

Abstract
The genetic changes and mechanisms underneath the progression of estrogen-dependent to estrogen-independent, antiestrogen-resistant and metastatic breast cancers are unclear despite it is a major problem of the endocrine therapy. To identify genes responsible for this progression, we carried out a genetic screening by an enhanced retroviral mutagen (ERM)-mediated random mutagenesis in the estrogen-dependent T47D breast cancer cells. We found that T47D cells contain only one p27kip1 (p27) allele coding for the p27 cyclin-dependent kinase inhibitor. An ERM insertion into the p27 locus of T47D cells disrupted the p27 gene and created estrogen-independent and antiestrogen-resistant breast cancer cells that still maintained functional estrogen receptors. Disruption of p27 in T47D cells resulted in several changes and most of these changes could be rescued by p27 restoration. First, CDK2 activity was increased in the absence of estrogen or presence of estrogen antagonists tamoxifen or ICI; second, AIB1, a cancer-overexpressed transcriptional coactivator, was hyper-phosphorylated, which made AIB1 a better coactivator for E2F1; and third, Gab2 and Akt activity were increased following E2F1 overactivation, leading to a significant enhancement of cell migration and invasion. Furthermore, the p27-deficient cells, but not their T47D control cells, developed lung metastasis in an ovarian hormone-independent manner when they were intravenously injected into nude mice. In sum, loss of p27 activated AIB1, E2F1, Gab2 and Akt, increased cell migration and invasion, caused antiestrogen insensitivity and promoted metastasis of breast cancer cells. These findings suggest that p27 plays an essential role in restriction of breast cancer progression.
In estrogen-dependent breast cancer cells, estrogen, through ERα, enhances c-Myc and cyclin D1 expression, downregulates p27 and activates cyclin E/CDK2 to promote G1/S transition (1, 2). Since most primary breast cancers express ERα and require estrogen to grow, estrogen antagonists and aromatase inhibitors such as tamoxifen and letrozole are used to treat estrogen-dependent breast cancers (3–5). Though these treatments are initially effective, acquired resistances are major problems. In most cases, development of drug resistance is not due to a loss or mutation of ERα (4, 6).
Overexpression or activation of receptor tyrosine kinases (RTK) and Ras oncoproteins is common in cancers. HER2/Neu overexpression happens in 20–30% breast cancers and correlates with more aggressive cancer phenotypes and tamoxifen resistance (7, 8). Ras and RTKs including HER2, IGFR and EGFR activate PI3K/Akt pathway (9–12). This pathway plays a pivotal role in cell survival, proliferation, motility, tumorigenesis and metastasis through phosphorylation and subsequent relocalization of key regulatory molecules such as p27 (13–15).
In cell nucleus, p27 associates with cyclin E/CDK2 and inhibits Rb hyper-phosphorylation to keep cells in G1 phase (16). Mitogenic stimuli cause p27 phosphorylation, ubiquitination, degradation and translocation to the cytoplasm and increase cyclin E-CDK2 activity, leading to G1/S transition through Rb phosphorylation and E2F activation (16). In breast cancers overexpressing HER2, Akt phosphorylates p27 and keeps p27 in the cytoplasm, which precludes p27-induced G1 arrest (13–15). Therefore, both total p27 reduction and p27 exclusion from the nucleus of breast cancer cells are associated with poor prognosis and estrogen independence (16, 17). Downregulation of p27 also enhances MCF-7 breast cancer cell growth in the presence of antiestrogens (18, 19). However, despite many studies correlating p27 levels and locations with tumorigenesis, the lack of p27 null mutation in human tumors has made it difficult to understand the exact role of p27 in breast cancer progression (20). Furthermore, the fact that oncogene-induced mammary tumorigenesis is accelerated in p27+/− mice but suppressed in p27−/− mice suggests a complex role of p27 in breast cancer (21).
E2F is the key transcriptional factor for cell cycle progression (16). E2F1 interacts with AIB1 (amplified in breast cancer 1) and this interaction potentiates E2F1 target gene expression (22, 23). AIB1 is a transcriptional coactivator for nuclear receptors and other transcription factors including E2F1 (22, 24). AIB1 is overexpressed in about 60% of human breast tumors (25). Overexpression of AIB1 in mouse mammary epithelium causes mammary carcinomas (26), while inactivation of AIB1 in mice suppresses oncogene and carcinogen-induced mammary tumorigenesis (27, 28). These findings indicate that AIB1 is an oncogene. AIB1 activity is not only determined by its concentration but also regulated by phosphorylation (29). In addition, E2F1, through upregulation of the adaptor protein Grb2-associated binder 2 (Gab2), strongly activates Akt (30) that promotes cell motility, invasion and cancer metastasis (31, 32). Thus, we hypothesize that the serial events initiated by p27 inactivation may promote breast cancer cell migration, invasion and metastasis.
In this study, we have performed a genetic screening by using the ERM system (33) and identified the p27 loss-of-function clone. We show that p27 deficiency in T47D cells causes estrogen-independent and antiestrogen-resistant growth, stimulates AIB1 phosphorylation and its coactivator activity for E2F1, promotes Akt activity, cell motility and invasion, and results in lung metastasis in ovariectomized nude mice injected intravenously with these cells.
Materials and Methods
Retroviral infection
T47D cells (104) were infected with MSCV-Eco-zeo and MSCV-SV-tTA viruses (MOI 10:1) and selected in medium with 200 μg/ml G418 (33). The survived T47DtTA cells were expanded to 107, transferred into 6-well plates (5 × 105 cells/well) and spin-infected with ERM retroviruses. Each of the three ERM retroviral vectors carried an ERM promoter, an expression tag in one of the three reading frames and a splice donor (SD) (Fig. 1b) (33). Two days after ERM infection, cells were transferred to 100-mm dishes for a 30-day selection in RPMI 1640 medium containing 10 μg/ml insulin, 10% charcoal dextran stripped fetal calf serum (CSFCS) and 1 μg/ml puromycin. For p27 expression, the C-terminally poly-histidine-tagged p27 cDNA was taken from the pCMV-p27 plasmid (a kind gift from Dr. Mong-Hong Lee) and subcloned into the pQCXIH retroviral plasmid (BD Biosciences). The pQCXIH-p27 retroviruses were produced in PT67 packaging cells.
Fig. 1.
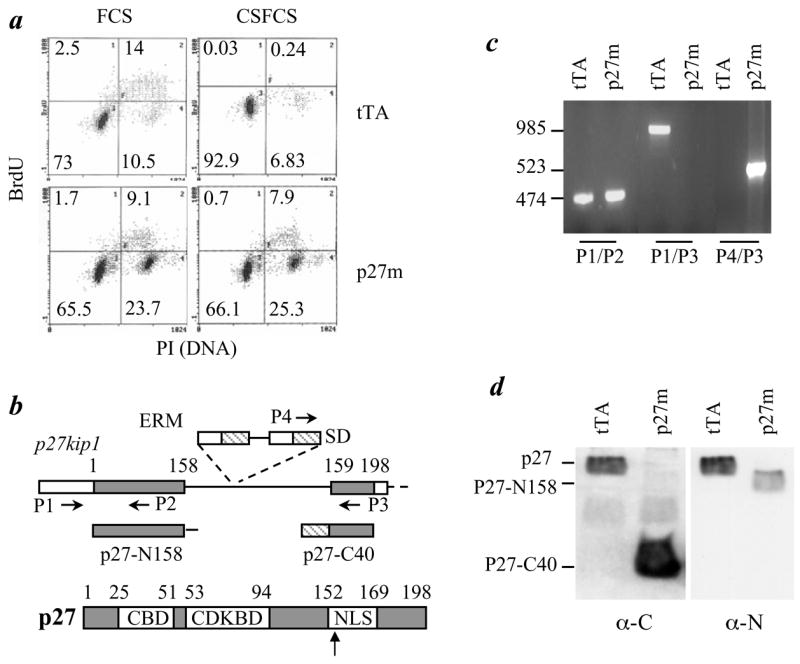
Disruption of p27 in T47D cells causes loss of G1/S checkpoint and estrogen-independent growth. a. T47DtTA (tTA) and T47DtTA/p27m (p27m) cells were cultured in medium containing BrdU and 10% FCS or 10% CSFCS. BrdU incorporation and DNA content per cell were analyzed by fluorescence labeling and flow cytometry. Cell fractions (%) are indicated. Note the increase in G2/M fraction of T47DtTA/p27m cells cultured in medium with FCS and the unchanged proliferation status of T47DtTA/p27m cells cultured in medium with CSFCS. b. The p27 gene structure and the insertional disruption of p27 by ERM. The first and second exon of p27 and p27 protein are outlined. The ERM insertion site and the locations of 4 primers (P1 – P4) used for PCR are indicated. White box, noncoding regions; gray box, amino acid coding regions; SD, splice donor; stretched box, the ERM tag. CBD, cyclin binding domain; CDKBD, cyclin dependent kinase binding domain; and NLS, nuclear localization signal. c. PCR analysis of the p27 gene. Note the absence of the 985-bp P1/P3 product of the wild type allele and the presence of 523-bp P4/P3 product of the ERM allele in T47DtTA/p27m cells. The 474-bp P1/P2 products were amplified from exon 1 and served as a positive control for the PCR. The ERM insertion was longer than 3 kb and thereby PCR using P1/P3 and Taq DNA polymerase was unable to amplify this long fragment in T47DtTA/p27m cells. d. Immunoblotting analyses using antibodies against p27 C-terminus (α-C) (BD Transduction Lab) and N-terminus (α-N) (Santa Cruz Biotech.). Note the absence of full-length p27 (lane 1 and 4) and the presence of p27-C40 (lane 2) and p27-N158 (lane 4) in T47DtTA/p27m cells.
