

Abstract
Over 80% of the body's activated B cells are located in mucosal sites, including the intestine. The intestine contains IgM+ B cells, but these cells have not been characterized phenotypically or in terms of their developmental origins. We describe a previously unidentified and unique subset of immunoglobulin M+ B cells that present with an AA4.1−CD21−CD23− major histocompatibility complex class IIbright surface phenotype and are characterized by a low frequency of somatic hypermutation and the potential ability to produce interleukin-12p70. This B cell subset resides within the normal mucosa of the large intestine and expands in response to inflammation. Some of these intestinal B cells originate from the AA4.1+ immature B2 cell pool in the steady state and are also recruited from the recirculating naive B cell pool in the context of intestinal inflammation. They develop in an antigen-independent and BAFF-dependent manner in the absence of T cell help. Expansion of these cells can be induced in the absence of the spleen and gut-associated lymphoid tissues. These results describe the existence of an alternative pathway of B cell maturation in the periphery that gives rise to a tissue-specific B cell subset.
BM-derived immature IgM+ B cells can mature in the BM itself or migrate to the spleen and mature into follicular and marginal zone B cells (1–7). Maturation proceeds through short-lived transitional B cell stages (2, 3, 8), and several specific signaling receptors and transcription factors are involved in this process (1–6). Newly formed B cells that have recently emerged from the BM represent the T1 transitional stage of development in which cells express high levels of CD24 and AA4.1 and low levels of CD23, CD21, and IgD. At the T2 stage, cells acquire the ability to recirculate and to express higher levels of CD23, CD21, and IgD, and they are capable of maturing into either marginal zone or follicular B cells (2, 5, 9). The T3 subset may represent a specific follicular B cell population that has recently matured (3), but it has also been recently suggested that T3 cells may represent anergic B cells (10).
In several systemic and organ-specific inflammatory disorders, ectopic neolymphoid follicles, which are sometimes called tertiary lymphoid tissue, are induced by the mobilization of B cell progenitors from the BM to the periphery (11–15). Interestingly, although the BM functions as a primary lymphoid organ, it may also serve as a secondary lymphoid organ in which activation of B cells may occur in response to circulating pathogens (7, 16). Although >80% of the body's B cells are located in gut-associated lymphoid tissues (GALTs) (17, 18), it is unknown whether these cells are derived from the activation of recirculating B cells or if they include novel populations of naive B cells that specifically home to these sites.
The mucosal immune system is constitutively exposed to a wide spectrum of commensal microflora (18–21). The effecter lymphoid component of GALT includes scattered lymphocytes throughout the lamina propria of the small and large intestine. The inductive sites consist of organized lymphoid tissues, such as Peyer's patches (PPs), which are formed in a lymphotoxin (LT)-α dependent manner (17, 19–22). Because the small intestine represents a major source of IgA+ plasma cells, the majority of studies on intestinal B cells have focused on small intestinal IgA+ plasma cells (17–20, 23–28). Three pathways have been identified for small intestinal plasma cell development. Approximately half of all IgA+ plasma cells in the intestine may originate from peritoneal B1 cells, whereas the remainder may be either derived from activated B cells that migrate from organized lymphoid tissues such as PPs, or originate from naive B cells through a distinct pathway for gut-primed B cells (17–20, 23–28). In addition to plasma cells, IgM+ B cells also exist within the normal intestine and significantly increase within large intestinal mucosal tissues during inflammatory responses (29–32). However, the phenotypic and developmental properties of IgM+ B cells in the intestine have not yet been fully explored.
We describe a previously unidentified, intestine-specific, IgM+ B cell subset that is characterized by an AA4.1−CD21−CD23−MHCIIbright phenotype, by a low frequency of somatic hypermutation (SHM), and by the potential ability to produce IL-12p70 after exposure to microbes. These cells appear to exist in a unique preactivated state, as indicated by the very high expression levels of MHC class II on these cells. This intestinal IgM+ B cell subset may originate from AA4.1+ immature transitional B cells in the steady state, and may be significantly increased in the inflamed intestine after recruitment from the recirculating naive B cell pool.
