of N-RasG12D is dependent on the apoptotic stimulus and specific cell death pathway(s) engaged (data not shown).
Figure 3.
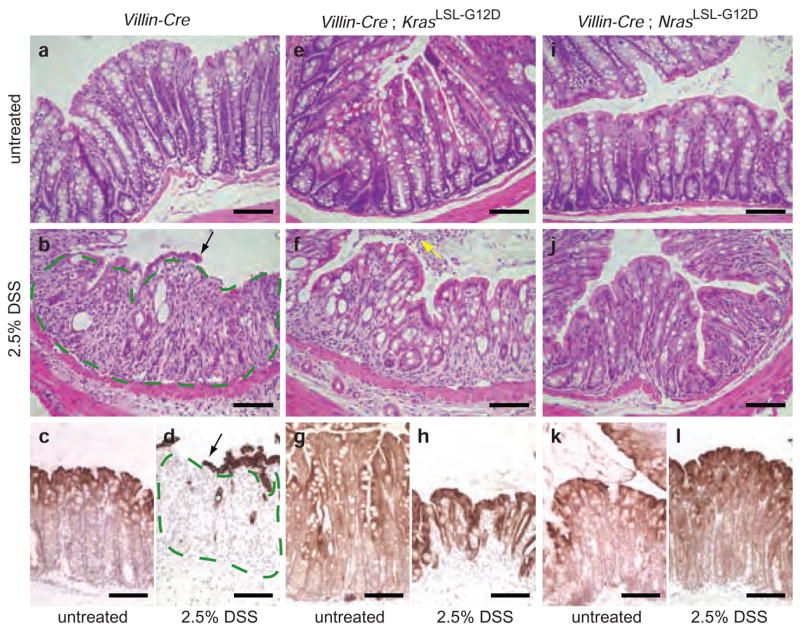
Activated N-Ras, but not K-Ras, suppresses DSS-induced apoptosis in the colonic epithelium. (a) Hematoxylin and eosin staining of colonic epithelium from an untreated Villin-Cre mouse. Villin-Cre directs expression of Cre throughout the entire small intestinal and colonic epithelia34. (b) Hematoxylin and eosin staining of colonic epithelium from a Villin-Cre mouse treated with 2.5% DSS in the drinking water for 7 days. The colonic epithelium is largely absent, having undergone extensive apoptosis. Small remnants of epithelium are left behind at this time point (black arrow). Where the epithelium is normally found, only residual connective tissue remains (outlined in green). These animals develop widespread ulceration of the colonic epithelium that results in rectal bleeding. (c,d) Cytokeratin 20 (CK20) staining of untreated and treated Villin-Cre colons. CK20 specifically marks the epithelial component of the colon. Compare c to d, where the area outlined in green contains stromal tissue rather than epithelium because of the extensive damage caused by DSS. (e) Hematoxylin and eosin staining of colonic epithelium from an untreated Villin-Cre;KrasLSL-G12D/+ mouse. (f) Animals expressing K-RasG12D, like control animals, are extremely sensitive to treatment with DSS. Note the apoptotic epithelial cells filling the colonic lumen (yellow arrow). (g,h) Treated and untreated Villin-Cre;KrasLSL-G12D/+ colons stained for CK20. (i) Hematoxylin and eosin staining of colonic epithelium from an untreated Villin-Cre;NrasLSL-G12D/+ mouse. (j) Expression of N-RasG12D exclusively in the intestinal epithelium confers resistance to DSS-induced apoptosis. After 7 d exposure to DSS, animals expressing activated N-Ras are largely unaffected, and their colonic epithelium remains intact. (k,l) Treated and untreated Villin-Cre;NrasLSL-G12D/+ colons stained for CK20 The treated sample looks almost identical to the untreated sample. Scale bar in all panels, 50 μm. Experimental animals were approximately 8 weeks of age at the beginning of treatment.
One explanation for the phenotypic differences between K-RasG12D and N-RasG12D is that they are expressed at different levels within specific cell types. For example, high expression of mutant K-Ras could promote proliferation, whereas low expression of mutant N-Ras could suppress apoptosis. Expression of mutant K-Ras from its endogenous promoter in colon cancer cell lines enhances growth, but it also sensitizes cells to apoptosis11,12. Using colon cancer cells that overexpress N-RasG12V (ref. 13), we found that high expression of mutant N-Ras did not promote proliferation, but did suppress apoptosis (Supplementary Note and Fig. 2e,f). These results reiterate that the functional differences between K-Ras and N-Ras are not determined by differences in expression, and they also suggest that mutation of Nras, which occurs in 5% of colon cancers, may function to suppress apoptosis in a developing tumor.