RT-PCR for identification of ERM-targeted genes
Colonies were isolated and cultured in RPMI 1640 medium with 10 μg/ml insulin and 10% CSFCS. RNA was prepared and reversely transcribed with primer 5′-GCAAATACGACTCACTATAGGGATCCNNNN(G/C)ACG. PCR was performed using ERM-tag primer (5′-ACCATGGGGAGCAGCAAGAGCAAACCAAAAGACCCCAGCCAACGC) and T7 primer. PCR products were sequenced and GenBank was searched to identify ERM-targeted genes.
Cell cycle analysis, colony formation assay, immunochemical analyses and kinas assay, transfection assay, migration and invasion assays
These assays were carried out by performing standard methods as described previously (28, 29, 34). For details, please refer to Supplementary Data for Materials and Methods.
Tumor growth and experimental metastasis assays
Mouse protocols were approved by Baylor College of Medicine Animal Care and Use and Committee. For tumor growth in mammary glands, 8 × 106 cells were mixed with matrigel and injected into both fourth mammary fat pads of ovariectomized athymic nude mice (35). Each type of cells was injected into 6 mice without pellets and 6 mice with 60-day estradiol-releasing pellets (1.7 mg/pellet from Innovative Research). Four weeks after injection, tumors were excised for histology and immunohistochemistry. Other organs including lung, liver and mesenteric lymph nodes were examined for metastasis. For experimental metastasis, 106 cells were injected into the tail lateral vein of ovariectomized nude mice. Three weeks after injection, their lungs were examined for metastasis and processed for histology and immunohistochemistry as described (28).
Results
Identification of estrogen-independent growth mutants (EIGMs)
T47D cells express ERα and depend on estrogen to grow and survive. A genome-wide screening using ERM was employed to identify genes with gain- or loss-of-function mutations that could make T47D cells survival in estrogen-free medium. To prepare cells for ERM infection, T47D cells were first infected with MSCV-Eco-zeo viruses to express the mouse ecotropic receptor and MSCV-SV-tTA to express tTA tetracycline-off regulator. The obtained stable T47DtTA cells were subsequently infected with ERM and growth-selected in estrogen-free medium. As expected, no colonies were found from uninfected T47DtTA cells after culturing in the estrogen-free medium. In contrast, about 150 colonies formed from a total of 5 million ERM-infected T47DtTA cells in the selection medium. Survived colonies were isolated and the ERM-targeted genes were identified in each clone using RT-PCR that amplified ERM expression tag and its trapped 3′ exon sequences (33). Out of 15 clones analyzed, we obtained 12 RNA sequences containing ERM tag. Each of the 12 clones only produced one ERM tag-fused RNA sequence, indicating that each clone contains a single ERM integration. Among these EIGMs, EIGM-1 was a loss-of-function mutation for p27, which was characterized in this study. The other 11 clones included 1 small GTPase (EIGM-2 to -4), 2 components of the Wnt-signaling pathway (EIGM-5 and -6), 1 transcription factor (EIGM-7), 1 Golgi protein (EIGM-8), 1 bone marrow stromal cell antigen (EIGM-9), 1 enolase (EIGM-10) and 2 hypothetical proteins (EIGM-11 and -12).
Disruption of the single p27kip1 allele in T47D cells results in estrogen-independent growth
The sequence of EIGM-1 matched the second exon of the p27 gene, so it was designated as T47DtTA/p27m. Bromodeoxyuridine (BrdU)-labeling assay confirmed the estrogen-independent growth feature of T47DtTA/p27m cells. About 93% T47DtTA parent cells cultured in the estrogen-free medium were arrested at G1 and less than 0.3% cells were labeled with BrdU. In contrast, only 66% T47DtTA/p27m cells cultured in the estrogen-free medium was in G1 and as many as 8.6% T47DtTA/p27m cells were labeled with BrdU, which were similar to T47DtTA and T47DtTA/p27m cells cultured in the growth medium with complete serum (Fig. 1a).
The p27 gene contains two exons separated by a 511-bp intron. Exons 1 and 2 encode for the N-terminal 158 and the C-terminal 40 (159–198) a.a. residues, respectively (Fig. 1b). PCR and sequence analyses revealed that the ERM integration site was between bp 174 and 175 of intron 1 (Fig. 1, b & c). In the RT-PCR product from T47DtTA/p27m cells, the ERM tag was fused in frame with the second exon of p27, indicating that the 337-bp intron sequence between the splice donor site of the ERM tag and the second exon of p27 was excised during RNA processing (Fig. 1b). Accordingly, the inserted ERM promoter generated a 10-kDa protein that was recognized by antibodies against p27 C-terminus and ERM tag, indicating that this fusion protein contains ERM tag (62 a.a.) and the C-terminal 40 a.a. of p27 (p27-C40) (Fig. 1d and data not shown). The p27 N-terminal antibody recognized a smaller and less abundant protein in T47DtTA/p27m cells than the full-length p27 in T47DtTA cells (Fig. 1d). Since this smaller p27 protein was not detectable by the p27 C-terminal antibody and its apparent molecular mass (~10 kDa) matched the predicted size translated from exon 1 and its extending intron sequence, we concluded that ERM insertion in the p27 intron interfered the splicing between exons 1 and 2. This resulted in a truncated p27 protein with the N-terminal 158 a.a. and the C-terminal 21 a.a. encoded by the extended intron sequence before meeting the TAA stop codon at 63–65 bp of intron 1 (Fig. 1, b & d). This N-terminal protein was designated as p27-N158. Interestingly, no wild type p27 allele and full-length p27 protein were detected in T47DtTA/p27m cells by PCR and immunoblotting (Fig. 1, c & d), indicating that T47D cells only contain a single p27 wild type allele and T47DtTA/p27m cells only contain a disrupted p27 allele.
T47DtTA/p27m cells are resistant to antiestrogens although they still express functional ERα
To test whether disruption of p27 reduces the antiproliferative effects of antiestrogens, we compared cell proliferation rates of T47DtTA/p27m cells with T47DtTA cells treated with 4-OH-tamoxifen (4-OH-TAM) and ICI 182780 (ICI) for 4 days. The vehicle-treated T47DtTA and T47DtTA/p27m cells exhibited similar S-phase fractions (~10%), while 4-OH-TAM treatment reduced the S-phase fraction of T47DtTA cells to ~3%. However, 4-OH-TAM treatment only slightly decreased the S-phase fraction of T47DtTA/p27m cells to 7%. ICI treatment diminished the S-phase fraction of T47DtTA cells to ~3%, but only partially reduced the-S phase fraction of T47DtTA/p27m cells to ~5.5%, indicating that T47DtTA/p27m cells were also less sensitive than T47DtTA cells to ICI (Fig. 2a). Consistent results were also obtained from cell number growth of T47DtTA and T47DtTA/p27m cells cultured in growth medium containing vehicle, 4-OH-TAM or ICI and in estrogen-free medium with CSFCS (Fig. 2b). To validate the specific role of p27 in these experiments, we restored stable p27 expression in T47DtTA/p27m cells by retroviral infection. The restored p27 was a poly-histidine-tagged bigger protein compared with the endogenous p27 in T47DtTA cells (Fig. 2b). Restoration of p27 in T47DtTA/p27m/+p27 cells rescued the growth-inhibitory sensitivities of these cells to 4-OH-TAM, ICI and CSFCS (Fig. 2b). These results indicate that p27 function is required for T47D cells to respond to the antiproliferative effects of antiestrogens.
