

Abstract
Computational modeling and its application in ligand screening and ligand receptor interaction studies play important roles in structure-based drug design. A series of sphingosine 1-phosphate (S1P) receptor ligands with varying potencies and receptor selectivities were docked into homology models of the S1P1-5 receptors. These studies provided molecular insights into pharmacological trends both across the receptor family as well as at single receptors. This study identifies ligand recognition features that generalize across the S1P receptor family, features unique to the S1P4 and S1P5 receptors, and suggests significant structural differences of the S1P2 receptor. Docking results reveal a previously unknown sulfur-aromatic interaction between the S1P4 C5.44 sulfur atom and the phenyl ring of benzimidazole as well as π-π interaction between F3.33 of S1P1,4,5 and aromatic ligands. The findings not only confirm the importance of a cation-π interaction between W4.64 and the ammonium of S1P at S1P4 but also predict the same interaction at S1P5. S1P receptor models are validated for pharmacophore development including database mining and new ligand discovery and serve as tools for ligand optimization to improve potency and selectivity.
Keywords: Sphingosine 1-phosphate (S1P), G protein-coupled receptor (GPCR), Endothelial differentiation gene (EDG), Computational Model, Ligand Recognition
INTRODUCTION
Members of the G protein-coupled receptor (GPCR) superfamily are involved in many major diseases such as cancer, cardiovascular disease, asthma and neurodegenerative diseases, and control fundamental aspects of human physiology and behaviors 1,2. With more than 800 unique members 3, GPCR ligands comprise over 50% of current drugs from which annual revenues exceed $50 billion 4. Bovine rhodopsin is the only GPCR that has been experimentally characterized at high resolution 5,6, demonstrating that three-dimensional structures for GPCR are very difficult to obtain. Knowledge about most other GPCR structures usually comes from homology models in combination with mutagenesis or pharmacological data 7. The accuracy of these models remains unclear, but their utility in ligand screening and structure-based drug design is undeniable.
The majority of S1P receptors belong to the endothelial differentiation gene (EDG) family GPCR. Five known members of the EDG family bind S1P specifically and with affinities in the nanomolar range, including S1P1/EDG1 8,9, S1P2/EDG5/H218 10, S1P3/EDG3 10, S1P4/EDG6 11,12 and S1P5/EDG8 13. S1P receptors have been discovered in almost every tissue tested, although expression levels vary significantly 14,15 indicating they have distinct functions. Cellular effects of S1P are mediated through cell-surface S1P receptors as well as uncharacterized intracellular targets. S1P is involved in angiogenesis and cell migration.16–21 S1P action as survival factor for a variety of cell lines such as human umbilical vein endothelial cells (HUVECs) 22,23, cardiac fibroblasts 24 and oocytes 25 suggests potential therapeutic applications for S1P receptor ligands in cancer, cardiovascular disease, wound healing and inflammation. In addition, S1P and the related phosphorylated metabolite of the immune modulator FTY720 have recently been demonstrated to interfere with lymphocyte trafficking by reversibly sequestering lymphocytes in lymph nodes 26, a response which leads to immunosuppression 27. Thus S1P analogs and their precursors may be useful agents to protect against transplant rejection or in the treatment of autoimmune disease. One effective way of altering S1P effects is providing substances that can block or mimic its activity under pathological or biological conditions through S1P receptors.
In this study, we developed or updated models of the S1P receptor family members and computationally validated the ability of the models to reflect pharmacological trends in agonist binding. The hybrid homology models with experimentally derived loop structures better reflect ligand binding affinity at individual receptors and will be the keys for understanding the mechanism of ligand-protein interaction, identifying S1P agonists and antagonists, and evaluating lead compounds for anticancer, cardiovascular and immunosuppressive drug design.
METHODOLOGY
Model Development
The homology model of the S1P1 receptor was developed as described in our recent paper 28–30, and modified in the first extracellular loop (E1) based on the NMR structure of a S1P4 E1peptidomimetic (Protein Databank 31 entry 2DCO 32. The S1P1 E1 structure was remodeled using MOE package 33 as follows. First, the sequence of the S1P1 receptor was aligned against the 2DCO sequence. A homology model of S1P1 E1, VAYTANLLLSGATTYKLTPAQWFLREGS, was generated based on 2DCO. Coordinates from A2 to E26 of the E1 model replaced corresponding residues in the S1P1 model. The structure was minimized with the MMFF94 force field 34 at splice points (AY and EG) to a root mean square gradient (RMSG) of 1 kcal/mol*Å. The entire structure was subsequently minimized to RMSG = 0.1 kcal/mol*Å.
Homology models of the S1P2-S1P5 receptors were generated from the revised model of the S1P1 receptor after sequence alignment. In addition, TM3 of S1P3 - LREGSMFVALGASTCSLLAIAIERHL - was remodeled as a standard helix, and then spliced into the receptor model after minimization with the MMFF94 force field 34. For S1P2, a single manual refinement in the agonist binding pocket was performed by rotating the ε-amine moiety of K7.34 such that the residue is positioned to interact with the phosphate functionality of S1P. All models were initially minimized to an RMSG of 1 kcal/mol*Å while the protein backbones were fixed. The process was repeated with flexible backbones for 1000 iterations of steepest descent, followed by conjugate gradient and Truncated Newton minimizations to an RMSG of 0.1 kcal/mol*Å.
Docking Studies
A series of S1P agonists (Table 1) were docked into all S1P receptor models to discover ligand binding specificity at the receptor subtype level. Phosphate and ammonium groups were assigned −2 and +1 charges, respectively. Azetidine was modeled in both cis and trans stereoisomers. All docking studies were performed with AUTODOCK version 3.0 35. This software allows stochastic exploration of ligand conformations and configurations through torsion angle rotations, molecular translations and rotations in a rigid protein. Docked complexes were rated based on energy. AUTODOCK calculates energies for various ligand atom type occupancies of grid points in a user-defined box within the protein to generate grid maps before docking ligands. The software generates grid maps for each type of atom of the ligand separately as well as an electrostatic map. Energies for various ligand poses are then computed by summing energy values from these grid maps based on the positions of each atom in the ligand. Only one set of grid maps were generated for each receptor and all studied ligands.
Table 1.
S1P receptor agonist potencies
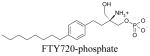
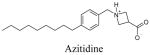
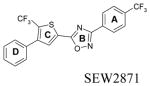
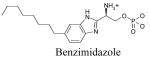
Default AUTODOCK parameters were used in our studies with three exceptions. The number of energy evaluations in the genetic algorithm search was increased to 9×1010; the optimization continued for 6×104 generations and 3000 iterations were used in the Solis and Wetts local search. A docking box of 95×51×51 grid points, equivalent to 35.625 Å × 19.125 Å × 19.125 Å, was used for all docking studies. This box contained most of the transmembrane domains and part of the extracellular loops. Docking results were analyzed based on visualized interactions and docked energy. The best conformations (lowest docked energy conformations) were minimized in the context of the fixed receptor after adding hydrogen and re-assigning charges with the MMFF force field.