K-RasG12D promotes colon cancer progression
Although our data indicated that mutational activation of K-Ras alone is insufficient to initiate cancer in the mouse colon, we suspected that it might have an important role in promoting cancer progression in this tissue. Because mutation of the Apc tumor suppressor gene is known to be among the earliest genetic events during tumorigenesis in the human colon, we combined our Kras and Nras alleles with a conditional allele of Apc (Apc2lox14) to generate colonic tumors that express activated Ras in the absence of Apc expression. Fapbl-Cre;Apc2lox14/+ animals developed focal, pedunculated adenocarcinomas of the colon upon stochastic loss of heterozygosity at the Apc locus (data not shown). These adenocarcinomas were typically heterogeneous lesions and showed areas of both adenomatous (low grade) and malignant (high grade) epithelium (Fig. 4a,b). The low-grade component of these tumors was well differentiated, as gauged by the presence of terminally differentiated goblet cells (Fig. 4b).
Figure 4.
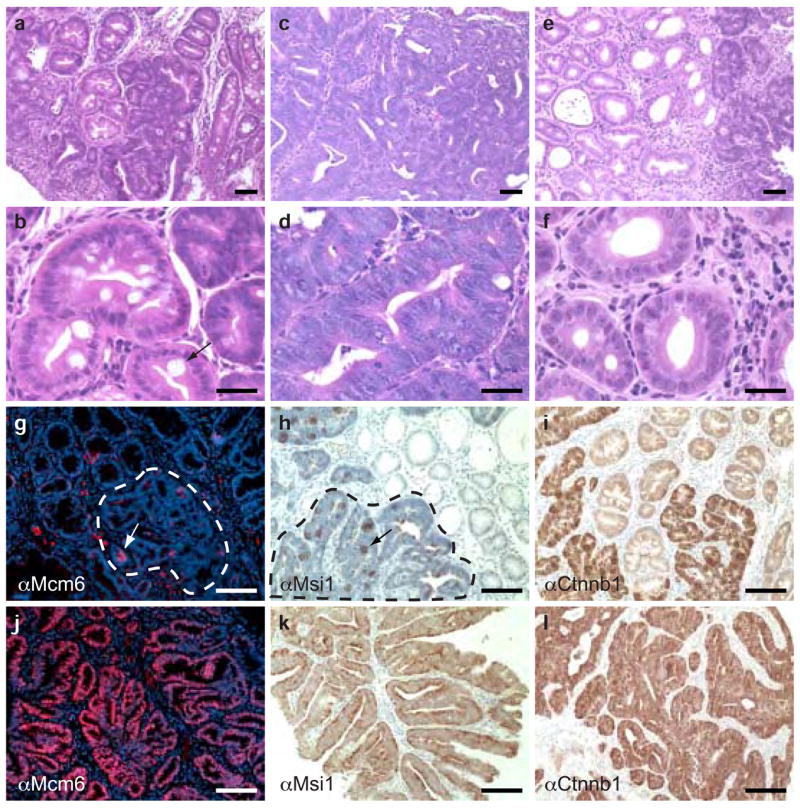
Mutationally activated K-Ras suppresses differentiation in Apc-mutant colon cancers. (a) Hematoxylin and eosin staining of a polyp expressing wild-type K-Ras and N-Ras. This lesion is a composed largely of well-differentiated epithelium. (b) High-magnification view of a tumor expressing wild-type K-Ras and N-Ras, showing a well-differentiated region with a terminally differentiated goblet cell (arrow). (c) Hematoxylin and eosin staining of a polyp expressing activated K-Ras. The tumor epithelium is highly dysplastic. (d) High-magnification view of a tumor expressing K-RasG12D. Note the stratified epithelium, serrated crypt borders, high nucleus to cytoplasm ratio, and lack of terminally differentiated goblet cells. (e) Hematoxylin and eosin staining of a tumor expressing activated N-Ras, which is histologically similar to a tumor expressing wild-type K-Ras and N-Ras. (f) High-magnification view of a tumor expressing activated N-Ras showing benign epithelium. (g) Immunohistochemistry for Mcm6 on a tumor expressing wild-type K-Ras and N-Ras. Small, focal areas are positive for Mcm6 (red cells, white arrow), but the poorly differentiated region of the tumor (outlined in white) is largely negative for this marker. (h) Immunohistochemistry for Msi1 on a tumor expressing wild-type K-Ras and N-Ras, showing focal expression (black arrow) within the poorly differentiated region of the tumor (outlined in black). (i) Immunohistochemistry for β-catenin (Ctnnb1) on a tumor expressing wild-type K-Ras and N-Ras, showing widespread expression throughout the poorly differentiated region of the tumor. (j) Immunohistochemistry for Mcm6 on a tumor expressing K-RasG12D, showing expression throughout the tumor epithelium. (k) Immunohistochemistry for Msi1 on a tumor expressing mutant K-Ras. As with Mcm6, the entire tumor epithelium is positive for this stem cell marker. (l) Immunohistochemistry for β-catenin on a tumor expressing activated K-Ras, showing widespread expression throughout the tumor. Scale bar in all panels, 50 μm. All histology is from animals at 3 months of age. αMcm6, antibody to Mcm6. αMsi1, antibody to Msi1. αCtnnb1, antibody to β-catenin.