Fig. 2.
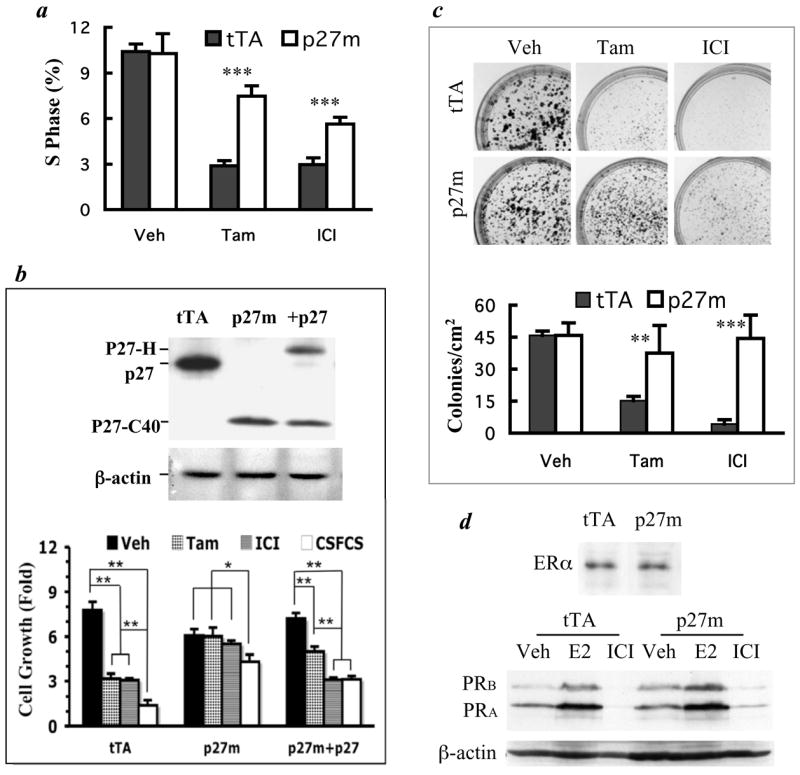
T47DtTA/p27m cells are resistant to antiestrogens although they express functional ERα. a. T47DtTA (tTA) and T47DtTA/p27m (p27m) cells in growth medium were treated with vehicle (Veh), 4-OH-TAM (Tam) or ICI for 4 days. The mean of the S phase fraction was calculated from 4 assays of flow cytometry. ***, p < 0.001, unpaired t-test. b. p27 restoration in T47DtTA/p27m cells rescues their sensitivity to antiestrogens. Immunoblotting assay using the p27 C-terminal antibody detected p27 in T47DtTA cells, the truncated p27-C40 in T47DtTA/p27m and the His-tagged p27 (p27-H) and p27-C40 in T47DtTA/p27m/+p27 (+p27) cells (upper panel). For growth assay (lower panel), T47DtTA, T47DtTA/p27m and T47DtTA/p27m/+p27 cells (105/well, n = 4) were cultured in medium containing 10% CSFCS overnight and then changed to either the medium containing 10% FCS and Veh, Tam or ICI or the medium with 10% CSFCS. Cells were cultured for 4 days before harvested and counted. Data are presented as fold of cell number increase (Mean ± S.D.). *, p < 0.05 and **, p < 0.01 by One-Way ANOVA. c. Photographs of crystal violet-stained colonies formed from T47DtTA and T47DtTA/p27m cells treated with Veh, Tam and ICI (upper panel). Note the significantly larger and more numerous colonies formed from T47DtTA/p27m cells than T47DtTA cells treated with Tam and ICI. The lower panel presents the quantitative data of colony formation assays in the upper panel. For Veh and Tam treated cells, colonies bigger than 1 mm in diameter were counted. For ICI treated cells, colonies bigger than 0.5 mm were counted. Data are presented as mean ± SD colony numbers in eight 1-cm2 areas of culture wells in two repeat experiments. ** and ***, p < 0.01 and p < 0.001, unpaired t-tests. d. Immunoblotting analysis of ERα in T47DtTA and T47DtTA/p27m cells cultured in growth medium and PRB and PRA in T47DtTA and T47DtTA/p27m cells treated with Veh, 17β-estradiol (E2) and ICI in estrogen-free medium. Note that the PRs were increased by estradiol treatment and reduced by ICI treatment in both types of cells. Protein loading amounts were reflected by immunoblotting analysis of β-actin.
To examine the long-term effects of antiestrogens on T47DtTA/p27m cell survival and growth, we compared the colony formation capability of T47DtTA/p27m cells with T47DtTA cells over a 3-week culture period. In growth medium T47DtTA/p27m and T47DtTA cells formed colonies equally well, giving ~45 colonies/cm2. 4-OH-TAM treatment significantly reduced the density of colonies formed from T47DtTA cells to 15 colonies/cm2. In contrast, 4-OH-TAM treatment did not significantly change the colony formation capability of T47DtTA/p27m cells (Fig. 2c). ICI treatment profoundly reduced both the number and sizes of the colonies formed from T47DtTA cells. However, ICI only reduced the colony sizes of T47DtTA/p27m cells but not their total number of colonies (Fig. 2c). These results demonstrate that T47DtTA/p27m cells are fully resistant to 4-OH-TAM and partially resistant to ICI.
Loss of ERα is a possibility for breast cancer cells to become insensitive to antiestrogens. However, immunoblotting analysis revealed that ERα levels in T47DtTA and T47DtTA/p27m cells were comparable. The estradiol-induced or ICI-inhibited transcription of PRA and PRB from the estrogen responsive PR gene also was comparable in these cells (Fig. 2d). Thus, the antiestrogen resistance observed in T47DtTA/p27m cells is not due to a loss of ERα expression or function.
The p27-N158 and p27-C40 proteins are loss-of-function mutants
Most p27 immunoreactivity was detected in the nucleus of T47DtTA cells by p27 N-terminal antibody, while the p27-N158 immunoreactivity was detected in both cytoplasm and nucleus of T47DtTA/p27m cells by the same antibody (Fig. 3a). Immunoblotting analyses further identified p27 in both cytoplasmic and nuclear fractions of T47DtTA cells, at the contrary, the mutant p27-N158 was mainly found in the cytoplasmic fraction of T47DtTA/p27m cells (Fig. 3b, upper panel). The later observation is consistent with the absence of p27 nuclear localization sequence (NLS) (a.a. 159 to a.a. 169) in p27-N158 (Fig. 1b). These results indicate that although p27-N158 contains the N-terminal domains for binding cyclins and CDKs, it mainly stays in the cytoplasm and does not function as a nuclear CDK inhibitor.
Fig. 3.

