

Abstract
The Pto gene encodes a serine/threonine kinase that confers resistance in tomato to Pseudomonas syringae pv. tomato strains that express the avirulence gene avrPto. Partial characterization of the Pto signal transduction pathway and the availability of transgenic tomato lines (± Pto) make this an ideal system for exploring the molecular basis of disease resistance. In this paper, we test two transgenic tomato cell suspension cultures (±Pto) for production of H2O2 following independent challenge with two strains of P. syringae pv. tomato (±avrPto). Only when Pto and avrPto are present in the corresponding organisms are two distinct phases of the oxidative burst seen, a rapid first burst followed by a slower and more prolonged second burst. In the remaining three plant–pathogen interactions, we observe either no burst or only a first burst, indicating that the second burst is correlated with disease resistance. Further support for this observation comes from the finding that both resistant and susceptible tomato lines produce the critical second oxidative burst when challenged with P. syringae pv. tabaci, a nonhost pathogen that elicits a hypersensitive response on both tomato lines. The Pto kinase is not required, however, for the oxidative burst initiated by non-specific elicitors such as oligogalacturonides or osmotic stress. A model describing a possible role for the Pto kinase in the overall scheme of oxidative burst signaling is proposed.
Keywords: hydrogen peroxide, plant defense, disease resistance gene, Fen, fenthion
Plants activate a variety of defense mechanisms upon pathogen infection. These responses include production of active oxygen species (1–3), biosynthesis of phytoalexins (4), induction of pathogenesis-related genes (5), strengthening of barriers to invasion (6, 7), and generation of the hypersensitive response (2). Resistance to a variety of microorganisms occurs in a “gene-for-gene” manner, wherein plant varieties expressing a specific resistance (R) gene withstand infection by pathogen strains expressing a corresponding avirulence gene (avr) (8). Absence of either the R gene or avirulence gene from the corresponding organisms results in susceptibility to disease. Recently, several R genes that conform to the gene-for-gene model have been cloned (9), the first being the Pto gene from tomato (10).
The Pto gene encodes a cytoplasmic serine/threonine kinase that confers resistance to strains of Pseudomonas syringae pv. tomato that express the avrPto avirulence gene (10–12). Recently, other signaling elements in this pathway leading to the hypersensitive response and, ultimately, disease resistance have been identified and probably include a serine/threonine kinase designated Pti1 (13). Another member of the Pto gene family, Fen, encodes a serine/threonine kinase that confers sensitivity in tomato to an organophosphorous insecticide, fenthion. Fen may also participate in signaling disease resistance, but neither the cognate avirulence gene nor its pathogenic host has been identified (14). Although the Pto kinase is one of the best-characterized R gene products, the molecular mechanisms by which Pto confers disease resistance are currently unknown. Nevertheless, the availability of transgenic lines (±Pto) and the partial characterization of the Pto signaling pathway make this system ideal for addressing the question of how plants resist infection.
Previous work has established the rapid production of active oxygen species (termed the oxidative burst) as an important plant response to pathogen infection (2, 15). The oxidative burst is an early localized defense response that involves the production of potentially cytotoxic quantities of H2O2 and O2 (1). Although genes known to be associated with generation of the oxidative burst have not yet been isolated, identification of signaling events leading to this response so far include receptor binding (16), activation of G proteins (17), stimulation of phospholipases C (18) and A (19), and changes in protein phosphorylation (20). Presumably, one or more of these essential signal transduction intermediates could constitute the product of an R gene if it were specifically activated by a corresponding avirulence gene product from an incompatible pathogen.
In plants, the oxidative burst commonly occurs in two distinct phases (2, 21). The initial rapid phase ends within an hour of its initiation and is followed by a second slower burst that may last for 3–6 hr (21). Several reports indicate that the second burst is only obtained in incompatible interactions between cultured plant cells and live pathogens (2, 22, 23). However, in these studies the responsible resistance and avirulence genes were not well-characterized and hence an unequivocal attribution of the oxidative burst to activation of a specific R gene could not be made. Nevertheless, the observation that disease resistance may be correlated with expression of the second phase of the oxidative burst prompted us to conduct a more detailed examination of this relationship in transgenic tomato plants that differ solely in expression of the Pto resistance gene. Using P. syringae pv. tomato (±avrPto), we show that the second phase of the oxidative burst is indeed dependent on coexpression of Pto in the tomato and avrPto in the pathogen. This study constitutes the first time that all four possible gene-for-gene interactions have been examined for induction of the oxidative burst, and it also is the first implication of a specific kinase in this defense response.