Colonic tumors from Fapbl-Cre;Apc2lox14/+;KrasLSL-G12D/+ animals were markedly different than those from Apc-mutant animals. Unlike tumors expressing wild-type K-Ras, adenocarcinomas expressing K-RasG12D invariably had uniform high-grade dysplasia throughout the tumor (Fig. 4c,d), suggesting that the presence of activated K-Ras accelerates the progression to malignancy in colonic tumors. Features of high-grade dysplasia included large, fused glands with serrated borders, pseudostratified epithelium, loss of apical–basal polarity, and high nucleus to cytoplasm ratio. Moreover, animals expressing mutant K-Ras throughout the colonic epithelium developed a greater number of tumors than those expressing wild-type K-Ras, leading to a dramatic reduction in lifespan (Supplementary Note and Fig. 3a online). In contrast to those expressing activated K-Ras, tumors expressing mutant N-Ras were virtually indistinguishable from those expressing wild-type K-Ras and N-Ras (Fig. 4e,f), indicating that K-RasG12D has a unique ability to promote tumor progression in the colon.
A notable feature of colonic adenocarcinomas expressing K-RasG12D was the complete lack of terminally differentiated cells in the tumor epithelium. To explore this phenotype in more detail, we used immunohistochemistry to stain tumors for markers of differentiation state (Fig. 4). Tumors expressing wild-type K-Ras showed patchy expression of Mcm6, but tumors expressing K-RasG12D were uniformly positive for this marker (Fig. 4g,j). Although portions of tumors expressing wild-type K-Ras appeared to be poorly differentiated, they did not express this marker of truly undifferentiated cells. To extend this analysis, we immunostained tumors for Musashi-1 (Msi1), a protein expressed in putative stem cells of the murine intestinal epithelium14,15. As was the case with Mcm6, tumors expressing wild-type K-Ras contained small clumps of Msi1-positive cells scattered throughout the poorly differentiated regions of the tumors (Fig. 4h). In contrast, most tumors expressing mutationally activated K-Ras (14 of 16) were uniformly positive for Msi1 (Fig. 4k). This difference caused by the expression of mutant K-Ras is even more surprising because the poorly differentiated regions of K-Ras+ and K-RasG12D tumors both expressed high levels of β-catenin (Fig. 4i,j). Thus, it seems that K-RasG12D cooperates with β-catenin upregulation to fully suppress differentiation. The widespread expression of Mcm6 and Msi in cancers expressing activated K-Ras suggests that this oncoprotein promotes colon cancer progression by locking the tumor epithelial cells in a stem-like state in which differentiation is inhibited.
Tumors expressing K-RasG12D show attenuated MAPK signaling
To understand the molecular pathways operating downstream of mutant K-Ras in the context of colon cancer, we began by analyzing signaling through Ras effector pathways in tumors. Mek was highly phosphorylated in tumors expressing K-RasG12D, but phospho-Erk was not detectably upregulated in those same tumors (Fig. 5a and Note and Supplementary Fig. 3b). Consistent with our observations of mouse cancers, we could not detect significant amounts of phospho-Erk by immunohistochemistry in primary human colon cancers (Supplementary Note and Fig. 4a online).
Figure 5.

Attenuated MAPK signaling in tumors expressing mutant K-Ras. (a) In the context of an autochthonous mouse colonic tumor, K-RasG12D activates Mek, but does not seem to affect the steady state levels of phospho-Erk. Tumors expressing mutant K-Ras also upregulate Mkp3, an Erk phosphatase. Isogenic human colon cancer cell lines also show this attenuated MAPK signaling. (Note that DLD cells express mutant K-Ras (KrasG13D/+) and DKs cells express only wild-type K-Ras (Kras+/−)) (b) Quantitation of phospho-Erk levels in Mkp3 knockdown cells. The steady state levels of phospho-Erk are increased in cells expressing mutant K-Ras (DLD-1) upon knockdown of Mkp3. *P < 0.05, Wilcoxon rank sum test. Phospho-Erk is unchanged when Mkp3 is knocked down in cells expressing wild-type K-Ras (DKs-8). For both DLD-1 and DKs-8, data was combined for two different Mkp3 knockdown lines. pSICOR represents cells infected with empty lentivirus. (c) Growth curves in Mkp3 knockdown cells. DLD-1 cells grow faster when the levels of Mkp3 are reduced.