Subcellular localization and function of p27, p27-N158 and p27-C40 proteins and insensitive inhibition of the CDK2 kinase activity in T47DtTA/p27m cells by 4-OH-TAM (Tam), ICI and CSFCS. a. Immunocytofluorescence labeling of T47DtTA (tTA) and T47DtTA/p27m (p27m) cells with antibodies against p27 N-terminus (α-P27-N) and C-terminus (α-P27-C). Note the cytoplasmic localization of immunoactivity in T47DtTA/p27m cells detected by both antibodies. DAPI staining was used to show cell nuclei. b. Immunoblotting analysis of cytosol and nuclear fractions of T47DtTA and T47DtTA/p27m cells using α-P27-N (upper panel) and α-P27-C (lower panel) antibodies. The subcellular fractions were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Chemical). c. Immunoblotting analysis of T47DtTA and T47DtTA/p27m cells using mixed α-P27-N and α-P27-C antibodies after cells were treated with or without tetracycline (Tet). All samples were assayed in the same blot; the image of the left two lanes and the image of the right two lanes were aligned together after the image of two unused lanes between them was deleted. β-actin was assayed as a loading control. d. Treatment of T47DtTA and T47DtTA/p27m cells with vehicle (Veh), Tam, ICI or CSFCS (estrogen-free medium) and immunoblotting assays of p27, p27-N158, cyclin E, CDK2, 32P-histone H1, ppRB, pRB, pp130, P130 and β-actin control. For CDK2 kinase activity assay, the cyclin E-CDK2 complexes were immunoprecipitated by a cyclin E antibody from equal amounts of cell lysates. CDK2 kinase activity was assayed by using [γ-32P]-ATP and histone H1 as substrates. Relative band intensity (Rel. Int.) of the phosphorylated histone H1 (32P-H1) in each lane is indicated by setting the first lane as 1. For immunoblotting analyses of the phosphorylated Rb and p130, the ratios of hyper-phosphorylated Rb (ppRb) and pp130 to hypo-phosphorylated Rb (pRb) and p130 are indicated for each lane, respectively. Note that the increases in pRb and p130 were observed only in T47DtTA cells but not in T47DtTA/p27m cells after these cells were treated with Tam, ICI and CSFCS.
The nuclear p27 in T47DtTA cells was also detected by the p27 C-terminal antibody. Interestingly, the p27-C40, detected by the same antibody, was mainly found in the cytoplasm of T47DtTA/p27m cells, although weaker immunoreactivity were also detected in their nuclei (Fig. 3a). Immunoblotting analyses confirmed that p27-C40 was distributed in both cytoplasmic and nuclear fractions of T47DtTA/p27m cells (Fig. 3b, lower panel). Since p27-C40 expression is controlled by the tetracycline-off ERM promoter, we examined the effect of p27-C40 on cell proliferation by treating cells with tetracycline. The p27-C40 level was significantly reduced in T47DtTA/p27m cells upon tetracycline treatment (Fig. 3c). However, the distribution of cell fractions in all proliferation phases, determined by flow cytometry, was identical between vehicle- and tetracycline-treated T47DtTA/p27m cells (data not shown). These results indicate that change of p27-C40 concentration does not affect cell proliferation.
p27 deficiency reduces the inhibitory effects of antiestrogens on CDK2 activity
In estrogen-dependent breast cancer cells, antiestrogens induce p27, inhibit cyclin E-CDK2 activity, reduce Rb and Rb-related p130 hyper-phosphorylation, suppress E2F and arrest cell cycle at G1 (16). In T47DtTA cells, 4-OH-TAM induced a slight and ICI and estrogen-free condition induced a significant p27 increase. In T47DtTA/p27m cells, these treatments failed to induce p27-N158 levels, suggesting that antiestrogens or estrogen-free condition do not induce p27 promoter activity in the estrogen-independent T47DtTA/p27m cells. The level of p27-C40 was decreased when T47DtTA/p27m cells were cultured in estrogen-free medium due to, presumably, lower ERM promoter activity under this culture condition (Fig. 3d and data now shown). Cyclin E and CDK2 levels were identical between T47DtTA and T47DtTA/p27m cells under all examined conditions (Fig. 3d).
To examine the effect of p27 mutation on the kinase activity of cyclin E-CDK2 complexes, cyclin E was immunoprecipitated from cell extracts with equal protein amount and the kinase activity of precipitates was assayed using histone H1 as substrates. The cyclin E-associated CDK2 kinase activity was reduced 33% and 66%, respectively, when T47DtTA cells were treated with 4-OH-TAM and ICI. When T47DtTA cells were cultured in estrogen-free medium, the cyclin E-associated CDK2 kinase activity was reduced 87%. In contrast, the cyclin E-CDK2 kinase activity in T47DtTA/p27m cells was not significantly reduced by 4-OH-TAM, and only reduced 37% and 57% by ICI and estrogen-free medium (Fig. 3d). In agreement with the increase of CDK2 activity, the ratios of hyper-phosphorylated Rb (ppRb) to hypo-phosphorylated Rb (pRb) were increased in T47DtTA/p27m cells treated with 4-OH-TAM, ICI and estrogen-free medium compared with the ratios of ppRb to pRb in T47DtTA cells receiving the same treatments (Fig. 3d). Similarly, the ratios of pp130 to p130 in T47DtTA/p27m cells treated with 4-OH-TAM, ICI and estrogen-free medium were higher than the ratios of pp130 to p130 in T47DtTA cells (Fig. 3d). The increase in these ratios was mainly due to the decrease in the hypo-phosphorylated inhibitory forms of Rb and p130. These results indicate that p27 deficiency desensitizes the inhibition of CDK2 activity by antiestrogens and estrogen depletion, causing sustained hyper-phosphorylation and inactivation of tumor suppressors including pRb and p130, even in the presence of antiestrogens or the absence of estrogen.
Loss of p27 stimulates AIB1 phosphorylation and E2F1 transcriptional activity
The elevated CDK2 activity is known to activate E2F transcriptional activity, which plays an essential role in G1/S transition of the cell cycle. To measure E2F1 activity, we transfected T47DtTA and T47DtTA/p27m cells with an E2F1-responsive luciferase reporter and measured the luciferase activity. The reporter activity was about two fold higher in T47DtTA/p27m cells compared with T47DtTA cells when these cells were treated with vehicle, 4-OH-TAM, ICI or estrogen-free medium (Fig. 4a). These results indicate that p27 deficiency significantly stimulates E2F1 activity in T47DtTA/p27m cells.
Fig. 4.
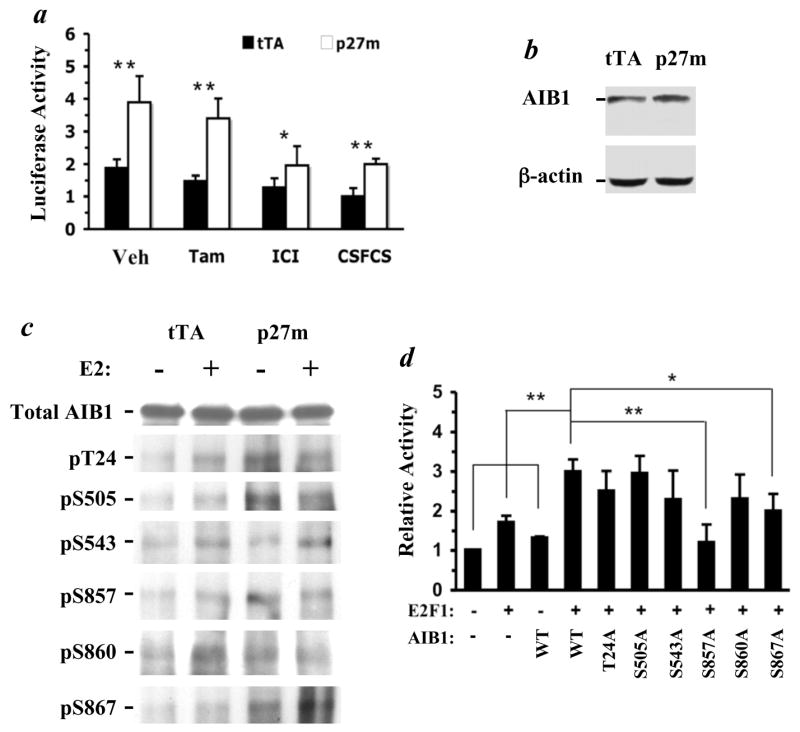
p27 deficiency enhances AIB1 phosphorylation and increases the transcriptional activity of E2F1 and AIB1. a. T47DtTA/p27m (p27m) cells have higher E2F1 activity compared with T47DtTA cells (tTA). E2F1 activity was measured by transfecting cells with an E2F1 luciferase reporter. Transfected cells were cultured in growth medium containing vehicle (Veh), tamoxifen (Tam) and ICI or in medium with 10% CSFCS for two days. Transfection efficiency was normalized to β-galactosidase activity, which was expressed by co-transfecting cells with the pRSV-β-gal vector. Data are presented by setting relative average luciferase activity in CSFCS-treated T47DtTA cells as 1 unit. b. Immunoblotting analysis, showing comparable AIB1 levels in T47DtTA and T47DtTA/p27m cells. Immunoblotting analysis of β-actin serves as a loading control. c. p27 deficiency and 17β-estradiol (E2) enhance AIB1 phosphorylation. T47DtTA and T47DtTA/p27m cells were cultured in the presence or absence of E2 for 1 hour in medium with 10% CSFCS. Total AIB1 was immunoprecipitated with AIB1 antibody and then subjected to immunoblotting analysis with antibodies specific to the phosphorylation sites as indicated. These antibodies were developed and affinity-purified as described (29). Note the total amount of precipitated AIB1 is similar among groups, but the different sites are differentially phosphorylated. d. Mutation of specific phosphorylation sites impairs AIB1 coactivator activity for E2F1-mediated target gene transcription. Cells were transfected with E2F1, E2F1-responsive luciferase reporter and AIB1 or its mutants as indicated. Luciferase activity was assayed as described under Methods. Data are presented as Mean ± S.D. (n = 4) by setting the first bar as 1 unit. *, p < 0.05; **, p < 0.01 by One-Way ANOVA.