MATERIALS AND METHODS
Materials.
Pyranine was obtained from Molecular Probes. Fensulfothion was obtained from Chemserve (West Chester, PA). All other chemicals were of reagent grade or higher purity and were obtained from major suppliers.
Tomato Lines.
Rio Grande-PtoR (Pto/Pto, Fen/Fen), is resistant to bacterial speck disease caused by avrPto-expressing P. syringae pv. tomato and sensitive to fenthion. Rio Grande (pto/pto, fen/fen) is a near isogenic line that is susceptible to bacterial speck and insensitive to fenthion (10). Transgenic lines differing only in the presence or absence of a 35S::Pto transgene were also used and were designated BC-R and BC-S, respectively (10). All tomato plants were maintained under greenhouse conditions in a 16-hr light/8-hr dark cycle at 25–30°C.
Plant Cell Culture.
Cell suspension cultures were generated from each tomato line by standard plant cell culture techniques (24). Briefly, tomato stems were surface sterilized and placed on agar plates (R-3 media with 5% agar) to allow callus formation. The calli were fragmented and transferred to liquid R-3 medium. The resulting cell cultures were maintained by transferring 3 ml of culture to 25 ml fresh medium every 14 days. Cells were generally responsive to bacteria or elicitor stimulation ≈20 hr after transfer to fresh medium and were used for oxidative burst evaluation within 5 hr of this time period.
Bacterial Strains.
P. syringae pv. tomato strains with or without avrPto [Pst(avrPto) and Pst, respectively] as well as P. syringae pv. tabaci strain 11528R (provided by Kyle Willis, University of Wisconsin, Madison) were grown on King’s medium B supplemented with antibiotics. Bacteria were grown for ≈45 hr at 30°C, washed twice, and resuspended into 10 mM MgCl2 before use. Colony-forming units/ml were determined spectrophotometrically (0.05 OD600 ≈2.3 × 107 colony-forming units/ml).
Elicitors.
An oligogalacturonic acid (OGA) fraction that elicits hydrogen peroxide production in cultured cells was purified, as described (1). The preparation used in this study contained 0.5 galacturonic acid equivalents/ml as determined by the method of Blumenkrantz and Asboe-Hansen (25).
Spectrofluorometric Determination of H2O2 Production.
H2O2 production in cultured tomato cells was detected by monitoring the oxidative quenching of the fluorescent peroxidase substrate, pyranine (8-hydroxy pyrene-1,3,6-trisulfonic acid trisodium salt/405 nm λex/512 nm λem), as described (1, 26). Briefly, 1.5 ml cells were mixed with 7 μl pyranine (0.2 mg/ml stock solution) in a fluorimetric cuvette and maintained in suspension by mild stirring. Following addition of bacteria or purified elicitor, the loss of fluorescence of the dye was continuously monitored and the initial slope of the quenching curve was used to determine the rate of H2O2 production.
RESULTS
The Pto Gene Is Required for the Second Oxidative Burst Initiated by P. syringae pv. tomato Expressing avrPto.
To determine whether the Pto kinase is required for the oxidative burst, we tested transgenic tomato lines (± Pto) for their ability to mount an oxidative burst when challenged by avirulent (+avrPto) or virulent strains of P. syringae pv. tomato. Fig. 1 shows the oxidative burst profiles of the four possible plant pathogen combinations. In whole plants, the BC-R tomato line (containing the Pto transgene) is able to recognize the avirulent pathogen (Pst [avr Pto]) and mount an effective resistance response. The remaining three combinations are susceptible interactions that result in disease symptoms. In the BC-R/Pst(avrPto) interaction, two distinct phases of the oxidative burst can be seen, the first burst ending within 1 hr and the second burst peaking at 3 hr postinoculation (Fig. 1A). The BC-S line, which is genetically identical to BC-R but lacks the Pto gene, generates neither the first nor the second burst on infection with Pst(avrPto) (Fig. 1C). Further, the disease-susceptible interaction between BC-R cells and Pst initiates the first but not the key second burst (Fig. 1B). Finally, BC-S cells upon treatment with Pst induced neither burst (Fig. 1D). Importantly, peroxidase activity was in vast excess in both resistant and susceptible tomato lines, indicating that the observed differences reflect actual differences in oxidant production.