RESULTS
S1P receptor models
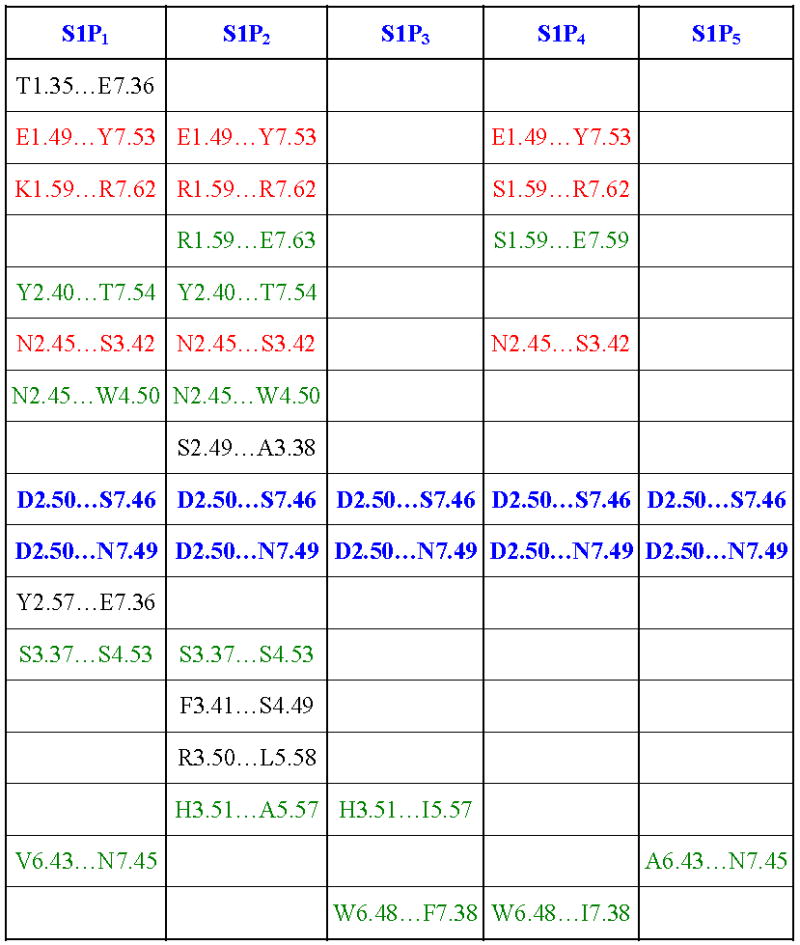
The first extracellular loop in our experimentally-validated S1P1 receptor model 28–30 was modified by analogy to the experimental NMR structure of a peptide mimetic of the first extracellular loop of S1P4 (pdb id 2DCO) 32. This refined S1P1 model was used as a template for the other S1P receptor models and in docking studies with a series of ligands in order to identify the contributions to and trends in molecular recognition. Models of S1P2-5 receptors were developed based on the refined S1P1 model using the sequence alignment shown in Figure 1. S1P receptor models share similarities in helical bends and several interhelical hydrogen bond patterns. Hydrogen bonds from D2.50 to both S7.46 and N7.49 were observed for all receptor models. Meanwhile, hydrogen bonds from E1.49 to Y7.53 and from x1.59 to R7.62 as well as from N2.45 to S3.42 were detected in S1P1, S1P2 and S1P4 only. A complete listing of interhelical hydrogen bonds is shown in Table 2. In comparison with bovine rhodopsin, our S1P receptor models have a shorter second extracellular loop and no disulfide bond between this loop and TM3. In addition, our models do not include an analog to helix 8 observed for bovine rhodopsin.
Figure 1. Sequence alignment of S1P receptors.

Red boxes indicate residues in transmembrane domains. Green boxes indicate residues at .50 positions. Yellow boxes show residues required for ligand binding and receptor activation. Purple boxes show residues required for ligand binding and receptor activation in one receptor but not others.
Table 2.
Interhelical hydrogen bonding
|
Binding pocket
The binding site of these receptor models consists of charged residues in TM3 (R3.28 and E3.29), TM5 (K5.38) and TM7 (R7.34) for S1P1-3 or W4.64 for S1P4-5 which interact with the polar head groups of ligands. The hydrophobic tails of ligands adopt different conformations according to the geometric shape of the binding pocket. The hydrophobic tails adopt extended conformations for all ligands docked into the S1P1 and S1P5 receptors (Figure 2A and B) and folded conformations in the S1P4 receptor (Figure 2D). This result reflects the 4 Å shorter binding pocket of S1P4 relative to S1P1. For the S1P3 receptor, most of the ligands’ hydrophobic tails adopted folded conformations (Figure 2C). Figure 3 shows the best docked complexes of selected ligands in S1P1. As expected, docking results of S1P, FTY702P, benzimidazole, phosphonate and azetidine (Figure 3A and B) revealed interactions between positively charged residues R3.28, K5.38 and R7.33 and the negatively charged groups (phosphate or carboxylate) of ligands, as well as interactions between E3.29 and the ammonium nitrogen of ligands. Docking studies of SEW2871 were consistent with our docking studies using the previous S1P1 receptor model 36. The salt-bridge interaction of the S1P phosphate group is replaced by ion-dipole interactions involving the SEW2871 trifluoromethyl group and salt bridge interactions of the S1P ammonium group are replaced by π-stacking interactions. In fact, the fluorine atoms of CF3 group were 2.7–3.9 Å away from positively charged nitrogen atoms of R3.28, K5.38 and R7.34 (Figure 3C). In addition, multiple hydrophobic residues, F3.33, W6.48 and F7.38, are close in contact with the aromatic rings of SEW2871 (not shown).
Figure 2. Electrostatic surfaces of S1P1 and S1P4 binding pockets.