RESULTS
Identification of a unique AA4.1−CD21−CD23−IgM+ B cell subset in the large intestine
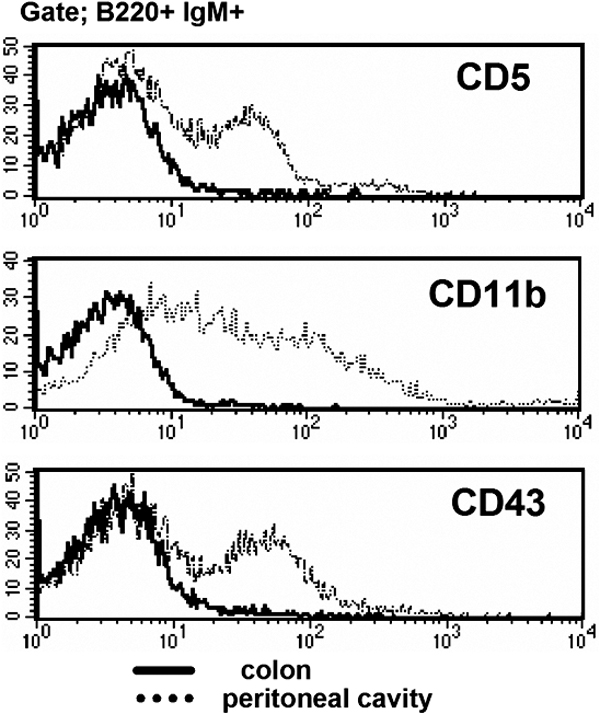
IgM+ B cells have been described in the large intestine, and these cells expand significantly and form B cell clusters in certain disease states involving intestinal inflammation (29–32). We herein demonstrate that the majority of IgM+ B cells in the large intestine of WT mice exhibit a unique surface phenotype that is distinct from that of recirculating splenic and mesenteric LN (MLN) B cells. In contrast to splenic, MLN, and PP B cells, the majority of intestinal IgM+ B cells are CD21−CD23−IgDlow (Fig. 1 A). Because a significant increase in intestinal B cells has been well documented in several colitis models (29–32), we next examined the phenotype of IgM+ B cells from the inflamed intestine of three colitis models, including TCRα KO mice (Th2 cell–dependent chronic colitis), IL-10 KO mice (Th1 cell–dependent chronic colitis), and dextran sodium sulfate (DSS)–induced acute colitis (29, 32, 33). As previously demonstrated (29, 32), a significant increase in IgM+ B cells was observed in the inflamed intestine of TCRα KO mice and IL-10 KO mice (Fig. 1 B). Interestingly, we found that in the DSS colitis model intestinal B cell expansion was specifically induced during the recovery phase (after termination of DSS treatment), but not during the acute phase when DSS was being administered. In comparison to day 0 (normal mucosa), the proportion as well as the absolute numbers of IgM+ cells in the large intestine were slightly decreased at day 4 (acute phase), but increased significantly at day 8 (during the recovery phase) of DSS colitis (Fig. 1, B and C). Like normal intestinal IgM+ B cells, the majority of intestinal IgM+ B cells in these colitis models were also of the CD21−CD23−IgDlow phenotype (Fig. 1 D). In addition, intestinal B cells from normal and inflamed mucosa were CD24intermediateCD62L−CD1d+ and did not express B1 cell markers (CD43, CD11b, and CD5; Fig. 1 E and Fig. S1). The intestinal B cells proliferated in vitro in the response to BCR ligation, as well as LPS stimulation (Fig. 1 F). These findings define a previously unidentified, unique B cell subset that exists in normal and inflamed large intestine.
Figure 1.