Figure 1.
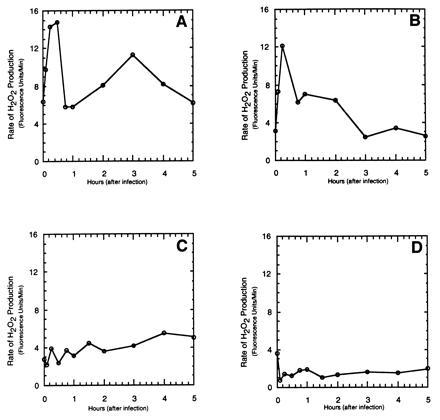
Dependence of the rate of H2O2 release by transgenic tomato suspension cultures on time following inoculation with P. syringae pv. tomato. Twenty-five milliliters of the desired tomato suspension culture (± Pto) was inoculated with 107 colony-forming units/ml Pst or Pst (avrPto). Aliquots (1.5 ml) were then taken out at the appropriate times, transferred to a fluorimeter cuvette and treated with 7 μl pyranine. The fluorescence quenching due to H2O2 catalyzed oxidation of pyranine was then monitored for 15 min. The initial rate of H2O2 production was calculated and plotted as a function of time after inoculation with bacteria. Ten units of fluorescence corresponds to 1 μmol of H2O2. Similar profiles were seen in at least two independent experiments. (A) BC-R + Pst (avrPto); (B) BC-R + Pst; (C) BC-S + Pst (avrPto); (D) BC-S + Pst.
While the data presented in Fig. 1 are representative of several trials with each combination, it should be noted that some variability was observed in the rates of H2O2 biosynthesis, mainly a decrease with cell age (e.g., 24-hr old cultures often showed a reduction in burst activity of 40%). Further, there was also some variation in the timing (peaking 45–90 min postinoculation) and magnitude (±30%) of the first burst in the case of the BC-R/Pst combination. Whether this latter variability is a function of cell age remains to be investigated. Nevertheless, in all cases, resistance to bacterial speck disease as mediated by the Pto kinase is correlated with expression of the slower, more prolonged second phase of the oxidative burst, and susceptible plant-pathogen combinations invariably failed to generate this latter phase of oxidant production.
P. syringae pv. tabaci Induces a Burst in Both Resistant and Susceptible Lines.
To further support the correlation between disease resistance and the oxidative burst, we challenged both transgenic cultures with P. syringae pv. tabaci, a pathogen of tobacco that elicits a nonhost hypersensitive resistance response on tomato. Consistent with expectations, we observed elevated levels of H2O2 production that lasted at least 5 hr and peaked 1.5 hr following inoculation (Fig. 2). Although the first oxidative burst was not always visible (Fig. 2B), the invariant expression of the second burst confirms that prolonged production of oxidants correlates well with the hypersensitive disease resistance response. Further, these data suggest that the Pto kinase is not required for other bacterially induced oxidative bursts and that both BC-R and BC-S tomato lines possess the signaling machinery necessary for induction of the second burst in interactions with other incompatible pathogens.
Figure 2.
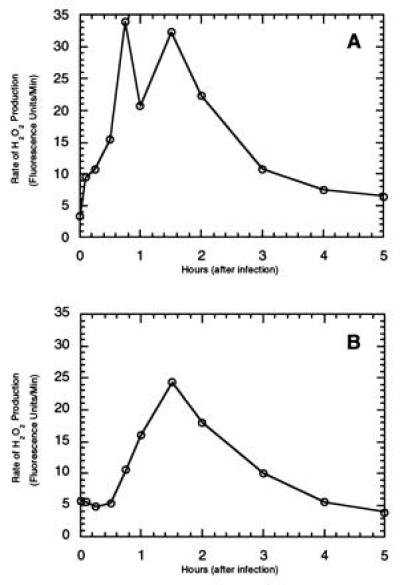
Time dependence of the rate of H2O2 release by transgenic tomato suspension cultures (±Pto) following inoculation with P. syringae pv. tabaci. BC-R cells (A) or BC-S cells (B) were treated with 107 colony-forming units/ml P. syringae pv. tabaci strain 11528R. Aliquots (1.5 ml) were removed at the appropriate times, transferred to a fluorimeter cuvette, and treated with 7 μl pyranine. The fluorescence transition due to peroxidase catalyzed oxidation of pyranine was then monitored for 15 min. The initial rate of H2O2 production was calculated and plotted as a function of time after inoculation with bacteria. Similar profiles were seen in three independent experiments.