AIB1 is a strong coactivator for E2F1 and plays an important role in breast cancer proliferation and antiestrogen resistance (22, 23, 36, 37). Because AIB1 cellular concentration and phosphorylation levels determine its coactivator activity, we assessed if p27 deficiency would alter AIB1 protein level and/or phosphorylation status using specific antibodies detecting total AIB1 and its individual phosphorylated sites known to be important for its coactivator activity (29). While total AIB1 remained unchanged in T47DtTA/p27m cells compared with T47DtTA cells (Fig. 4, b & c), its phosphorylation levels on five of the six known sites, with the exception of serine 543, were significantly enhanced in T47DtTA/p27m cells, including phosphorylated threonine 24 (pT24), phosphorylated serine 505 (pS505), pS857, pS860 and pS867 in the absence of estrogen treatment (Fig. 4c). Interestingly, 17β-estradiol treatment stimulated the phosphorylation of these sites in T47DtTA cells but not in T47DtTA/p27m cells (Fig. 4c), suggesting that the phosphorylation levels of these sites induced by p27 deficiency are similar to the levels induced by estrogen in T47DtTA cells.
Next, we analyzed the impact of each AIB1 phosphorylation site on E2F1 reporter by comparing the coactivator activity of AIB1 with its mutants, containing a threonine (T) or serine (S) to alanine (A) mutation. These mutant and wild type AIB1 vectors expressed at similar levels in MCF-7 breast cancer cells. MCF-7 cells were used because they gave a better efficiency than T47D cells when multiple plasmids were co-transfected. Our experiments demonstrated that expression of E2F1 or AIB1 alone had limited stimulation on E2F1 reporter activity, but coexpression of E2F1 and AIB1 robustly potentiated E2F reporter activity (Fig. 4d). Expression of the T24A, S505A, S543A and S860A AIB1 mutants also coactivated E2F1 activity as wild type AIB1 did. Nevertheless, coactivator activities were partially impaired for the S867A mutant and completely diminished for the S857A mutant (Fig. 4d). These results suggest that phosphorylation on two of the six sites of AIB1 has a positive effect on E2F1-mediated transcription. Since disruption of p27 increased the phosphorylation on five of the six known sites, including pS857 and pS867, the hyper-phosphorylated AIB1 in T47DtTA/p27m cells should serve as a stronger coactivator for E2F1 compared with the hypo-phosphorylated AIB1 in T47DtTA cells.
Disruption of p27 increases Akt activity
The adaptor protein Gab2 is a direct E2F target (30). Indeed, Gab2 was significantly increased in T47DtTA/p27m cells compared with T47DtTA cells; restoration of p27 caused a reversible decrease in Gab2 in T47DtTA/p27m/+27 cells (Fig. 5a). The E2F-induced Gab2 plays an essential role in mediating E2F-dependent activation of the PI3-K/Akt signaling pathway in U-2OS osteosarcoma cells (30). To address whether p27 deficiency-induced hyperactive E2F and overexpressed Gab2 would also potentiate Akt activation in breast cancer cells, we assayed total Akt and active Akt, the phospho-serine473-Akt (pS473-Akt) (38), by immunoblotting. The total Akt was comparable in T47DtTA/p27m and T47DtTA cells (Fig. 5a). However, the pS473-Akt was about 3 fold higher in T47DtTA/p27m cells than T47DtTA cells. Restoration of p27 in T47DtTA/p27/+27 cells did not change total Akt but decreased active Akt (Fig. 5a). Accordingly, Akt protein immunopurified from T47DtTA/p27m cells showed higher kinase activity than Akt from T47DtTA cells as determined by in vitro kinase assays using the glycogen synthesis kinase-3β (GSK-3β) as substrates (39) (Fig. 5a). These results demonstrate that inactivation of p27 in T47D cells causes super activation of Akt, probably through enhancing the transcriptional capability of E2F.
Fig. 5.

Inactivation of p27 increases Gab2 protein and Akt activity, cell motility and cell invasive ability. a. p27 deficiency increases Gab2 protein and Akt activity. Gab2 levels in T47DtTA (tTA), T47DtTA/p27m (p27m) and T47DtTA/p27m/+27 (+27) cells were assayed by immunoblotting. Total Akt and phosphorylated active Akt in these cells were detected by respective Akt antibody and phosphorylation site-specific antibody by immunoblotting. Akt activity in T47DtTA and T47DtTA/p27m cells was assayed using GSK-3β as substrates and the phosphorylated GSK-3β (p-GSK-3β) was detected by immunoblotting using a p-GSK-3β-specific antibody. The β-actin assay served as a loading control. b. Measurement of cell migration. T47DtTA and T47DtTA/p27m cells were cultured on plates coated with FN, LN, Col I or Col IV and cell migration was traced by using blue fluorescence beads. Individual cells (arrows) were stained with Rhodamine-Phalloidin. Dark areas indicate cell migration tracks. Cells migrated faster on FN and Col IV. c. Measurement of migration areas. Cell migration tracks of 25 to 30 cells were analyzed by using NIH image software. Data are presented as average (relative migration area) ± standard deviation. The migration areas of T47DtTA/p27m cells on all coated matrices examined are significantly bigger compared with T47DtTA cells (p < 0.01, unpaired t test). d. Cell invasion assay. The invasive capability of T47DtTA, T47DtTA/p27m and T47DtTA/p27m/+27 in the matrigel was measure as describe in Materials and Methods. Data represent the mean of 3 independent experiments.
Inactivation of p27 promotes cell mobility and invasion
The PI3-K/Akt signaling pathway plays a central role in cancer cell motility, invasion and metastasis (31, 32). To assess the effects of p27 deficiency-triggered Akt activation on cell motility, we measured the extent of individual cell migration on extracellular matrix proteins by using blue fluorescent beads to determine the track area cells had moved through in 18 hours. On culture plates coated with each of the four extracellular matrix proteins, the track area of T47DtTA/p27m cells was significantly larger than the track area of T47DtTA cells (Fig. 5b). Quantitative analysis of the track areas revealed that T47DtTA/p27m cells migrated 2.1, 4.6, 4.5 and 3 fold faster than T47DtTA cells on FN-, LN-, Col I- and Col IV-coated culture plates, respectively (Fig. 5c). These results indicate that inactivation of p27 in T47D cells remarkably promotes cell motility.
We also evaluated the capability of T47DtTA, T47DtTA/p27m and T47DtTA/p27m/+p27 cells to invade through the extracellular matrix by using Matrigel Invasion Chambers. The average number of T47DtTA/p27m cells that invaded through the Matrigel layer increased more than 3 fold compared with T47DtTA and T47DtTA/p27m/+p27 cells (Fig. 5d). These results indicate that p27 deficiency significantly enhances the invasive capability of breast cancer cells.
T47DtTA/p27m and T47DtTA cells showed similar cell adhesion capability on FN, LN, Col I or Col IV-coated culture plates (data not shown). Immunoblotting and immunocytochemistry revealed that the levels and distribution patters of E-cadherin, an epithelial marker for mammary epithelial association (40), were similar between T47DtTA and T47DtTA/p27m cells (data not shown). These results indicate that p27 deficiency does not alter E-cadherin expression and location as well as mammary epithelial cell-cell interaction.