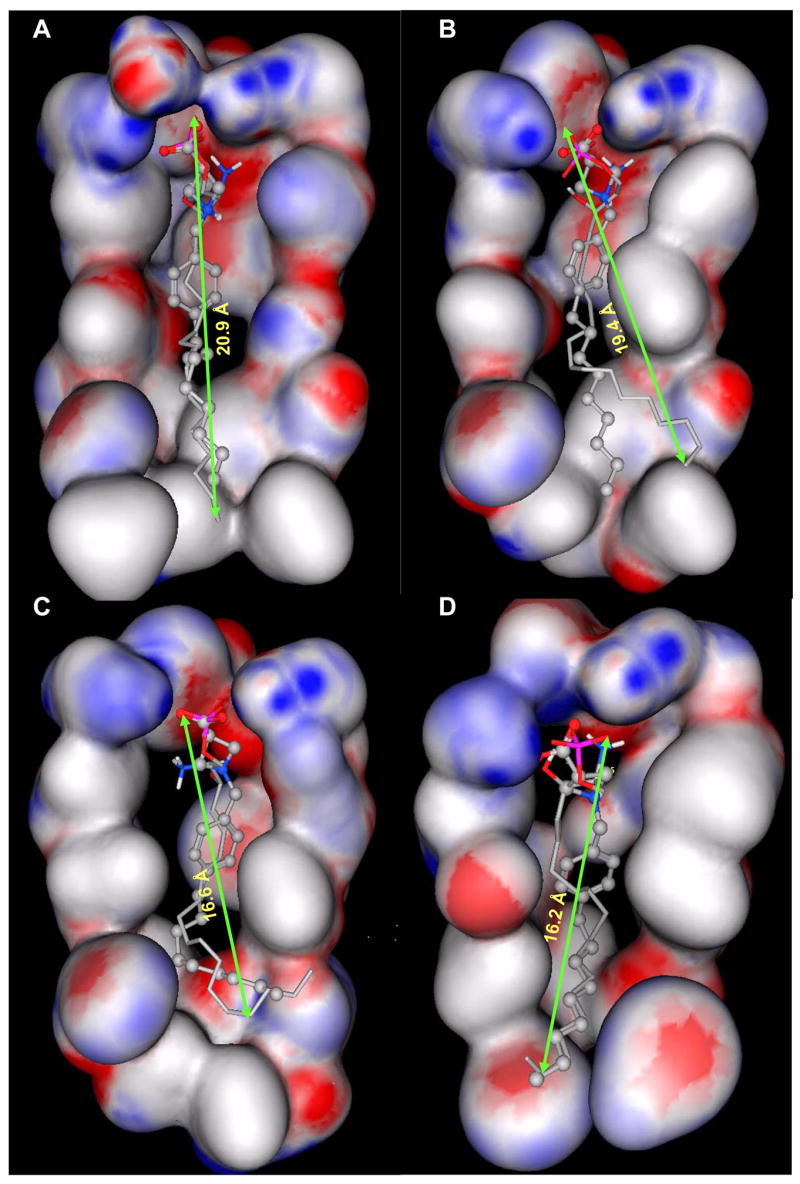
Gauss Connelly surfaces colored by atomic partial charge (red indicates negative, white indicates neutral and blue indicates positive partial charge) for atoms within 4.5 Å of S1P in TM3-5 were computed with the MOE program. The bound position of S1P is shown as a stick model and azetidine as stick and ball model. S1P1 binding pocket with docked position of S1P (A) is more than 4 Å longer than S1P3 binding pocket with docked position of S1P (C) and S1P4 binding pocket with docked position of S1P (D), and 1 Å longer than S1P5 binding pocket with docked position of S1P (B).
Figure 3. Refined S1P1 receptor model.
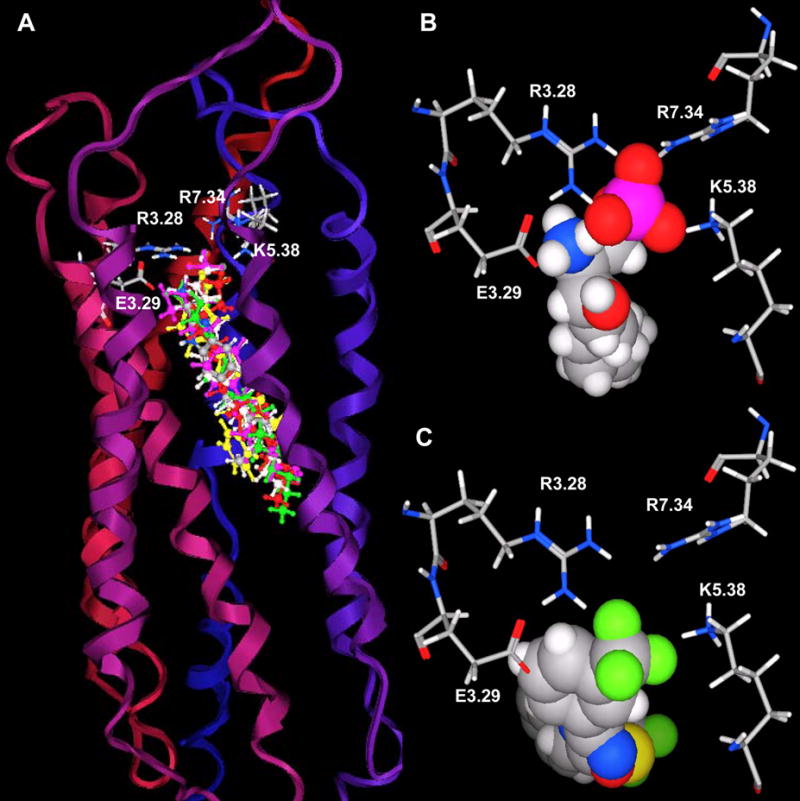
Panel A. S1P1 complex with FTY720P, S1P, benzimidazole, phosphonate, azetidine and SEW2871. The model is shown with extracellular loops at the top. Ribbons colored from red at the amino terminus to blue at the carboxy terminus represent the backbone of S1P1. The stick atoms represent residues required for ligand binding (red: oxygen, blue: nitrogen, grey: carbon, white: hydrogen and magenta: phosphorus). The ball & stick atoms represent ligands; FTY720P (magenta), S1P (green), benzimidazole (element), phosphonate (red), azetidine (light grey) and SEW2871 (yellow). Panel B. View of the S1P1 complex with S1P from the extracellular space. Selected residues are shown as stick models and labeled. Panel C. View of the S1P1 complex with SEW2871 from the extracellular space. Selected residues are shown as stick models and labeled.
S1P receptor models: Reflections of ligand binding affinity
Docking results of selected ligands (Table 1) in S1P receptor models reflect the experimental trend in ligand affinity. Ligand potency has been reported for S1P1 and S1P4 receptors in order of FTY720P> S1P > phosphonate > azetidine > SEW2871 37. Docking studies indicate the numbers of complementary polar interactions within 3 Å (Table 3) are in the same order for S1P1 and S1P4 receptors. Additionally, the number of close complementary interactions is lower for all ligands at the S1P4 receptor, consistent with the lower potencies observed for each agonist at this receptor. Complementary polar interactions are measured between the oxygen atoms of the carboxylate/phosphate groups of most agonists or fluorine atoms of SEW2871 to the nitrogen atoms of R3.28, K5.38 or K/R7.34, as well as between the ammonium nitrogen and the oxygen atoms of E3.29, the centroid of the 5-membered ring of W4.64 and the centroid of the F3.33 phenyl ring.
Table 3.
Number of interactions within 3 Å between the polar head groups of ligands and N/O atoms of charged residues in TM 3, 5 and 7
| Ligand/Receptor | S1P1 | S1P2 | S1P3 | S1P4 | S1P5 |
|---|---|---|---|---|---|
| FTY720P | 7 | 5 | 7 | 6 | 5 |
| S1P | 5 | 6 | 7 | 4 | 5 |
| Benzimidazole | 5 | 7 | 7 | 4 | 4 |
| Phosphonate | 4 | 6 | 6 | 3 | 4 |
| Azetidine(cis) | 3 | 4 | 5 | 2 | 3 |
| Azetidine(trans) | 3 | 4 | 5 | 2 | 3 |
| SEW2871 | 2 | 0 | 0 | 0 | 2 |
Figure 4 illustrates key interactions that explain the difference in ligand affinity for FTY720P, phosphonate and SEW2871 at the S1P4 receptor. S1P4 binds FTY720P through ion-pair interactions between the phosphate group and R3.28 and K5.38 as well as between the ammonium group and E2.39, and a cation-π interaction between the ammonium group and the 5-membered ring of W4.64 (Figure 4A) as observed for S1P. Interactions with the phosphonate ligand are less optimal at the ammonium group characterized by weak interactions with E3.29 (5 Å away) and negligible interaction with W4.64 (6.8 Å away) (Figure 4B). In addition, a hydrogen bond between the hydroxyl group of FTY720P and the E3.29 oxygen atoms and a π-π interaction between the FTY720P phenyl ring and F3.33 comprise a molecular rationale for the greater potency of FTY720P at S1P4 compared to S1P and phosphonate. Docking SEW2871 into the S1P4 model reveals no complementary interactions at all (Figure 4C). This is consistent with the fact that S1P4 has no affinity for SEW2871. For the S1P5 receptor, the number of complementary interactions within 3 Å observed are 5, 5, 4, 3 and 2 for FTY720P, S1P, phosphonate, azetidine and SEW2871, respectively. The docking data agree with the ligand affinity trend FTY720P, S1P > phosphonate > azetidine > SEW2871 for the S1P5 receptor 37.