Fensulfothion Induces an Oxidative Burst.
Tomato cultivars that contain the Pto locus develop small necrotic lesions, similar to a hypersensitive response upon exposure to the organophosphorous insecticide fenthion (14). Fen, a member of the Pto gene family, is responsible for this fenthion sensitivity (14). The Fen protein shares 80% predicted sequence identity with Pto, and since transgenic tomato plants that stably overexpress the Pto gene display mild sensitivity to fenthion, it has been hypothesized that Pto and Fen participate in the same signal transduction pathway (14). To further test this hypothesis, we assayed tomato lines that are isogenic for the Pto region (Pto/Pto, Fen/Fen; or pto/pto, fen/fen) for their ability to generate an oxidative burst upon exposure to fenthion. Since fenthion is insoluble in water, we substituted fensulfothion, a more water soluble analog that elicits responses similar to those of fenthion in tomato plants expressing a Fen transgene (14). As seen in Fig. 3, fensulfothion indeed induces a prolonged oxidative burst in cell cultures containing the Pto locus but not in the isogenic line lacking the locus. These data indicate that the Fen gene also mediates expression of an oxidative burst. Whether the prolonged nature of the burst derives from a merger of the first and second phases of active oxygen production, or instead, reflects a slow inactivation of the pathway could not be deduced from these studies.
Figure 3.
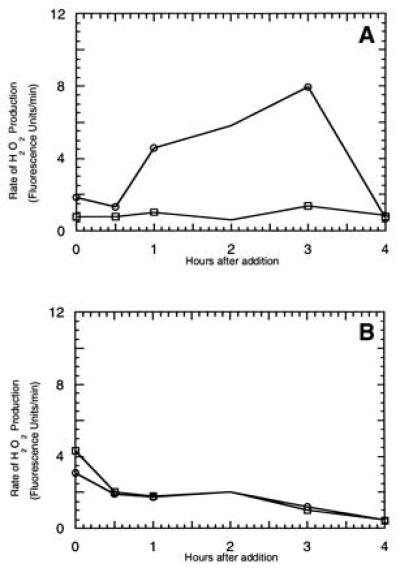
Analysis of the ability of fensulfothion to induce an oxidative burst in tomato cell suspension cultures expressing or lacking the Fen gene. Tomato line Rio Grande-PtoR (Pto/Pto, Fen/Fen) (A) or Rio Grande (pto/pto, fen/fen) (B) isogenic cell lines were treated either with dimethyl sulfoxide (□) or 0.02% fensulfothion in dimethyl sulfoxide (○) and at the appropriate times, 1.5 ml aliquots were transferred to fluorimeter cuvette and treated with 7 μl pyranine. H2O2-mediated quenching of pyranine fluorescence was followed spectrofluorimetrically as a function of time. Similar results were obtained in three independent experiments.
The Pto Kinase Is Not Required for the Oxidative Burst Induced by Other Elicitors.
Earlier work has shown that the oxidative burst can be initiated by a wide variety of elicitors (15). These elicitors include plant cell wall degradation products such as OGA, pathogen derived polypeptides, and even mechanical stress. Several of these elicitors appear to initiate different signal transduction pathways that culminate at the same oxidase complex in the plasma membrane (15, 27). We were therefore interested in ascertaining whether the Pto kinase plays a role in oxidative bursts initiated by these elicitors. Transgenic tomato lines were osmotically stressed or treated with OGA, and then H2O2 production was followed spectrofluorimetrically. As can be seen in Fig. 4, both BC-R and BC-S cell lines were able to respond to both stimuli, indicating that the Pto kinase is not required for the OGA- or osmotically induced oxidative burst. Thus, tomato plants lacking the Pto kinase may be compromised in mounting a sustained oxidative burst only when attacked by pathogenic strains of P. syringae pv. tomato.
Figure 4.