Intravenous injection of p27-deficient T47DtTA/p27m cells develops lung metastasis in ovariectomized nude mice
T47D cells are estrogen-dependent human breast cancer cells, which can only form non-metastatic local tumors in ovariectomized nude mice treated with estrogen (35). To test whether p27 deficiency in estrogen-dependent breast cancer cells could promote tumor formation in an ovarian hormone-independent manner, we compared tumor development and progression of T47DtTA/p27m cells with T47DtTA cells in ovariectomized nude mice. Both T47DtTA and T47DtTA/p27m cells developed local tumors in the mammary fat pats of all nude mice (n = 6) treated with estradiol pellets, but failed to develop tumors in the mammary fat pads of nude mice treated with placebo (Fig. 6a and data not shown). Histological examination revealed that T47DtTA tumors had a smooth surface and a relatively tight cell-cell association (Fig. 6a). In contrast, the edge of T47DtTA/p27m tumors frequently protruded into the surrounding stromal tissues (Fig. 6a). These results suggest that loss of p27 function may make T47DtTA/p27m tumors more invasive.
Fig. 6.

Primary and metastatic tumor formation in ovariectomized nude mice. a. The upper panels show the primary tumors (arrow heads) formed from T47DtTA (tTA) and T47DtTA/p27m (p27m) cells in nude mice with 17β-estradiol pellets. The lower panels show the images of H&E stained tumor sections prepared from T47DtTA and T47DtTA/p27m tumors. Note the different morphologies of the tumor edges indicated by arrowheads in the two types of tumors. The scale bars in the lower panels represent 50 μm. b. Photographs of lungs from different ovariectomized nude mice receiving T47DtTA/p27m cells from their tail veins. Arrowheads indicate visible metastatic lung tumors. c. H&E stained lung sections prepared from ovariectomized nude mice receiving intravenous injection of T47DtTA and T47DtTA/p27m cells. An invasive big lung tumor is outlined in the right upper panel. The lower panels are larger images of the boxed areas in the upper panels. The scale bars for the upper and lower panels represent 500 and 50 μm, respectively. d. Immunohistochemical staining of K8 (brown color) on lung sections prepared from ovariectomized nude mice injected with T47DtTA or T47DtTA/p27m cells as indicated. The scale bars represent 50 μm.
To address if the local tumors formed from T47DtTA/p27m cells might develop systemic metastasis, we examined the lung and other internal organs of mice bearing T47DtTA/p27m tumors in their mammary fat pats. However, we did not find any metastasis developed in any secondary organs, suggesting that T47DtTA/p27m tumors formed in the mammary gland are unable to accomplish the entire process of distant metastasis.
To determine whether p27 deficiency-induced cellular alterations could potentiate metastasis of breast cancer cells after intravasation, we compared the capability of T47DtTA/p27m with T47DtTA cells to form metastatic tumors in the lung using a well-established experimental metastasis method (41). T47DtTA cells did not develop any tumors in the lung of ovariectomized nude mice (n = 5) 3 weeks after injected intravenously (Fig. 6c). In contrast, all of the 7 ovariectomized nude mice developed lung tumors 3 weeks after receiving T47DtTA/p27m cells intravenously. In these mice lacking ovarian steroids, many tumors were visible on the lung surfaces, and big invasive tumors were observed on lung sections (Fig. 6, b&c). In these tumor cells, strong immunoreactivity of p27-C40 was detected in the cytoplasm, which was similar to that seen in T47DtTA/p27m cells in Fig. 3a. In addition, the mammary epithelial marker K8 was also detected in the lung tumor cells of nude mice injected with T47DtTA/p27m cells, but not in the lung of nude mice injected with T47DtTA cells (Fig. 6d), indicating that these metastatic tumors originated from T47DtTA/p27m cells. These results demonstrate that loss of p27 in T47D estrogen-dependent breast cancer cells is sufficient to permit metastatic tumor formation in the lung in an ovarian hormone-independent manner, once these tumor cells enter the circulation.
Discussion
Development of estrogen independence and tamoxifen resistance is a major problem of breast cancer endocrine therapy. Characterization of the genetic changes responsible for the transition from estrogen dependence to independence and from tamoxifen responsiveness to resistance is essential for understanding the mechanisms for this key step progression. In this study, we found that the estrogen-dependent T47D cells contain only one p27 allele, suggesting that p27 is already decreased in the original T47D tumor. Our genome-wide screening using ERM identified the p27-deficient T47DtTA/p27m cells that still express functional ERα, but grow in an estrogen-independent manner and are fully resistant to tamoxifen. These observations recapitulate the similar features of ERα-positive breast cancers that progress from tamoxifen sensitive to tamoxifen resistant and are consistent with an important role of p27 reduction and cytoplasmic retention in breast cancer initiation and progression (13–15, 17–19). Since our results demonstrate that disruption of p27 is sufficient to permit estrogen-dependent and tamoxifen-sensitive breast cancer cells to grow in the absence of estrogen and presence of tamoxifen and these growth phenotypes can be rescued by p27 restoration, we conclude that functional p27 is required for tamoxifen-mediated inhibition of ERα-positive breast cancer growth.
T47DtTA/p27m cells also exhibited a partial resistance to ICI compared with T47DtTA cells. ICI only reduced the sizes of T47DtTA/p27m cell colonies and inhibited 37% of CDK2 activity. Previous studies have shown that both p27 and p21cip1 can contribute to antiestrogen-mediated cell cycle arrest in breast cancer cells (18). Our analyses also reveal that p21 is more obviously induced by ICI in T47DtTA/p27m cells than in T47DtTA cells and more cyclin D1-CDK4 and cyclin E-CDK2 were associated with p21cip1 in T47DtTA/p27m cells (data not shown). Therefore, the partial ICI resistance of T47DtTA/p27m cells may be accredited to the compensatory role of p21cip1 when p27 function is lost.
E2F1 transcriptional activity is significantly elevated in T47DtTA/p27m cells because p27 deficiency activates CDK2, leading to Rb phosphorylation and E2F1 activation. Recently, AIB1 has been identified as a major coactivator for E2F1; E2F1 interacts with AIB1 N-terminus and recruits AIB1 to the promoter of E2F1 target gene (22). Other studies also have shown that growth factors, cytokines and estrogen can stimulate AIB1 phosphorylation at specific sites and AIB1 phosphorylation usually alters AIB1 coactivator activity for steroid receptors (29, 42). Nevertheless, the mechanisms leading to AIB1 phosphorylation are complex and many kinases including p38, IKKs, ERK, JNK and GSKs have been shown to phosphorylate AIB1 (29). Since CDK2 and Akt are stimulated in p27-deficient T47DtTA/p27 cells, these two kinases might also play a role in AIB1 phosphorylation. Our data demonstrate that loss of p27 in T47DtTA/p27m cells enhances AIB1 phosphorylation on 5 out of the 6-mapped sites, and the phosphorylation on 4 out of these 5 sites was also enhanced by estrogen in T47DtTA cells (Fig. 5c), suggesting that p27 deficiency causes an AIB1 phosphorylation pattern similar to that caused by estrogen. Importantly, mutation of two of these sites (S857 and S867) either attenuated or diminished AIB1 coactivator activity for E2F1. These results indicate that the phosphorylated AIB1 is a better coactivator for E2F1. Thus, p27 deficiency-induced AIB1 phosphorylation further potentiates E2F1 target gene transcription following CDK2 activation and Rb phosphorylation. This double enhancement of E2F1 activity may play a crucial role in breast cancer progression into estrogen independence and tamoxifen resistance.
Intriguingly, T47DtTA/p27m cells can grow in estrogen-free medium, but are unable to develop tumors in the mammary fat pads of ovariectomized nude mice with or without tamoxifen treatment, suggesting that tumor formation in the mammary fat pads in the absence of ovarian hormones is more difficult than cell growth in estrogen-free medium for T47DtTA/p27m cells. With estrogen replacement, both T47DtTA and T47DtTA/p27m cells developed solid tumors in the mammary fat pads. Although T47DtTA/p27m tumors are morphologically more invasive, both T47DtTA and T47DtTA/p27m tumors formed in the mammary fat pads are not metastatic, suggesting that p27 deficiency alone is not sufficient for these tumor cells to progress into fully metastatic cancer cells.