In our analysis of MAPK signaling in tumors, we noted that tumors expressing K-RasG12D showed elevated concentrations of Mkp3, an Erk phosphatase16. We suspected that the inability of mutant K-Ras to upregulate phospho-Erk may have been as a result of, at least in part, this concomitant upregulation of Mkp3. To address this issue, we used a set of isogenic human colon cancer cell lines. DLD-1 cells express K-RasG13D from its endogenous promoter, whereas their isogenic derivatives, DKs-8, express only wild-type K-Ras by virtue of the mutant allele being deleted by homologous recombination11. In this isogenic pair, mutant K-Ras transmitted downstream signals identical to our autochthonous mouse colon tumors (Fig. 5a and Supplementary Note and Fig. 4b). We used shRNA-mediated knockdown to suppress Mkp3 expression in DLD-1 and DKs-8 cells (Supplementary Table 1 and Fig. 4c). These shRNAs were specific to Mkp3 and did not affect the expression of the related gene Mkp1 (Supplementary Fig. 4d). Knockdown of Mkp3 increased the steady state expression of phospho-Erk, but only in DLD-1 cells expressing K-RasG13D and not in DKs-8 cells (Fig. 5b). This increase in phospho-Erk expression correlated with an increase in the growth rate of the Mkp3 knockdown cells, again only in those cells that expressed mutant K-Ras (Fig. 5c and Supplementary Fig. 4e). Taken together, our data indicate that upregulation of Mkp3 is responsible, at least in part, for the inability of K-Ras to increase the steady state levels of phospho-Erk in colon cancer cells.
Raf is a mediator of K-Ras signaling in colon cancers
Although Erk did not seem to be an important mediator of K-Ras signaling in colon cancers, it remained unclear whether upstream members of the pathway were transmitting the oncogenic K-Ras signal. Indeed, we showed that Mek was a critical mediator of the K-RasG12D phenotype in non-neoplastic colonic epithelium (Fig. 2c). To determine whether Mek has a central role in the ability of K-RasG12D to suppress proliferation and differentiation in the context of an Apc-mutant colonic tumor, we treated tumor-bearing animals with CI-1040. Even though the drug was able to inhibit colonic hyperplasia in Fapbl-Cre;Apc2lox14/+;KrasLSL-G12D/+ animals, CI-1040 treatment did not decrease proliferation in tumors (Fig. 6a). Moreover, tumors from mice treated with CI-1040 retained their widespread expression of Mcm6 and Msi1 (Fig. 6b). These results suggest that Mek is not an important mediator of K-Ras signaling during colon cancer progression, and they are consistent with the results of a Phase II clinical trial in which CI-1040 was ineffective against several types of cancer, including colorectal cancer17.
Figure 6.
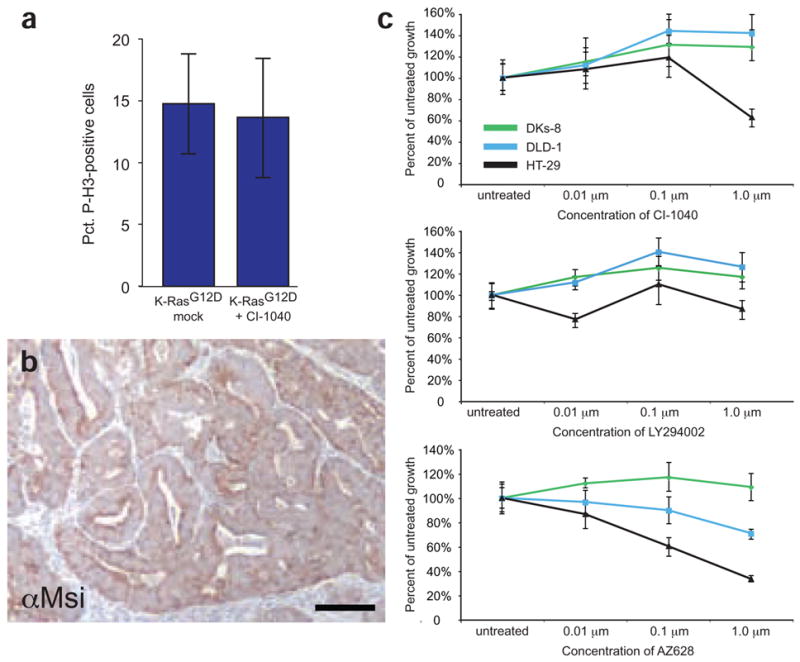
K-RasG12D signals through Raf, but not Mek, to promote tumor proliferation. (a) Inhibition of Mek with CI-1040 does not suppress proliferation in tumors from Fabpl-Cre;Apc2lox14/+;KrasLSL-G12D animals. (b) Msi1 staining of a tumor expressing K-RasG12D from a mouse treated with CI-1040 for one week. The epithelium remains uniformly Msi1-positive, suggesting that the expansion of tumor stem cells by mutant K-Ras is independent of Mek. (c) Inhibition of Ras effectors with small molecules. DLD-1 cells expressing mutant K-Ras are not hypersensitive (compared to DKs-8) to inhibition of Mek by CI-1040 or PI3K by LY294002. However, cells expressing K-RasG13D are sensitive to inhibition of Raf by AZ628 (P = 0.001 at 0.1 μM and P = 0.0007 at 1 μM). As a control in this experiment, HT-29 cells expressing mutant B-Raf were included. These cells are sensitive to CI-1040 and AZ628. Error bars in panels a and c represent standard deviations for each sample class.