Figure 4. Interaction vs. ligand binding affinity at S1P4.
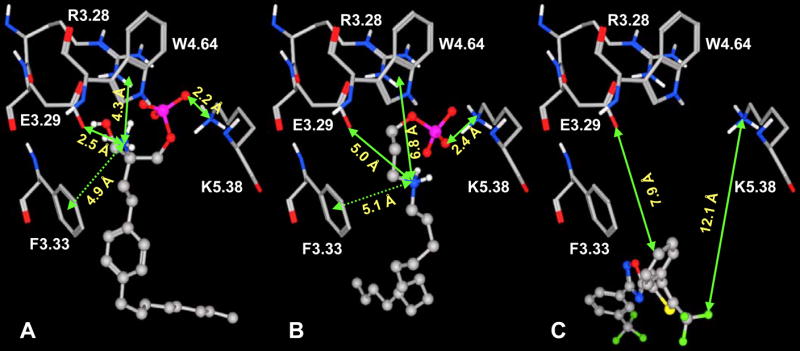
S1P4 complex with FTY720P (A), phosphonate (B) and SEW2871 (C). Ball and stick atoms represent ligands and stick atoms represent residues required for ligand binding. Non-polar hydrogens are omitted for clarity. S1P4 interactions with ligands in order FTY720P (A) > S1P ~ benzimidazole > Phosphonate (B) > Azetidine > SEW2871 (C) agree with binding affinity observed for these ligands (Table 1).
Docking studies with S1P receptor models not only rationalize binding affinity of various ligands at one receptor but also binding affinities of one ligand at different S1P receptors. For example, analysis of S1P docking results indicates the number of strong interactions characterized by distances less than 3 Å between the S1P head group and complementary sites in S1P1-5 are 5, 6, 7, 4 and 5, respectively. This result is consistent with affinities reported for S1P across the receptor family: S1P3 > S1P2 > S1P5 ~ S1P1 > S1P4.
Strengths/weaknesses of the S1P2 receptor model
Although docking S1P and SEW2871 into the S1P2 model resulted in good agreement with other receptor models and experimental trends, data on other ligands failed to reflect relative binding affinities. Binding assays carried out by Hale et al.37 show S1P2 does not bind FTY720P, phosphonate and azetidine. In contrast to experimental studies, docking results with the S1P2 receptor model indicated this receptor interacts favorably with all studied ligands, except SEW2871 (Figure 5A). The greatest inconsistency between the model and experimental observation is observed for phosphonate. The ammonium group of phosphonate interacts weakly with E3.29 of the S1P1 (Figure 5B) and S1P3-5 receptors. The distance from the ammonium nitrogen atom to the E3.29 oxygen atom is about 4.7–5.0 Å. Meanwhile the corresponding distance observed for S1P2 receptor is 2.4 Å (Figure 5C). Due to inconsistent docking results on this receptor, we are in the process of experimentally characterizing the binding pocket to refine the model.
Figure 5. Failure of S1P2 homology model to reflect pharmacological trend.
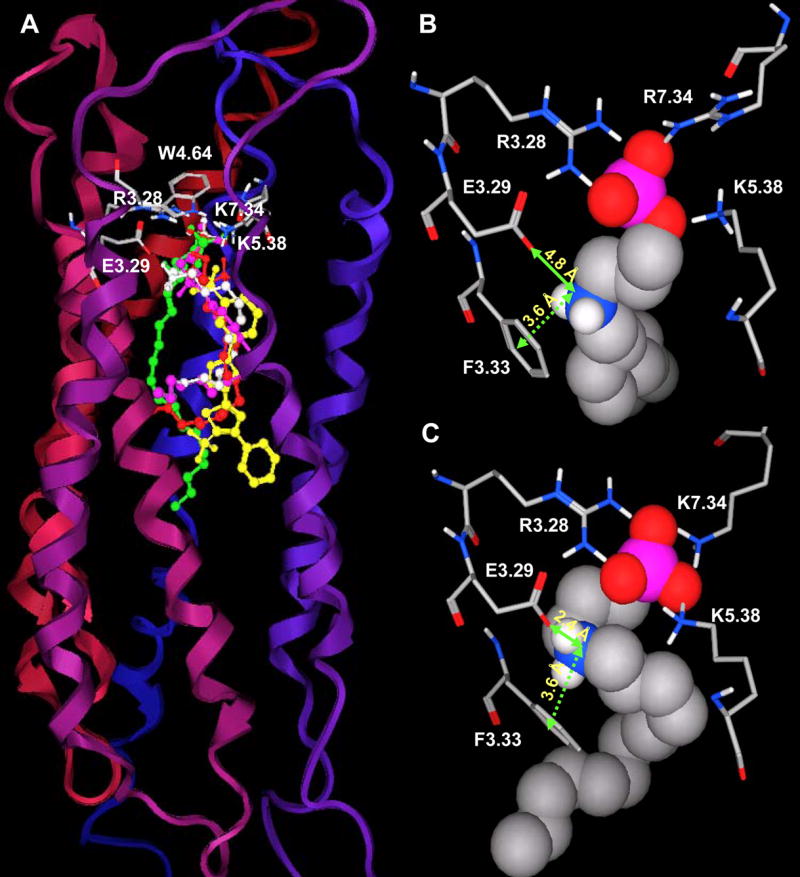
Panel A. A homology model of S1P2 receptor was generated using the refined S1P1 model. S1P2 complexes with S1P, phosphonate and SEW2871 are colored as described for Figure 3A. Panels B and C illustrate the failure of S1P2 model to accurately reflect the pharmacological trend based on the distances between the ammonium group of phosphonate and E3.29 and F3.33 aromatic centroid for S1P1 (B) and S1P2 (C). Residues required for ligand binding are shown in stick with non-polar hydrogens omitted for clarity. Phosphonate is shown in space filling atoms. The models suggest stronger phosphonate interactions with S1P2 (shorter distances) than S1P1 (longer distances) which is contrary to the fact that S1P2 has no affinity to phosphonate.