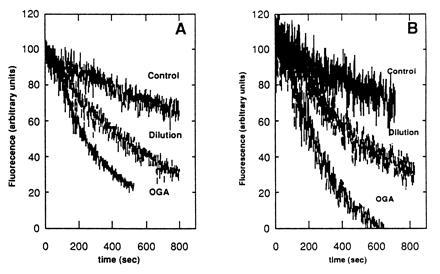
Evaluation of the ability of transgenic tomato suspension cultures (±Pto) to respond to generic elicitors of the oxidative burst. Transgenic tomato cells expressing the Pto transgene (BC-R) (A) or lacking Pto (BC-S) (B) were cultured for 18 hr and either left untreated (control), treated with 50 μl OGA (0.5 mg/ml) or diluted with an equal volume of distilled water (1:1 dilution) to induce a mechanically stimulated oxidative burst. The quenching of pyranine fluorescence due to H2O2 production was then followed spectrofluorimetrically as a function of time. Similar results were obtained in three independent experiments.
DISCUSSION
Significant strides have been made in identifying and cloning R genes from a variety of plants that confer resistance to pathogens in a gene-for-gene manner (9, 11). The next challenge is to determine the molecular mechanisms by which the R gene products mediate disease resistance. While Pto has been shown to be a functional serine/threonine kinase that phosphorylates Pti1, another serine/threonine kinase, the remainder of the signal transduction pathway and the ultimate defense mechanisms activated by the pathway remain to be elucidated (13). Our present findings suggest that the Pto kinase plays an essential role in generation of the second oxidative burst seen in the incompatible BC-R/Pst(avrPto) interaction. Studies with near-isogenic tomato lines suggest that the closely related Fen kinase plays a similar role in the oxidative burst seen in tomato cells after exposure to fensulfothion.
The second oxidative burst is observed only in the interaction of the BC-R resistant tomato line and the avirulent pathogen expressing avrPto. Although the interactions with the susceptible transgenic line exhibited neither phase of the burst, we have commonly observed the first burst (but not the second burst) in similar studies with isogenic lines (data not shown). Further, the compatible interaction between BC-R and Pst invariably yielded the first phase of oxidant production (Fig. 1B). Together, these data suggest that the first burst may be a generic or nonspecific response to a developing interaction not yet identified by the plant. They also indicate that all cells have the basic machinery for initiating this first burst and that the second burst is the principal determinant of disease resistance. Based on these data, it is reasonable to propose that plants may respond to many stress/defense-related signals by generating the first burst. However, only when the plant cell receives a second confirming signal that a pathogen is indeed present will it initiate the second oxidative burst. This more stringent requirement for induction of the latter phase of oxidant production may exist to prevent tissue damage in response to benign stimuli.
Earlier work has suggested that the second oxidative burst is essential of the resistant reaction, namely, hypersensitive cell death (2, 3). In contradiction to these studies, Glazener et al. (28) have recently shown that the second burst is not sufficient for induction of the hypersensitive response. Whether the second burst observed in the BC-R/Pst(avrPto) interaction is essential for the hypersensitive response awaits further study.
We have also shown that fensulfothion can elicit an oxidative burst in tomato plants carrying the Fen gene, suggesting that Pto and Fen may converge on a common signal transduction pathway. Other evidence for this contention, aside from their homology and location in the same gene family cluster, includes the observation that transgenic plants overexpressing Pto exhibit mild sensitivity to fenthion (14). Furthermore, Salmeron et al. (29) have identified a tomato locus (Prf) that controls both Pseudomonas resistance and fenthion sensitivity. How the Prf gene product participates with the Pto and Fen kinases in signaling an oxidative burst will be an important area for future research.
Previous work from our lab has shown that OGA activates an oxidative burst via a pathway involving receptor binding, activation of a G-protein, Ca2+ influx, induction of phospholipase C, and stimulation of protein kinases (15, 30). Preliminary evidence suggests that mechanical induction of the oxidative burst may involve some of the same intermediates (31) The observation that absence of Pto does not impair development of the oxidative burst in response to the above elicitors obviously requires that Pto function in an independent signaling pathway. Use of multiple signaling cascades is, in fact, not new to the oxidative burst system, since a Verticillium-derived elicitor has already been shown to transduce its signal via activation of phospholipase A, with no participation of phospholipase C (19). Assuming that all pathways culminate in assembly of the same oxidase complex (15, 27), it is tempting to speculate that the various pathways might converge upon kinases that control complex assembly. Activation of a mitogen-activating protein kinase pathway has, in fact, already been noted in wounded and fungally infected plants (32, 33). However, building on the growing observations of similarity with the neutrophil oxidase system (27, 34) and considering the above findings, the scheme presented in Fig. 5 can be offered as a working model of plant oxidase activation in response to pathogen attack. In addition to its role in the oxidative burst the Pto kinase is likely to activate other defense responses. Further scrutiny will obviously be required to verify this model.