To carry out additional mechanistic studies on potential mediators of the K-Ras phenotype, we again used the isogenic pair of human colon cancer cell lines. We designed a medium-throughput assay that allowed us to screen for the relative sensitivity of the cell lines to specific small molecule inhibitors. As a confirmation of our in vivo results with autochthonous mouse cancers, we found that DLD-1 cells expressing mutant K-Ras were not sensitized to inhibition of Mek when compared to the K-Ras wild-type DKs-8 cells, even though CI-1040 was clearly inhibiting Erk activation (Fig. 6c and Note and Supplementary Fig. 4f). As a control in our analysis, we used HT-29 cells, which express mutationally activated B-Raf (B-RafV600E). Cells expressing mutant B-Raf were previously shown to be sensitive to CI-1040 (ref. 18). We screened our panel of cell lines for sensitivity to a variety of small molecule inhibitors of other Ras effector pathways. Cells expressing mutant K-Ras were not sensitized to inhibition of any of the canonical Ras effector pathways, including the PI3K pathway (Fig. 6c). Of note, when we exposed cells to small molecule inhibitors of Raf (AZ628 and Sorafenib)19,20, we found DLD-1 cells to be hypersensitive to Raf inhibition compared to DKs-8 cells (Fig. 6c and Supplementary Fig. 4g). Overall, our data suggest that K-Ras exerts its oncogenic properties in colon cancers through a noncanonical pathway that includes Raf, but not Mek or Erk.
DISCUSSION
Here we demonstrate clear phenotypic differences associated with activating the endogenous Kras and Nras genes in vivo in the colonic epithelium. We find that activated Kras affects proliferation and differentiation, whereas activated Nras suppresses apoptosis. Our studies address the important question of why human colon cancers show a preponderance of Kras mutations, and provide insight into the complexity of signaling downstream of mutationally activated K-Ras.
We found that expression of mutant K-Ras from its endogenous locus induced hyperproliferation in the colonic epithelium (Fig. 1 and 2), but did not seem to be sufficient to induce neoplasia. When combined with a mutation in the Apc tumor suppressor gene, however, mutant Kras promoted tumor progression (Fig. 4). These observations are consistent with data suggesting that mutations in Kras arise after loss of Apc during colon cancer progression in humans21. Previously, mutant K-Ras was found to be sufficient to induce the formation of adenocarcinomas in the murine intestine22. In that model, however, mutant Kras was expressed from the exogenous Villin promoter, which probably led to overexpression and/or ectopic expression. In a second study, mutant K-Ras was expressed from its endogenous promoter, yet failed to induce any detectable histologic or biochemical changes in the mouse small intestine23. At this point, it remains unclear how our conditional Kras allele compares to that used in this previous study23. Although the integrity of the endogenous K-Ras locus has been preserved in our own model, the Kras 3′ UTR has been replaced by an IRES-LacZ cassette in the other conditional allele24. We speculate that the deletion of the endogenous 3′ UTR affects the expression, processing and/or translation of the Kras mRNA, leading to changes in protein expression and signaling. Indeed, clear phenotypic differences have been described when these two alleles are activated in the lung24,25.