Ligand recognition differences among the S1P receptor family
Docking studies with S1P receptor models revealed significant differences in ligand recognition among this family, especially for benzimidazole. Benzimidazole activates 3 out of 5 S1P receptors and fully agonizes only the S1P4 receptor 38. Docking results show that the benzimidazole headgroup interactions with S1P1, S1P4 and S1P5 are similar to those of S1P. More favorable cation-π and π-π interactions involving the benzimidazole ammonium nitrogen and 5-membered ring, respectively, and F3.33 phenyl ring (Figure 6A and B) may account for the activation response at S1P1, S1P4 and S1P5 receptors but not S1P2 and S1P3 receptors. Table 4 presents the measured distances for these interactions and those involving the sulfur atom of C5.44. Interestingly, the distance between C5.44-S of S1P4 and the six-membered ring of benzimidazole (D = 5.4 Å) is unique across the receptor family in meeting the criterion for favorable interaction (D ≤6 Å) 39. This ligand-protein recognition feature distinguishes S1P4 from other members of S1P receptor family.
Figure 6. Ligand recognition differences in the S1P receptor family.
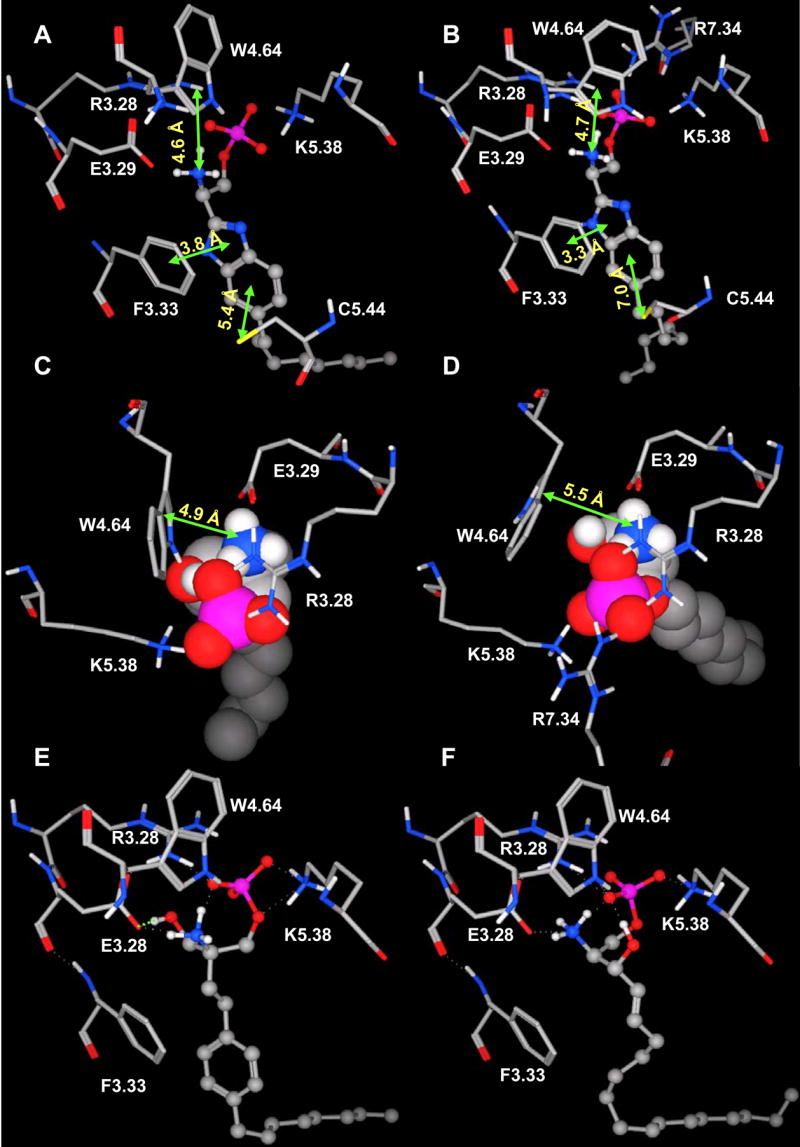
Panels A and B. Benzimidazole interacts selectively with C5.44 of S1P4 (A) but not S1P1 (B). The S1P4 C5.44 sulfur atom is closed to the optimized position for sulfur-aromatic interaction (5.4 Å); while the S1P1 C5.44 sulfur atom more distant from the imidazole ring (7.0 Å). Views and labels are similar to Figure 5. Panels C and D. S1P (space filling atoms) interacts selectively with W4.64 of S1P4 (C) but S1P1 (D). The distance between ammonium nitrogen and W4.64 pyrrolic centroid is < 5 Å for S1P4 and > 5 Å for S1P1. Views are the same for C and D but different from A and B. Panels E and F. FTY720P (E) and S1P (F) recognize the S1P4 receptor differently. The hydroxyl group of FTY720P forms a hydrogen bond with the E3.29 oxygen atom (E). This extra interaction explains the higher binding affinity of FTY720P compared to S1P.
Table 4.
Observed distances (Å) between ammonium nitrogen (N) atom or centroid of 5-membered ring (5R)/phenyl ring (6R) of benzimidazole and F3.33-6R, W4.64-5R or C5.44 sulfur atom (S)
| Ligand/Receptor Site | S1P1 | S1P2 | S1P3 | S1P4 | S1P5 |
|---|---|---|---|---|---|
| N···W4.64-5R | 4.7 | 4.6 | 5.4 | 4.6 | 5.0 |
| N···F3.33-6R | 4.9 | 4.8 | 5.1 | 4.9 | 4.9 |
| 5R···F3.33-6R | 3.3 | 4.7 | 5.0 | 3.8 | 4.2 |
| 6R···C5.44-S | 7.0 | 10.7* | 9.5 | 5.4 | 7.6 |
Distance between the centroid of phenyl ring of benzimidazole and C5.43-S instead of C5.44-S.
Ligand recognition differences were also detected for S1P. S1P is the endogenous ligand for the S1P receptors, but only a subset of the complementary interaction sites are conserved across the entire receptor family. Docking studies revealed a cation-π interaction between the ammonium group of S1P and the W4.64 pyrrole ring in the S1P4 and S1P5 receptors with distances of 4.9 (Figure 6C) and 4.4 Å. The corresponding distances in S1P1-3 are 5.5 (Figure 6D), 7.9 and 6.7 Å, respectively, suggesting a minimal contribution to overall S1P binding affinity at these receptors. In addition, the hydroxyl group of S1P is probably involved in ligand recognition only at S1P3. In contrast to S1P, the hydroxyl group of FTY720P is involved in ligand binding observed for all receptor models. This feature may contribute to higher affinity of FTY720P compared to S1P (Figure 6E and F).