Figure 5.
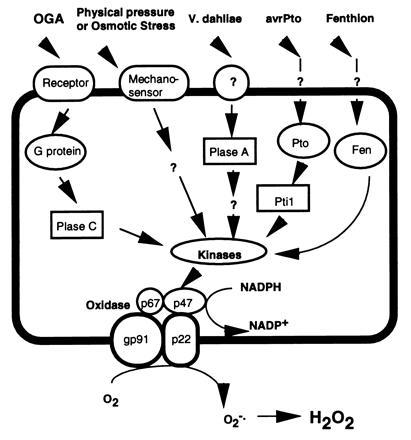
Hypothetical model showing the integration of the Pto kinase with other known signaling intermediates in the oxidative burst signal transduction pathways. See text for details and references. Plase A, phospholipase A; Plase C, phospholipase C; oxidase subunits include p47, p67, gp91, and p22.
Finally, our results add to the growing body of evidence supporting a role for the oxidative burst in disease resistance (3, 15, 21). However, while oxidant generation may be critical to successful repulsion of microbial attacks, the mechanism(s) by which H2O2 and O2⨪ exert their protective properties are still uncertain. Aside from their direct microbicidal effects (35) such oxidants may promote cell wall stabilization (6), enhance phytoalexin biosynthesis (36), generate new second messengers (37), or stimulate defense gene induction (3). Whether any or all of these effects are responsible for the observed linkage to disease resistance is an important question that also requires more detailed examination.
Acknowledgments
We thank Dr. P. M. Hasegawa for helping us generate tomato cell suspension cultures. This work was supported by National Science Foundation Grants MCB-93-03929 (P.S.L.) and MCB-93-03359 (G.B.M.), and the David and Lucille Packard Foundation (G.B.M.).
Footnotes
Abbreviations: OGA, oligogalacturonic acid; BC-R and BC-S, transgenic lines differing only in the presence or absence of a 35S::Pto transgene, respectively.
References
- 1.Legendre L, Rueter S, Heinstein P F, Low P S. Plant Physiol. 1993;102:233–240. doi: 10.1104/pp.102.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine A, Tenhaken R, Dixon R, Lamb C. Cell. 1994;79:583–593. doi: 10.1016/0092-8674(94)90544-4. [DOI] [PubMed] [Google Scholar]
- 3.Mehdy M C. Plant Physiol. 1994;105:467–472. doi: 10.1104/pp.105.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholson R L. Annu Rev Phytopathol. 1992;30:369–389. [Google Scholar]
- 5.Dixon R A, Lamb C J. Annu Rev Plant Physiol Plant Mol Biol. 1990;41:339–367. doi: 10.1146/annurev.arplant.48.1.251. [DOI] [PubMed] [Google Scholar]
- 6.Bradley D J, Kiellbom P, Lamb C J. Cell. 1992;70:21–30. doi: 10.1016/0092-8674(92)90530-p. [DOI] [PubMed] [Google Scholar]
- 7.Olson P D, Varner J E. Plant J. 1993;4:887–892. [Google Scholar]
- 8.Keen N T. Annu Rev Genet. 1990;24:447–463. doi: 10.1146/annurev.ge.24.120190.002311. [DOI] [PubMed] [Google Scholar]
- 9.Staskawicz B J, Ausubel F M, Baker B J, Ellis J G, Jones J D G. Science. 1995;268:661–667. doi: 10.1126/science.7732374. [DOI] [PubMed] [Google Scholar]
- 10.Martin G B, Brommonschenkel S H, Chunwongse J, Frary A, Ganal M W, Spivey R, Wu T, Earle E D, Tanksley S D. Science. 1993;262:1432–1436. doi: 10.1126/science.7902614. [DOI] [PubMed] [Google Scholar]
- 11.Lamb C J. Cell. 1994;76:419–422. doi: 10.1016/0092-8674(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 12.Loh Y-T, Martin G B. Proc Natl Acad Sci USA. 1995;92:4181–4184. doi: 10.1073/pnas.92.10.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J, Loh Y-T, Bressan R A, Martin G B. Cell. 1995;83:925–935. doi: 10.1016/0092-8674(95)90208-2. [DOI] [PubMed] [Google Scholar]