Our data suggest that K-RasG12D exerts its phenotypic effects via slightly different mechanisms in neoplastic versus non-neoplastic tissues. When K-Ras is mutationally activated on an otherwise wild-type background, it signals through Mek to promote expansion of the proliferative zone and hyperproliferation at the bottom of the crypt (Fig. 2). Coupled with the lack of phospho-Erk in the proliferative zone of animals expressing K-RasG12D, the data suggest that Mek may be acting in an Erk-independent manner in this setting. Although Erk is the only known downstream target of Mek, Erk-independent functions of Mek have been described, for example, in the insulin-stimulated phosphorylation of Sos26. In the context of an Apc-mutant colon cancer, Mek does not seem to act as a critical mediator of K-Ras signaling (Fig. 6). Notably, Raf does seem to mediate, at least in part, the oncogenic function of K-Ras (Fig. 6). The Raf family of kinases consists of three members, A-Raf, B-Raf and C-Raf (also called Raf1)27. At this time, it is not clear which of the Raf proteins is the relevant target of AZ628 and Sorafenib in colon cancer cells expressing mutant K-Ras20. Regardless of which Raf family member is the relevant target of the small molecule inhibitors, the primary function of the Raf kinases is thought to be to phosphorylate Mek. Thus, it is currently unclear how Raf is transmitting the oncogenic K-Ras signal if not through Mek. In order to fully elucidate the complexity of signaling through this pathway, further studies are needed to identify previously unknown downstream targets of Raf and Mek.
Mutated Nras produced a phenotype in the colonic epithelium that was distinct from that of mutated Kras. Instead of promoting proliferation and suppressing differentiation, N-RasG12D suppressed apoptosis after treatment with DSS (Fig. 3). In vitro studies demonstrated that mutant N-Ras also confers resistance to butyrate-induced apoptosis (Supplementary Fig. 2). It is of note that Nras mutations seem to arise quite late in colon cancer progression, similar to the stage at which mutations in p53 appear to arise1. Given that Nras mutations are found in a small subset of colon cancers, we hypothesize that they may arise in tumors that develop in a background of constant apoptotic stimulus, for example, in cancers that arise in individuals with chronic inflammation of the gastrointestinal tract.
A key question raised by these studies is whether the phenotypic differences between Kras and Nras are merely due to gene expression or whether they are truly reflective of functional differences between the protein isoforms. There is evidence to support the notion that Ras function is regulated by gene expression, especially during development. Knockout alleles of Kras are embryonic lethal, whereas null alleles of Nras and Hras are viable28,29. Homozygous mutation of Nras is lethal when Kras is heterozygous, suggesting that these oncogenes have partially overlapping functions28. Moreover, it has been shown that Hras can functionally substitute for Kras during development when expressed from the Kras locus30. Nevertheless, several lines of evidence argue against gene expression as the sole origin of the phenotypic differences between Kras and Nras in adult tissues. In vitro studies have demonstrated that the four Ras enzymes exert variable effects on downstream effector pathways31. We have previously analyzed K-Ras and N-Ras expression in a panel of human colon cancer cell lines and found that all lines tested express both oncoproteins, suggesting that the cell type that gives rise to colon cancer expresses both Kras and Nras12. shRNA-mediated knockdown of N-Ras suppresses growth (K.M.H., unpublished data), and expression of mutant Nras suppresses apoptosis in these cell lines (Supplementary Fig. 2), suggesting that N-Ras is not functionally inert in this context. Studies done in other in vivo systems also point toward unique functions for each of the Ras isoforms, although no study has examined mutant Ras isoforms expressed from their endogenous loci. For example, retrovirus-mediated expression of mutant Kras, Nras or Hras in hematopoetic cells produced unique leukemic phenotypes, even though they were expressed at similar levels32. On the basis of our own data, we suspect that the selection for Kras over Nras mutations in human colon cancers derives primarily from the unique ability of mutationally activated K-Ras to signal through Raf.
In summary, we have used genetically engineered mice to dissect the unique phenotypes associated with mutation of two highly homologous oncogenes. Our studies have yielded novel insights into the distinct molecular and cellular defects that arise in response to oncogenic activation of K-Ras and N-Ras and have identified Raf as a critical mediator of the K-RasG12D phenotype.
METHODS
Mouse strains and treatments
All mouse experiments were reviewed and approved by the Massachusetts Institute of Technology Committee on Animal Care (CAC). Fabpl-4X@132-Cre33 and Villin-Cre34 transgenic mice were obtained from the Mouse Models of Human Cancer Consortium (MMHCC.) KrasLSL-G12D/+ and Apc2lox14/+ mice have been described previously5,358. Primers used for genotyping of all mouse strains are listed in Supplementary Table 1. Targeting of the Nras locus was done in V26.2 embryonic stem cells derived from C57BL/6 embryos (Supplementary Fig. 1b). Additional information regarding animal experiments can be found in Supplementary Methods online.
For each animal, the entire intestinal tract was removed, flushed with 1× PBS, and fixed overnight in 10% neutral-buffered formalin. Tissue for histologic analysis was removed from the medial colon, approximately 4 cm proximal to the rectum.