DISCUSSION
Our S1P receptor models define important features of ligand recognition in this family and provide a molecular rationale for the trends in ligand binding affinity for a series of ligands at a single receptor and for a single ligand across the receptor family. The general trend observed experimentally for binding affinity is in order of FTY720P, S1P > phosphonate > azetidine > SEW2871, except S1P337. Our docking studies indicated ligands with phosphate (−2 charge) head groups (FTY720P and S1P) have stronger ion pair interactions with positively charged residues in TM3, 5 and 7 than ligands containing carboxylate (−1 charge) head groups (azetidine cis and trans) (Table 3). The phosphonate ligand interacts somewhat between the previous two functional groups. SEW2871 lacks an anionic head group and exhibits the weakest interaction (Table 3). Among dianionic head groups, interactions are in the order of FTY720P ≥ S1P >phosphonate (Table 3), except at S1P2 and S1P3. The S1P3 receptor has higher affinity to S1P than FTY720P. Docking results indicate S1P3 is the only receptor with a specific hydrogen-bonding interaction with the S1P hydroxyl group. The average distance from the S1P hydroxyl hydrogen atom to S1P3 E3.29 oxygen atoms is 2.4 Å. In addition, the S1P C4 = C5 double bond is located near the S1P3 F3.33 phenyl ring (< 4 Å), suggesting possible π–π interaction. Previous studies on S1P stereoisomers and analogs examined the difference between C3 hydroxyl, C3 oxo, and C3 dehydroxy compounds 40,41. The C3 dehydroxy findings are not particularly useful for direct comparison to our docking results as the dehydroxy compound was saturated, and two carbons shorted than S1P. However, the binding data on these compounds suggested that the C3 hydroxyl group has some effect on ligand binding at S1P1,3,5. Removing of this functional group while concurrently saturating C4–C5 and reducing the carbon chain length by two atoms results in reducing binding affinity compared to S1P approximately 18% and 16% for S1P3 and S1P5. With other S1P receptors, FTY720P interacts more strongly than S1P due to an extra hydrogen bonding interaction between the FTY720P hydroxyl group and the E3.29 oxygen atom (Figure 6). Meanwhile, the phosphonate ligand has much weaker ion pair interaction between the phosphonate ammonium group and E3.29 (Figure 4).
A case study of benzimidazole revealed interesting observations on ligand binding with potential impact on receptor activation. Our docking results support a π-π interaction between the aromatic rings of benzimidazole and F3.33 as well as weak cation-π interactions between the ammonium nitrogen and aromatic rings of W4.64 and F3.33 of S1P1, S1P4 and S1P5 (Table 4) that likely contribute to receptor activation of S1P1, S1P4 and S1P5. Considerably weaker interactions of these functional groups are observed for S1P2 and S1P3 receptors (Table 4, observed distances [D] ~ 5 Å). Moreover, an additional sulfur-aromatic interaction was observed only in the S1P4 receptor. The distance from the C5.44 sulfur atom to the centroid of the benzimidazole phenyl ring is 5.4 Å, only 0.1 Å longer than the optimized distance for sulfur-aromatic interactions in proteins 39. The corresponding distances observed for other receptors are at least 7 Å, indicating negligible interaction between the functional groups (favorable interaction requires D ≤ 6 Å). This distinct interaction is expected to be a key contribution to the full agonism of benzimidazole at the S1P4 receptor.
C5.44 is present in 4 out of 5 S1P receptors. S1P2 has cysteine at position 5.43 instead of 5.44. With the exception at S1P4, those cysteine sulfur atoms are at least 7 Å away from the aromatic rings of FTY720P, benzimidazole, azetidine and SEW2871 and, therefore, may not involve in ligand binding at S1P1,2,3,5 receptors. In contrast, the S1P4 C5.44 sulfur is in the range of interaction with the aromatic rings of FTY720P, benzimidazole and azetidine. This sulfur-aromatic interaction is unique feature of S1P4 receptor, probably arising due to the folded shape of the binding pocket (Figure 2D).
Our previous studies 28,29,42 and current S1P receptor models indicate that the binding pocket of S1P4 (Figure 2D) is shorter than that of S1P1 (Figure 2A). S1P1 prefers a linear binding mode and has a higher affinity to unsaturated ligands while S1P4 prefers a bent binding mode and saturated ligands 28,29,42. Docking results show the hydrophobic tails of all studied ligands are extended when docked into S1P1 and S1P5 models, but folded in the S1P4 binding pocket. Differences in binding modes and geometry of the binding pockets lead to differential binding across the S1P receptors. For example, S1P4 has much lower binding affinities to azetidine, approximately 300–500 fold compared to S1P1 and S1P5. This may be due to a combined effect of shorter binding pocket and bent binding mode required for S1P4 28. The distance between negatively and positively charged groups of azetidine is one atom less than phosphate ligands. In addition, the nitrogen atom of azetidine is involved in the ring system. Both factors prevent azetidine from adopting an optimized conformation for the bent binding mode that results in lower binding affinity of azetidine at S1P4. S1P3 has similar binding affinity to azetidine as S1P437 and, therefore, is expected to interact with azetidine similarly to S1P4. In fact, docked conformations of azetidine at both S1P3 and S1P4 have a comparable length about 3 Å shorter than those at S1P1 and S1P5. Moreover, S1P3 and S1P4 have analogous electrostatic distributions at the top of the binding pockets with anionic charge concentrated in a small area (Figure 2C and D) that prevents them from effectively interacting with the nitrogen atom in the ring system. In contrast, negative charge on the top of the S1P1 and S1P5 binding pockets distributes into an extended surface, (Figure 2A and B) allowing these receptors to interact with the azetidine nitrogen atom more effectively than S1P3 and S1P4.
It is known that S1P head groups bind S1P receptors via ion pair interactions with residues R3.28, E3.29, K5.38 and R/K7.33 28–30. S1P4 and S1P5 receptors lack the positively charged residue in TM7. Instead, S1P4 interacts with S1P through an extra cation-π interaction between the W4.64 pyrrole ring and the ammonium group of S1P (Figure 6). A similar result was observed for the S1P-S1P5 complex, but not the S1P-S1P1 (Figure 6), S1P-S1P2 or S1P-S1P3 complexes. Previous model-driven mutagenesis studies of S1P receptors showed that W4.64 is required for S1P binding to the S1P4 receptor, but not to S1P1 28 Our docking studies suggest that a cation-π interaction with W4.64 is also required for S1P binding to the S1P5 receptor to compensate for the lack of a positively charged residue in TM7, and is not necessary for S1P binding to the S1P2 and S1P3 receptors.
Docking studies of SEW2871 into S1P1 receptor models reveal significant interactions with S1P1 that overlap those of S1P. The results suggest ion-dipole interactions between the CF3 group attached on ring A of SEW2871 and R3.28, E3.29, K5.38 and R7.34 residues that support ion pair interactions with S1P polar head groups. In addition, multiple hydrophobic interactions between residues F3.33, W6.48 and F7.38 and the aromatic rings of SEW2871 probably contribute to SEW2781 potency at S1P1. Our previous studies on this agonist indicate that mutation of any of R3.28, E3.29 and R7.34 residues to alanine severely reduced the phosphorylation of both Akt and ERK2 by both S1P and SEW287136. SEW2871 has been shown to be a full agonist of S1P1 43. Its binding affinity at S1P1 is about 50 fold less than S1P,37 implying that the strength of dipole-ion interactions is not adequate to replace salt bridge interactions. However, these interactions certainly contribute to the potency of SEW2871 at the S1P1 receptor. Replacement of the CF3 group on ring D with a methyl group significantly diminishes Akt and ERK2 responses in the E3.29A and R7.34A mutants 36 confirming an important role for this functional group.