- 14.Martin G B, Frary A, Wu T, Brommonschenkel S, Chunwongse J, Earle E D, Tanksley S D. Plant Cell. 1994;6:1543–1552. doi: 10.1105/tpc.6.11.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Low P S, Merida J R. Physiol Plant. 1996;96:533–542. [Google Scholar]
- 16.Horn M A, Heinstein P F, Low P S. Plant Cell. 1989;1:1003–1009. doi: 10.1105/tpc.1.10.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Legendre L, Heinstein P F, Low P S. J Biol Chem. 1992;267:20140–20147. [PubMed] [Google Scholar]
- 18.Legendre L, Yueh Y G, Crain R, Haddock N, Heinstein P F, Low P S. J Biol Chem. 1993;268:24559–24563. [PubMed] [Google Scholar]
- 19.Chandra S, Heinstein P F, Low P S. Plant Physiol. 1996;110:979–986. doi: 10.1104/pp.110.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandra S, Low P S. Proc Natl Acad Sci USA. 1995;92:4120–4123. doi: 10.1073/pnas.92.10.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baker C J, Orlandi E W. Annu Rev Phytopathol. 1995;33:299–321. doi: 10.1146/annurev.py.33.090195.001503. [DOI] [PubMed] [Google Scholar]
- 22.Baker C J, O’Neill N R, Keppler L D, Orlandi E W. Phytopathology. 1991;81:1504–1507. [Google Scholar]
- 23.Baker C J, Mock N M, Glazener J A, Orlandi E W. Physiol Mol Plant Pathol. 1993;43:81–94. [Google Scholar]
- 24.Bressan R A, Hasegawa P M, Handa A. Plant Sci Lett. 1981;21:23–30. [Google Scholar]
- 25.Blumenkrantz N, Asboe-Hansen G. Anal Biochem. 1973;54:484–489. doi: 10.1016/0003-2697(73)90377-1. [DOI] [PubMed] [Google Scholar]
- 26.Apostol I, Heinstein P F, Low P S. Plant Physiol. 1989;90:109–116. doi: 10.1104/pp.90.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dwyer S C, Legendre L, Low P S, Leto T L. Biochim Biophys Acta. 1996;1289:231–237. doi: 10.1016/0304-4165(95)00156-5. [DOI] [PubMed] [Google Scholar]
- 28.Glazener J A, Orlandi E W, Baker C J. Plant Physiol. 1996;110:759–763. doi: 10.1104/pp.110.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salmeron J M, Barker S J, Carland F M, Mehta A Y, Staskawicz B J. Plant Cell. 1994;6:511–520. doi: 10.1105/tpc.6.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Low P S, Schroeder A T. In: Plant Microbe Interactions. Stacey G, Keen N, editors. Vol. 3. New York: Chapman Hall; 1996. in press. [Google Scholar]
- 31.Yahraus T, Chandra S, Legendre L, Low P S. Plant Physiol. 1995;109:1259–1266. doi: 10.1104/pp.109.4.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki K, Shinshi H. Plant Cell. 1995;7:639–647. doi: 10.1105/tpc.7.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seo S, Okamoto M, Seto H, Ishizuka K, Sano H, Ohashi Y. Science. 1995;270:1988–1992. doi: 10.1126/science.270.5244.1988. [DOI] [PubMed] [Google Scholar]
- 34.Low, P. S. & Dwyer, S. C. (1994) Proceedings of 1994 Korean Botanical Society Symposium on Plant Science, pp. 75–87.
- 35.Peng M, Kuc J. Phytopathology. 1992;82:696–699. [Google Scholar]
- 36.Degousee N, Triantaphylides C, Montillet J-L. Plant Physiol. 1994;104:945–952. doi: 10.1104/pp.104.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fialkow L, Chan C K, Grinstein S, Downer G P. J Biol Chem. 1993;268:17131–17137. [PubMed] [Google Scholar]