The next question is why T47DtTA/p27m cells lacking p27, but not T47DtTA cells with p27, develop lung metastasis when injected into the circulation of ovariectomized mice. To develop lung metastasis, tumor cells after intravasation or injection into the circulation must attach to the endothelium of small lung vessels, migrate or invade through the vessel wall, and form tumors in the lung (41). Our analyses demonstrate that T47DtTA and T47DtTA/p27m cells have similar adhesion capability to extracellular matrix (ECM) proteins, suggesting that p27 deficiency does not change cell adhesion properties and the increase in T47DtTA/p27m cell metastatic potential is unlikely due to the change of endothelial attachment. On the other hand, cell migration and invasion abilities are correlated with metastasis. Our data demonstrate that p27 deficiency in T47DtTA/p27m cells dramatically promotes their motility and invasiveness compared with T47DtTA cells. Thus, p27 deficiency-induced increase in cell motility and invasion may be responsible for the ability of T47DtTA/p27m cells to develop lung metastasis.
The mechanisms for p27 deficiency-induced cell motility, invasiveness and lung metastasis may be very complex. We showed that T47DtTA/p27m cells have an enhanced AIB1 phosphorylation, which makes AIB1 a stronger coactivator for E2F1. Previous studies have established that E2F1 strongly activates Akt through a direct upregulation of Gab2 expression (30). In this study, p27 deficiency in T47DtTA/p27m cells indeed increases E2F1 transcriptional activity, Gab2 protein and Akt activation following AIB1 phosphorylation. Akt plays a central role in cancer cell motility, invasion and metastasis (31, 32). Therefore, p27 deficiency-enhanced AIB1 phosphorylation may significantly contribute to cell motility, invasion and metastasis through activating Akt. This interpretation is consistent with the lower Akt activity and less lung metastasis of the mammary gland tumors induced by oncogenes and carcinogens in AIB1 null mice compared with wild type mice (27, 28).
Furthermore, it has been shown that overexpression of a p27 cytosolic mutant increases total Akt by inhibiting its turnover (43). Nevertheless, it is unlikely that the disrupted p27 fragments in T47DtTA/p27m cells can play the same role that the full-length cytosolic p27 mutant does, since there is no change in total Akt in T47DtTA/p27m cells. In addition, p27 deficiency may release its direct inhibition on cell motility. Although p27 has been reported as both an inhibitor and a stimulator of cell migration (44–47), a recent article has suggested that the controversial observations about the role of p27 in cell motility may be due to the type of cell motility assayed (48). This study demonstrated that p27 binds and impairs the function of the microtubule-destabilizing protein stathmin and thereby plays an inhibitory role in amoeboid cell migration, a type of cell movement used by tumor cells in tissues (48). Disruption of p27 in T47DtTA/p27m cells may impair the direct inhibitory role of p27 in cell motility and tumor cell metastasis. Finally, down-regulation of p27 causes an up-regulation of GPR48 (G protein-coupled receptor 48) and GPR48 can enhance cancer cell invasiveness and metastasis (49). Therefore, multiple pathways may be involved in the p27 deficiency-induced increase in breast cancer cell motility, invasiveness and metastasis.
Although p27 null mutations are rare, about 50% human tumors exhibit decreased p27. The p27 reduction and cytoplasmic mislocalization correlate with more aggressive phenotypes, including HER2/Neu overexpression, high proliferating indices, active invasive behavior and high mortality (13–15, 50). These observations suggest that p27 abundance inversely correlates with breast cancer progression. However, studies using transgenic mice suggest that a threshold of p27 seems required for normal mammary epithelial proliferation and mammary tumorigenesis induced by oncoproteins. For example, p27+/- mice exhibit a faster mammary epithelial growth and MMTV-neu-induced tumorigenesis; but p27−/− mice exhibit a delayed mammary gland morphogenesis and MMTV-neu-induced tumorigenesis (21). Molecular analysis suggests that certain p27 is required for stabilization of cyclin D1 and assembly and nuclear translocation of cyclin D1-CDK4 complexes. Thus, CDK4 in p27−/− mammary epithelial cells can not be activated, which in turn suppresses the neu-induced, cyclin D1-dependent mammary epithelial transformation (21). Similarly, cyclin D1 is decreased and CDK4 activity is reduced many folds in our T47DtTA/p27m cells (data not shown). However, T47DtTA/p27m cells exhibit comparable proliferation as T47DtTA cells when cultured in growth medium. These results indicate that T47DtTA/p27m cells are no longer dependent on cyclin D1-CDK4 for survival and growth. Although it is unknown whether the T47D parent tumor has acquired cyclin D1/CDK4-independent growth, the increase in cyclin E/CDK2 activity after loss of p27 in T47DtTA/p27m cells is cyclin D1/CDK4 independent and should be responsible for the antiestrogen insensitivity of these cells.
Based on this and previous studies discussed above, the impact of p27 gene dosage on breast cancer is likely dependent on the specific stages of tumor initiation and progression. It seems reasonable to propose that p27 reduction in the mammary epithelium and early stages of tumorigenesis may promote cell proliferation and accelerate hormone, oncoprotein and carcinogen-induced transformation while still maintaining cyclin D1 and CDK4 activity. Once these transformed cells acquire a cyclin D1-CDK4-independent growth feature, a further reduction or loss or mislocalization of p27 may facilitate breast tumor cells to develop more aggressive cancer phenotypes including estrogen-independent growth, antiestrogen insensitivity and metastasis. During this process, p27-insufficiency or deficiency-induced increase in AIB1, E2F1, Gab2 and Akt activities and decrease in p27 inhibitory role in cell motility may have a major contribution to breast cancer progression.
Supplementary Material
Acknowledgments
We thank Lan Liao for experimental assistance. This work was partially supported by National Institutes of Health grants DK58242, CA119689, CA112403 (to J.X.) and CA84208 (to Z.S.) and the RSG-05-082-01 American Cancer Society Research Scholar Award to J.X.