Several models of S1P receptors have been published in the literature by at least four different research groups 41,44,45. Models generated from the TASSER program 45 have similar overall folding as ours. However, in the TASSER models, the sidechains of R3.28, E3.29, K5.38 and R/K7.33 point away from the binding pockets. Our published mutagenesis studies indicate these residues are necessary for ligand binding and receptor activation 28,30. A model of S1P1 developed by Lim et al 41 includes ion pairing interactions between the S1P phosphate and R3.28, R7.34 as well as an ionic interaction between the S1P ammonium group and E3.29 analogous to our model. This group suggests hydrogen bonds from the Y2.57 (98) sidechain hydroxyl group and the F7.38 (296) backbone carbonyl group to the S1P hydroxyl group, which are not observed in our studies. A model of human S1P4 has been reported in the literature that differs from our current human S1P4 and previously published mouse28 and human42 S1P4 models. Vaidehi et al 44 indicated S1P interacts with human S1P4 at residues T3.34(127), E7.30(284) and W7.37(291). All these residues are located toward the extracellular loops in our model, and; therefore, are unlikely interact with S1P. In addition, the hydrophobic region of the ligand binding pocket of our S1P1 model has been experimentally validated 29.
CONCLUSIONS
This study identifies ligand recognition features that generalize across the S1P receptor family as well as features unique to the S1P4 and S1P5 receptors. Docking results highlight the previously unknown sulfur-aromatic interaction between the S1P4 C5.44 sulfur atom and the phenyl ring of benzimidazole as well as π-π interactions between F3.33 and ligand aromatic rings. The findings not only confirm the importance of cation-π interaction between W4.64 and the ammonium of S1P at S1P4 but also predict the same interaction at S1P5. These docking studies provide molecular insights into pharmacological trends both across the receptor family as well as at single receptors.
Consistency with pharmacological trends qualitatively validates the S1P1 and S1P3-5 models for pharmacophore development including database mining and new ligand discovery. The S1P1 model, in fact, has already identified experimentally confirmed hits on the basis of partial matches to an agonist pharmacophore 29. These models can also serve as tools for ligand optimization to improve potency and selectivity. For quantitative applications, such as free energy perturbation or thermodynamic integration, quantum mechanical studies and mutagenesis have validated the quantitive accuracy of the headgroup binding site46 additional evaluations are necessary to validate the quantitative accuracy of the models.
Acknowledgments
This work was supported in part by grants from NIH-NHLBI and NIH-NCI (R01 HL084007 and R01 CA92160). We acknowledge the Chemical Computing Group for generously donating the MOE program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spiegel AM, Weinstein LS. Inherited diseases involving G proteins and G protein-coupled receptors. Annu Rev Med. 2004;55:27–39. doi: 10.1146/annurev.med.55.091902.103843. [DOI] [PubMed] [Google Scholar]
- 2.Schoneberg T, Schulz A, Biebermann H, Hermsdorf T, Rompler H, Sangkuhl K. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104:173–206. doi: 10.1016/j.pharmthera.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 4.Staff MAN. World’s best-selling medicines. Med Ad News. 2004;23:60–64. [Google Scholar]
- 5.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp R, Yamamoto M, Miyano M. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 6.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 7.Becker OM, Shacham S, Marantz Y, Noiman S. Modeling the 3D structure of GPCRs: advances and application to drug discovery. Curr Opin Drug Discov Devel. 2003;6:353–361. [PubMed] [Google Scholar]
- 8.Okamoto H, Takuwa N, Gonda K, Okazaki H, Chang K, Yatomi Y, Shigematsu H, Takuwa Y. EDG1 Is a Functional Sphingosine-1-phosphate Receptor That Is Linked via a Gi/o to Multiple Signaling Pathways, Including Phospholipase C Activation, Ca2+ Mobilization, Ras-Mitogen-activated Protein Kinase Activation, and Adenylate Cyclase Inhibition. J Biol Chem. 1998;273:27104–27110. doi: 10.1074/jbc.273.42.27104. [DOI] [PubMed] [Google Scholar]
- 9.Lee MJ, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S, Hla T. Sphingosine-1-Phosphate as a Ligand for the G Protein-Coupled Receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- 10.An S, Bleu T, Huang W, Hallmark OG, Coughlin SR, Goetzl EJ. Identification of cDNAs encoding two G protein-coupled receptors for lysosphingolipids. FEBS Lett. 1997;417:279–282. doi: 10.1016/s0014-5793(97)01301-x. [DOI] [PubMed] [Google Scholar]
- 11.Van Brocklyn JR, Gräler MH, Bernhardt G, Hobson JP, Spiegel S. Sphingosine-1-phosphate is a Ligand for the G Protein-Coupled Receptor EDG-6. Blood. 2000;95:2624–2629. [PubMed] [Google Scholar]
- 12.Yamazaki Y, Kon J, Sato K, Tomura H, Sato M, Yoneya T, Okazaki H, Okajima F, Ohta H. Edg-6 as a putative sphingosine 1-phosphate receptor coupling to Ca(2+) signaling pathway. Biochem Biophys Res Commun. 2000;268:583–589. doi: 10.1006/bbrc.2000.2162. [DOI] [PubMed] [Google Scholar]
- 13.Im DS, Heise CE, Ancellin N, O’Dowd BF, Shei GJ, Heavens RP, Rigby MR, Hla T, Mandala S, McAllister G, George SR, Lynch KR. Characterization of A Novel Sphingosine 1-Phosphate Receptor, Edg-8. J Biol Chem. 2000;275:14281–14286. doi: 10.1074/jbc.275.19.14281. [DOI] [PubMed] [Google Scholar]
- 14.MacLennan AJ, Browe CS, Gaskin AA, Lado DC, Shaw G. Cloning and characterization of a putative G-protein coupled receptor potentially involved in development. Mol Cell Neurosci. 1994;5:201–209. doi: 10.1006/mcne.1994.1024. [DOI] [PubMed] [Google Scholar]
- 15.Hla T, Maciag T. An Abundant Transcript Induced in Differentiating Human Ensothelial Cells Encodes a Polypeptide with Structural Similarities to G-protein-coupled Receptors. J Biol Chem. 1990;265:9308–9313. [PubMed] [Google Scholar]
- 16.Wang F, Van Brocklyn JR, Hobson JP, Movafagh S, Zukowska-Grojec Z, Milstien S, Spiegel S. Sphingosine 1-Phosphate Stimulates Cell Migration through a Gi-coupled Cell Surface Receptor, A Potential Involvement in Angiogenesis. J Biol Chem. 1999;274:35343–35350. doi: 10.1074/jbc.274.50.35343. [DOI] [PubMed] [Google Scholar]
- 17.Kupperman E, An S, Osborne N, Waldron S, Stainier DYR. A Sphingosine-1-phosphate Receptor Regulates Cell Migration During Vertebrate Heart Development. Nature. 2000;406:192–195. doi: 10.1038/35018092. [DOI] [PubMed] [Google Scholar]