Colon cancer cell lines and in vitro experiments
All human colon cancer cell lines were grown in DMEM supplemented with 10% FCS (FCS). We carried out shRNA-mediated knockdown as described36. We selected cells expressing shRNAs in 7.5 μg/ml puromycin. We carried out viability assays by growing cells in 96-well plates in the presence or absence of specific small molecule inhibitors, and we quantified viability after 72 h by staining with Syto60 (Invitrogen), an infrared nucleic acid stain. Plates were scanned and analyzed on a LiCor Odyssey Infrared Imaging System.
Laser capture
To analyze gene expression in the mouse colonic epithelium, we cut 10 μm frozen sections to obtain longitudinal sections of crypts. Tissues corresponding to the top and bottom 30 μm of crypts were isolated by laser capture using an Arcturus microdissection system. We isolated RNA using the RNAqueous-Micro kit (Ambion), and our Taqman analysis has been described previously4. The experimental protocol for analyzing gene expression in primary human samples can be found in Supplementary Methods online.
Immunohistochemistry
We carried out immunohistochemistry according to manufacturer’s recommendations, typically using a modified citric acid unmasking protocol followed by standard detection with 3,3-diaminobenzidine (DAB) using a kit from Vector Laboratories. Samples were counter-stained with hematoxylin. In some cases, secondary antibodies were conjugated to AlexaFluor 594 (Invitrogen) and nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI). We used the following primary antibodies: rabbit α-phospho-histone H3 (Cell Signaling Technology), rabbit α-phospho-Erk1/2 (Cell Signaling Technology), rabbit α-phospho-Mek (Cell Signaling Technology), rabbit α-cleaved caspase 3 (Cell Signaling Technology), goat α-Mcm6 (Santa Cruz), rabbit α-Musashi-1 (Cell Signaling Technology), mouse α-β-catenin (BD Biosciences), and mouse anti-cytokeratin 20 (Sigma).
Quantitative analysis of tissue kinetics
We determined the height of the Mcm6-positive zone by counting the number of cell positions from the bottom of the crypt that the highest staining cell occupied. For phosphohistone H3 (PH3) staining, we determined the total number of positive cells per crypt. For each assay, we scored at least 50 crypts—originating from tissue sections of at least five different mice—for each genotype, and we only scored crypts for which a good longitudinal section was obtained. Statistical analysis was done using the Mstat computer program (see URLs section below). The Wilcoxon rank sum test (also called the Mann-Whitney U test) is a nonparametric test for assessing whether two sets of data points come from the same numerical distribution. It is ideal for biological data, because it does not assume that the data are normally distributed. We determined the total percentage of PH3-positive cells in tumor cells. In these experiments, each tumor represented a single data point.
Protein blotting and pulldowns
Protein blots and pulldown experiments were done on lysates derived from the entire colon or from colon tumors dissected away from nontumor tissue. Tissue was homogenized in 1× MLB buffer (Millipore). All pulldowns were done according to manufacturer’s instructions.
We loaded polyacrylamide gels (8% or 12%) with 25 or 50 μg of purified epithelial protein. Standard protein blots were detected with ECL Plus (Amersham Biosciences), and quantitative protein blots were visualized on a LiCor Odyssey Infrared Imaging System (Fig. 1f). We used the following primary antibodies: mouse α-β-tubulin (clone TUB 2.1) (Sigma), mouse α-K-Ras (F234) (Santa Cruz), mouse α-N-Ras (F155) (Santa Cruz), and chicken α-Dusp6a/Mkp3 (Abcam). The following antibodies were purchased from Cell Signaling Technology: mouse α-Mek1/2, rabbit α-phospho-Mek1/2 (Ser217/Ser221), rabbit α-Erk1/2, mouse α-phospho-Erk1/2 (Thr202/Tyr204), rabbit α-Jnk, mouse α-phospho-Jnk (Thr183/Tyr185), rabbit α-Akt, and mouse α-phospho-Akt (Ser473).
Supplementary Material
Note: Supplementary information is available on the Nature Genetics website.
Acknowledgments
The authors thank A. Charest and A. Shaw for critically reading the manuscript, A. Ventura, J. Keller and U. McDermott for technical advice and D. Lauffenburger and M. McMahon for helpful discussion. This work was supported by US National Institutes of Health grants U01-CA84306 and U54-CA112967 to T.J., K01-CA118425 to K.H., and U01-CA84221 and R01-CA72614 to K.S., grants from the Association pour la Recherche sur le Cancer, Ligue Nationale Contre le Cancer, and Inserm to M.G., and partially by the Cancer Center Support (Core) grant P30-CA14051 from the National Cancer Institute. K.H. was a Robert Black Fellow of the Damon Runyon Cancer Research Foundation and was supported by a Career Development award from the Harvard GI SPORE grant from the National Cancer Institute (P50-CA127003). T.J. is a Howard Hughes Medical Institute Investigator and a Daniel K. Ludwig Scholar. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
URLs. Mstat computer program, http://www.mcardle.wisc.edu/mstat.