References
- 1.Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer. 2003;10:179–86. doi: 10.1677/erc.0.0100179. [DOI] [PubMed] [Google Scholar]
- 2.Foster JS, Fernando RI, Ishida N, Nakayama KI, Wimalasena J. Estrogens down-regulate p27Kip1 in breast cancer cells through Skp2 and through nuclear export mediated by the ERK pathway. J Biol Chem. 2003;278:41355–66. doi: 10.1074/jbc.M302830200. [DOI] [PubMed] [Google Scholar]
- 3.Jordan VC. Third annual William L. McGuire Memorial Lecture “Studies on the estrogen receptor in breast cancer”--20 years as a target for the treatment and prevention of cancer. Breast Cancer Res Treat. 1995;36:267–85. doi: 10.1007/BF00713399. [DOI] [PubMed] [Google Scholar]
- 4.Schiff R, Massarweh S, Shou J, Osborne CK. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003;9(Pt 2):447S–54S. [PubMed] [Google Scholar]
- 5.Brodie A. Aromatase inhibitor development and hormone therapy: a perspective. Semin Oncol. 2003;30(Suppl 14):12–22. doi: 10.1016/s0093-7754(03)00303-8. [DOI] [PubMed] [Google Scholar]
- 6.Robertson JF. Oestrogen receptor: a stable phenotype in breast cancer. Br J Cancer. 1996;73:5–12. doi: 10.1038/bjc.1996.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piccart M, Lohrisch C, Di Leo A, Larsimont D. The predictive value of HER2 in breast cancer. Oncology. 2001;61(Suppl 2):73–82. doi: 10.1159/000055405. [DOI] [PubMed] [Google Scholar]
- 8.Lupu R, Cardillo M, Cho C, et al. The significance of heregulin in breast cancer tumor progression and drug resistance. Breast Cancer Res Treat. 1996;38:57–66. doi: 10.1007/BF01803784. [DOI] [PubMed] [Google Scholar]
- 9.Lobenhofer EK, Huper G, Iglehart JD, Marks JR. Inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase activity in MCF-7 cells prevents estrogen-induced mitogenesis. Cell Growth Differ. 2000;11:99–110. [PubMed] [Google Scholar]
- 10.Tsai EM, Wang SC, Lee JN, Hung MC. Akt activation by estrogen in estrogen receptor-negative breast cancer cells. Cancer Res. 2001;61:8390–2. [PubMed] [Google Scholar]
- 11.Lenferink AE, Busse D, Flanagan WM, Yakes FM, Arteaga CL. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001;61:6583–91. [PubMed] [Google Scholar]
- 12.Yang HY, Shao R, Hung MC, Lee MH. p27 Kip1 inhibits HER2/neu-mediated cell growth and tumorigenesis. Oncogene. 2001;20:3695–702. doi: 10.1038/sj.onc.1204472. [DOI] [PubMed] [Google Scholar]
- 13.Viglietto G, Motti ML, Bruni P, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med. 2002;8:1136–44. doi: 10.1038/nm762. [DOI] [PubMed] [Google Scholar]
- 14.Liang J, Zubovitz J, Petrocelli T, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002;8:1153–60. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 15.Shin I, Yakes FM, Rojo F, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002;8:1145–52. doi: 10.1038/nm759. [DOI] [PubMed] [Google Scholar]
- 16.Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol. 2003;13:41–7. doi: 10.1016/s1044-579x(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 17.Catzavelos C, Bhattacharya N, Ung YC, et al. Decreased levels of the cell-cycle inhibitor p27Kip1 protein: prognostic implications in primary breast cancer. Nat Med. 1997;3:227–30. doi: 10.1038/nm0297-227. [DOI] [PubMed] [Google Scholar]
- 18.Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A. 2000;97:9042–6. doi: 10.1073/pnas.160016897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donovan JC, Milic A, Slingerland JM. Constitutive MEK/MAPK activation leads to p27(Kip1) deregulation and antiestrogen resistance in human breast cancer cells. J Biol Chem. 2001;276:40888–95. doi: 10.1074/jbc.M106448200. [DOI] [PubMed] [Google Scholar]
- 20.Hwang HC, Martins CP, Bronkhorst Y, et al. Identification of oncogenes collaborating with p27Kip1 loss by insertional mutagenesis and high-throughput insertion site analysis. Proc Natl Acad Sci U S A. 2002;99:11293–8. doi: 10.1073/pnas.162356099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musgrove EA, Davison EA, Ormandy CJ. Role of the CDK inhibitor p27 (Kip1) in mammary development and carcinogenesis: insights from knockout mice. J Mammary Gland Biol Neoplasia. 2004;9:55–66. doi: 10.1023/B:JOMG.0000023588.55733.84. [DOI] [PubMed] [Google Scholar]
- 22.Louie MC, Zou JX, Rabinovich A, Chen HW. ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol. 2004;24:5157–71. doi: 10.1128/MCB.24.12.5157-5171.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mussi P, Yu C, O’Malley BW, Xu J. Stimulation of steroid receptor coactivator-3 (SRC-3) gene overexpression by a positive regulatory loop of E2F1 and SRC-3. Mol Endocrinol. 2006;20:3105–19. doi: 10.1210/me.2005-0522. [DOI] [PubMed] [Google Scholar]
- 24.Xu J, Li Q. Review of the in vivo functions of the p160 steroid receptor coactivator family. Mol Endocrinol. 2003;17:1681–92. doi: 10.1210/me.2003-0116. [DOI] [PubMed] [Google Scholar]
- 25.Anzick SL, Kononen J, Walker RL, et al. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–8. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 26.Torres-Arzayus MI, De Mora JF, Yuan J, et al. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004;6:263–74. doi: 10.1016/j.ccr.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 27.Kuang SQ, Liao L, Wang S, Medina D, O’Malley BW, Xu J. Mice lacking the amplified in breast cancer 1/steroid receptor coactivator-3 are resistant to chemical carcinogen-induced mammary tumorigenesis. Cancer Res. 2005;65:7993–8002. doi: 10.1158/0008-5472.CAN-05-1179. [DOI] [PubMed] [Google Scholar]
- 28.Kuang SQ, Liao L, Zhang H, Lee AV, O’Malley BW, Xu J. AIB1/SRC-3 deficiency affects insulin-like growth factor I signaling pathway and suppresses v-Ha-ras-induced breast cancer initiation and progression in mice. Cancer Res. 2004;64:1875–85. doi: 10.1158/0008-5472.can-03-3745. [DOI] [PubMed] [Google Scholar]
- 29.Wu RC, Qin J, Yi P, et al. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. 2004;15:937–49. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Chaussepied M, Ginsberg D. Transcriptional regulation of AKT activation by E2F. Mol Cell. 2004;16:831–7. doi: 10.1016/j.molcel.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Chau NM, Ashcroft M. Akt2: a role in breast cancer metastasis. Breast Cancer Res. 2004;6:55–7. doi: 10.1186/bcr739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 33.Liu D, Yang X, Yang D, Songyang Z. Genetic screens in mammalian cells by enhanced retroviral mutagens. Oncogene. 2000;19:5964–72. doi: 10.1038/sj.onc.1203992. [DOI] [PubMed] [Google Scholar]
- 34.Musgrove EA, Hunter LJ, Lee CS, Swarbrick A, Hui R, Sutherland RL. Cyclin D1 overexpression induces progestin resistance in T-47D breast cancer cells despite p27(Kip1) association with cyclin E-Cdk2. J Biol Chem. 2001;276:47675–83. doi: 10.1074/jbc.M106371200. [DOI] [PubMed] [Google Scholar]
- 35.Sartorius CA, Shen T, Horwitz KB. Progesterone receptors A and B differentially affect the growth of estrogen-dependent human breast tumor xenografts. Breast Cancer Res Treat. 2003;79:287–99. doi: 10.1023/a:1024031731269. [DOI] [PubMed] [Google Scholar]
- 36.Oh A, List HJ, Reiter R, et al. The nuclear receptor coactivator AIB1 mediates insulin-like growth factor I-induced phenotypic changes in human breast cancer cells. Cancer Res. 2004;64:8299–308. doi: 10.1158/0008-5472.CAN-04-0354. [DOI] [PubMed] [Google Scholar]
- 37.Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 38.Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 39.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 40.Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol. 2005;17:499–508. doi: 10.1016/j.ceb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 41.Kauffman EC, Robinson VL, Stadler WM, Sokoloff MH, Rinker-Schaeffer CW. Metastasis suppression: the evolving role of metastasis suppressor genes for regulating cancer cell growth at the secondary site. J Urol. 2003;169:1122–33. doi: 10.1097/01.ju.0000051580.89109.4b. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, Rose DW, Hermanson O, et al. Regulation of somatic growth by the p160 coactivator p/CIP. Proc Natl Acad Sci U S A. 2000;97:13549–54. doi: 10.1073/pnas.260463097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu FY, Wang SE, Sanders ME, et al. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66:2162–72. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- 44.Goukassian D, Diez-Juan A, Asahara T, et al. Overexpression of p27(Kip1) by doxycycline-regulated adenoviral vectors inhibits endothelial cell proliferation and migration and impairs angiogenesis. Faseb J. 2001;15:1877–85. doi: 10.1096/fj.01-0065com. [DOI] [PubMed] [Google Scholar]
- 45.McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–28. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun J, Marx SO, Chen HJ, Poon M, Marks AR, Rabbani LE. Role for p27(Kip1) in Vascular Smooth Muscle Cell Migration. Circulation. 2001;103:2967–72. doi: 10.1161/01.cir.103.24.2967. [DOI] [PubMed] [Google Scholar]
- 47.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–76. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baldassarre G, Belletti B, Nicoloso MS, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 49.Gao Y, Kitagawa K, Hiramatsu Y, et al. Up-regulation of GPR48 induced by down-regulation of p27Kip1 enhances carcinoma cell invasiveness and metastasis. Cancer Res. 2006;66:11623–31. doi: 10.1158/0008-5472.CAN-06-2629. [DOI] [PubMed] [Google Scholar]
- 50.Chiarle R, Pagano M, Inghirami G. The cyclin dependent kinase inhibitor p27 and its prognostic role in breast cancer. Breast Cancer Res. 2001;3:91–4. doi: 10.1186/bcr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.