Identification of a unique IgM+ B cell subset in the large intestine. (A) Expressions of IgM versus IgD, CD21, or CD23 of the cells from colon, spleen (SP), MLN, and PP of WT mice are shown. (B) For DSS colitis, intestinal inflammation was induced by administration of 4% DSS in drinking water, and the treatment was terminated by changing DSS water to normal water at day 4. (top) Proportions of IgM versus CD3ε-expressing cells in the large intestine from normal mice (Day 0, n = 21) and DSS-induced colitis mice (Day 4 [acute phase], n = 11; Day 8 [recovery phase], n = 24). (bottom) Proportions of IgM versus CD3ε-expressing cells in the large intestine of chronic colitis models, TCRα KO mice (n = 3), and IL-10 KO mice (n = 3). (C) Absolute numbers of IgM+ cells in the large intestine of DSS colitis at day 0, 4, 8, and 30 are shown. (D) Expressions of IgM versus CD23, CD21, or IgD on the cells from large intestine of three colitis models (DSS colitis, TCRα KO mice, and IL-10 KO mice) are shown. The data are representative of 3–11 individual experiments. (E) Expression of several surface makers on the gated IgM+ B220+ cells from the large intestine of DSS at day 8 (solid lines) and from spleen (dot lines) is shown. The data are representative of 11 individual experiments. (F) Proliferative responses of purified B cells (>95% are IgM+) from the colon (shaded bars) and spleen (open bars) to stimulation without (No) or with LPS (5 μg/ml) or F(ab')2 anti-Igμ Abs (anti-μ; 50 μg/ml) are shown. The results represent the mean ± the SEM of triplicate data from three individual mice.
Intestinal B cells are derived from AA4.1+ immature and transitional B cells
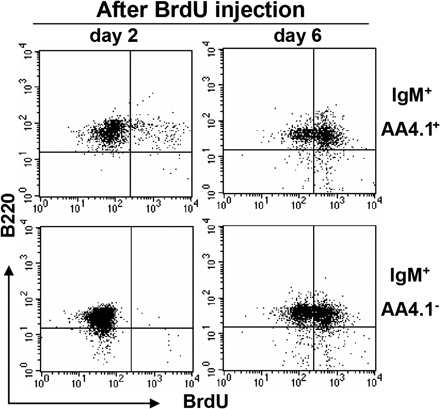
Intestinal B cells with their unique phenotype could either represent a naive or an activated B cell population. These cells could develop in the intestine in situ from uncommitted lymphoid progenitors, or they may be derived from newly formed B cells that not only seed the spleen but also home to the intestine, or they could represent a population that differentiates from recirculating follicular phenotype B cells or from B1 B cells. The rapid expansion of intestine-specific CD21−CD23− B cells in the DSS colitis model provided us an opportunity to more closely examine how these cells expand either by proliferation of existing cells or by enhanced recruitment or development from progenitors. Because intestinal B cells share some phenotypic features with T1 B cells, but the latter also express AA4.1, a marker for recently generated B cells, (34), we next examined AA4.1 expression on intestinal B cells. Less than 10% of intestinal B cells expressed AA4.1 (Fig. 2 A). Unlike AA4.1− intestinal B cells, the AA4.1+ intestinal B cells consisted of two subsets, T1-like CD23− and T2-like CD23+ B cells (Fig. 2 A). To examine whether AA4.1−CD21−CD23− intestinal B cells could potentially mature from the AA4.1+ immature B cells, in vivo BrdU pulse labeling was performed as previously described (35). In the AA4.1+ IgM+ population from the normal large intestine of WT mice, BrdU+ B220+ cells were observed on both day 2 and 6 after the initial BrdU injection (Fig. 2 B, top). In contrast, BrdU+ B220+ cells were not detectable at day 2 in the AA4.1−IgM+ population, but became clearly detectable at day 6 (Fig. 2 B, bottom). Similar findings were also observed in the inflamed intestine after DSS treatment (Fig. S2). To confirm whether AA4.1+ immature B cells actually possess the ability to differentiate into intestinal IgM+ B cells, we purified AA4.1+ B220+ B cells from the spleen of GFP transgenic mice, and then transferred them (5 × 106) intravenously into WT mice, with or without induction of DSS colitis. GFP+ IgM+ B cells were detected in the colon and spleen of both recipient mouse groups (Fig. 2 C, right). The reconstituted GFP+ IgM+ B cells in the spleen expressed CD21 and CD23 (Fig. 2 D). In contrast, the GFP+ IgM+ B cells reconstituted in the colon did not express CD21 and CD23 (Fig. 2 D).