AUTHOR CONTRIBUTIONS
K.M.H. and T.J. conceived and designed the experiments. K.M.H., K.R.K., Y.W., A.C., M.C.H., and A.S-C. performed the experiments. J.N.G. consulted on pathological analysis of samples. K.M.H. and T.J. analyzed the data. M.N-K. and M.G. generated and provided conditionally mutant Apc mice. J.S-L. and K.M.S. provided CI-1040. J.S. provided AZ628 and Sorafenib. K.M.H. and T.J. wrote the manuscript.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
Published online at http://www.nature.com/naturegenetics
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Vogelstein B, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 3.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 4.Haigis K, Sage J, Glickman J, Shafer S, Jacks T. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J Biol Chem. 2006;281:638–647. doi: 10.1074/jbc.M509053200. [DOI] [PubMed] [Google Scholar]
- 5.Tuveson DA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 6.Wong MH, Saam JR, Stappenbeck TS, Rexer CH, Gordon JI. Genetic mosaic analysis based on Cre recombinase and navigated laser capture microdissection. Proc Natl Acad Sci USA. 2000;97:12601–12606. doi: 10.1073/pnas.230237997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 8.Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. Bioessays. 2002;24:91–98. doi: 10.1002/bies.10028. [DOI] [PubMed] [Google Scholar]
- 9.Sebolt-Leopold JS, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 10.Wolfman JC, Palmby T, Der CJ, Wolfman A. Cellular N-Ras promotes cell survival by downregulation of Jun N-terminal protein kinase and p38. Mol Cell Biol. 2002;22:1589–1606. doi: 10.1128/mcb.22.5.1589-1606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 12.Keller JW, et al. Oncogenic K-RAS subverts the antiapoptotic role of N-RAS and alters modulation of the N-RAS:gelsolin complex. Oncogene. 2007;26:3051–3059. doi: 10.1038/sj.onc.1210103. [DOI] [PubMed] [Google Scholar]
- 13.Keller JW, et al. Oncogenic KRAS provides a uniquely powerful and variable oncogenic contribution among RAS family members in the colonic epithelium. J Cell Physiol. 2007;210:740–749. doi: 10.1002/jcp.20898. [DOI] [PubMed] [Google Scholar]
- 14.He XC, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39:189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Potten CS, et al. Identification of a putative intestinal stem cell and early lineage marker; musashi-1. Differentiation. 2003;71:28–41. doi: 10.1046/j.1432-0436.2003.700603.x. [DOI] [PubMed] [Google Scholar]
- 16.Pouyssegur J, Volmat V, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem Pharmacol. 2002;64:755–763. doi: 10.1016/s0006-2952(02)01135-8. [DOI] [PubMed] [Google Scholar]
- 17.Rinehart J, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 18.Solit DB, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDermott U, et al. Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007;104:19936–19941. doi: 10.1073/pnas.0707498104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilhelm SM, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 21.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 22.Janssen KP, et al. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. [DOI] [PubMed] [Google Scholar]
- 23.Sansom OJ, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci USA. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guerra C, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 25.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holt KH, Kasson BG, Pessin JE. Insulin stimulation of a MEK-dependent but ERK-independent SOS protein kinase. Mol Cell Biol. 1996;16:577–583. doi: 10.1128/mcb.16.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leicht DT, et al. Raf kinases: function, regulation and role in human cancer. Biochim Biophys Acta. 2007;1773:1196–1212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson L, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esteban LM, et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 2001;21:1444–1452. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potenza N, et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005;6:432–437. doi: 10.1038/sj.embor.7400397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voice JK, Klemke RL, Le A, Jackson JH. Four human ras homologs differ in their abilities to activate Raf-1, induce transformation, and stimulate cell motility. J Biol Chem. 1999;274:17164–17170. doi: 10.1074/jbc.274.24.17164. [DOI] [PubMed] [Google Scholar]
- 32.Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 2007;67:7139–7146. doi: 10.1158/0008-5472.CAN-07-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saam JR, Gordon JI. Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem. 1999;274:38071–38082. doi: 10.1074/jbc.274.53.38071. [DOI] [PubMed] [Google Scholar]
- 34.el Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 35.Colnot S, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab Invest. 2004;84:1619–1630. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- 36.Ventura A, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci USA. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Genetics website.