- 18.English D, Garcia JG, Brindley DN. Platelet-released phospholipids link haemostasis and angiogenesis. Cardiovasc Res. 2001;49:588–599. doi: 10.1016/s0008-6363(00)00230-3. [DOI] [PubMed] [Google Scholar]
- 19.Erl W, Siess W. Sphingosine-1-Phosphate and the Leading Edg-1 of Vascular Smooth Muscle Cells. Circ Res. 2001;89:474–476. [PubMed] [Google Scholar]
- 20.Rosenfeldt HM, Hobson JP, Macyka M, Olivera A, Nava VE, Milstien S, Spiegel S. EDG-1 Links the PDGF Receptor to Src and Focal Adhesion Kinase Activation Leading to Lamellipodia Formation and Cell Migration. FASEB J. 2001;15:2649–2659. doi: 10.1096/fj.01-0523com. [DOI] [PubMed] [Google Scholar]
- 21.Tamama K, Kon J, Sato K, Tomura H, Kuwabara A, Kimura T, Kanda T, Ohta H, Ui M, Kobayashi I, Okajima F. Extracellular mechanism through the Edg family of receptors might be responsible for sphingosine-1-phosphate-induced regulation of DNA synthesis and migration of rat aortic smooth-muscle cells. Biochem J. 2001;353:139–146. [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon YG, Min JK, Kim KM, Lee DJ, Billiar TR, Kim YM. Sphingosine 1-phosphate protects human umbilical vein endothelial cells from serum-deprived apoptosis by nitric oxide production. J Biol Chem. 2001;276:10627–10633. doi: 10.1074/jbc.M011449200. [DOI] [PubMed] [Google Scholar]
- 23.Hisano N, Yatomi Y, Satoh K, Akimoto S, Mitsumata M, Fujino MA, Ozaki Y. Induction and suppression of endothelial cell apoptosis by sphingolipids: a possible in vitro model for cell-cell interactions between platelets and endothelial cells. Blood. 1999;93:4293–4299. [PubMed] [Google Scholar]
- 24.Cavallini L, Venerando R, Miotto G, Alexandre A. Ganglioside GM1 protection from apoptosis of rat heart fibroblasts. Arch Biochem Biophys. 1999;370:156–162. doi: 10.1006/abbi.1999.1378. [DOI] [PubMed] [Google Scholar]
- 25.Morita Y, Perez GI, Paris F, Miranda SR, Ehleiter D, Haimovitz-Friedman A, Fuks Z, Xie Z, Reed JC, Schuchman EH, Kolesnick RN, Tilly JL. Oocyte Apoptosis is Suppressed by Disruption of the acid sphingomyelinase Gene or by Sphingosine-1-phosphate Therapy. Nat Med. 2000;6:1109–1114. doi: 10.1038/80442. [DOI] [PubMed] [Google Scholar]
- 26.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 27.Brinkmann V, Pinschewer DD, Feng L, Chen S. FTY720: Altered Lymphocyte Traffic Results in Allograft Protection. Transplantation. 2001;72:764–769. doi: 10.1097/00007890-200109150-00002. [DOI] [PubMed] [Google Scholar]
- 28.Inagaki Y, Pham TT, Fujiwara Y, Kohno T, Osborne DA, Igarashi Y, Tigyi G, Parrill AL. Sphingosine 1-phosphate analogue recognition and selectivity at S1P4 within the endothelial differentiation gene family of receptors. Biochem J. 2005;389:187–195. doi: 10.1042/BJ20050046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujiwara Y, Osborne DA, Walker MD, Wang DA, Bautista DA, Liliom K, Van Brocklyn JR, Parrill AL, Tigyi G. Identification of the hydrophobic ligand binding pocket of the S1P1 receptor. J Biol Chem. 2007;282:2374–2385. doi: 10.1074/jbc.M609648200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang DA, Lorincz Z, Bautista DL, Liliom K, Tigyi G, Parrill AL. A single amino acid determines lysophospholipid specificity of the S1P1 (EDG1) and LPA1 (EDG2) phospholipid growth factor receptors. J Biol Chem. 2001;276:49213–49220. doi: 10.1074/jbc.M107301200. [DOI] [PubMed] [Google Scholar]
- 31.Berman H, Henrick K, Nakamura H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 32.Pham TC, Kriwacki RW, Parrill AL. Peptide design and structural characterization of a GPCR loop mimetic. Biopolymers. 2007;86:298–310. doi: 10.1002/bip.20745. [DOI] [PubMed] [Google Scholar]
- 33.MOE. Chemical Computing Group; Montreal: 2003. [Google Scholar]
- 34.Halgren TA. Merck Molecular Force Field. I. Basis Form, Scope Parameterization, and Preformance of MMFF94*. J Comput Chem. 1996;17(56):490–519. [Google Scholar]
- 35.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated Docking Using a Lamarckian Genetic Algorithm and an Empirical Binding Free Energy Function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 36.Jo E, Sanna MG, Gonzalez-Cabrera PJ, Thangada S, Tigyi G, Osborne DA, Hla T, Parrill AL, Rosen H. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12:703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 37.Hale JJ, Lynch CL, Neway W, Mills SG, Hajdu R, Keohane CA, Rosenbach MJ, Milligan JA, Shei GJ, Parent SA, Chrebet G, Bergstrom J, Card D, Ferrer M, Hodder P, Strulovici B, Rosen H, Mandala S. A rational utilization of high-throughput screening affords selective, orally bioavailable 1-benzyl-3-carboxyazetidine sphingosine-1-phosphate-1 receptor agonists. J Med Chem. 2004;47:6662–6665. doi: 10.1021/jm0492507. [DOI] [PubMed] [Google Scholar]
- 38.Clemens JJ, Davis MD, Lynch KR, Macdonald TL. Synthesis of benzimidazole based analogues of sphingosine-1-phosphate: discovery of potent, subtype-selective S1P4 receptor agonists. Bioorg Med Chem Lett. 2004;14:4903–4906. doi: 10.1016/j.bmcl.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 39.Reid KSC, Lindley PF, Thornton JM. Sulfur-aromatic interactions in proteins. FEBS Lett. 1985;190:209–213. [Google Scholar]
- 40.Lim HS, Oh YS, Suh PG, Chung SK. Syntheses of sphingosine-1-phosphate stereoisomers and analogues and their interaction with EDG receptors. Bioorg Med Chem Lett. 2003;13:237–240. doi: 10.1016/s0960-894x(02)00893-4. [DOI] [PubMed] [Google Scholar]
- 41.Lim HS, Park JJ, Ko K, Lee MH, Chung SK. Syntheses of sphingosine-1-phosphate analogues and their interaction with EDG/S1P receptors. Bioorg Med Chem Lett. 2004;14:2499–2503. doi: 10.1016/j.bmcl.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Holdsworth G, Osborne DA, Pham TT, Fells JI, Hutchinson G, Milligan G, Parrill AL. A single amino acid determines preference between phospholipids and reveals length restriction for activation of the S1P4 receptor. BMC Biochem. 2004;5:12. doi: 10.1186/1471-2091-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 44.Vaidehi N, Floriano WB, Trabanino R, Hall SE, Freddolino P, Choi EJ, Zamanakos G, Goddard WAI. Prediction of Structure and Function of G Protein-Coupled Receptors. Proc Nat Acad Sci USA. 2002;99:12622–12627. doi: 10.1073/pnas.122357199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Devries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol. 2006;2:e13. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naor MM, Walker MD, Van Brocklyn JR, Tigyi G, Parrill AL. Sphingosine 1-phosphate pK(a) and binding constants: Intramolecular and intermolecular influences. J Mol Graph Model. 2007;26:519–528. doi: 10.1016/j.jmgm.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]