Figure 2.

Direct maturation of AA4.1+ immature B cells to AA4.1− intestinal B cells. (A) Intestinal inflammation was induced by administration of 4% DSS in drinking water and the treatment was terminated by changing the DSS-containing water to normal water at day 4. Expression of AA4.1 versus B220 (top) in total large intestinal cells (day 8) and CD23 versus IgM expression on the gated AA4.1+ B220+ cells (bottom; n = 12) is shown. (B) 1 mg BrdU was administered (intraperitoneally) into intact WT mice twice with a 24-h interval between injections, and the mice were killed at 2 and 6 d after initial BrdU injection. The B220 versus BrdU expressions on the gated AA4.1+ (top) or AA4.1− (bottom) IgM+ cells from large intestine are shown. The data are representative of three individual experiments. (C and D) Purified AA4.1+ B220+ B cells from the spleen of GFP transgenic mice (left) were intravenously transferred into WT mice with or without prior DSS exposure. 6 d after cell transfer, the recipient mice were killed. GFP- and IgM-expressing B cells in the recipient colon and spleen were identified (C). The expression of CD21 and CD23 versus IgM on the gated GFP+ cells in the spleen and colon of recipient mice without DSS treatment is shown in D. The data are representative of three to six individual recipients/group. (E and F) Purified IgDhigh B cells from the spleen of GFP transgenic mice were intravenously transferred into WT mice that had being treated without (top) or with DSS (bottom). 4 d after cell transfer, the recipient mice were killed. The expressions of GFP versus IgM in recipient spleen (left) and colon (right) are shown in (E). The expressions of CD21 or CD23 on the gated GFP+ cells from the spleen (left) and large intestine (right) of DSS treated mice are shown in (F). The data are representative of three individual recipients.
Naive follicular B cells, which mature from AA4.1+ immature B cells, have the ability to recirculate in search of antigens (36). Therefore, we next examined the involvement of recirculating naive B cells in the development of intestinal B cells. To do so, we purified IgDhigh naive recirculating B cells from the spleen of GFP transgenic mice and transferred them (5 × 106) intravenously into WT mice with or without induction of DSS colitis. 4 d after cell transfer, donor-derived GFP+ IgM+ B cells were detected in the spleen of both recipient mouse groups (Fig. 2 E, left). In contrast, GFP+ IgM+ B cells were detectable in the inflamed colon (DSS-treated), but not in normal colon (Fig. 2 E, right). In addition, the reconstituted GFP+ IgM+ B cells in the spleen had a CD21+ CD23+ phenotype, whereas the GFP+ IgM+ B cells in the colon were CD21−CD23− (Fig. 2 F). These data, together with Fig. 2 (B and C) and Fig. S2, support the view that AA4.1− intestinal B cells directly or indirectly mature from AA4.1+ immature B cells in the steady state and that intestinal inflammatory stimuli induce the recruitment of IgDhigh naive recirculating B cells to the intestine, resulting in the significant increase in IgM+ B cells within the inflamed intestine.
Intestinal B cells exhibit a unique activated phenotype
Because some intestinal B cells originate from the recirculating naive B cell pool, particularly under inflammatory conditions, we next tested whether the intestinal B cells phenotypically resemble naive or activated B cells. The expression of several activation markers (16) was analyzed. Interestingly, the IgM+ intestinal B cells were characterized by remarkably high expression levels of MHC class II in comparison to splenic B cells (Fig. 3 A). The expression levels of MHC class II were comparable on B cells from normal and inflamed intestine (Fig. 3 A). In addition, a slight increase in the expression of CD69 was observed on the intestinal B cells in comparison to splenic B cells (Fig. 3 A). In contrast, no significant up-regulation of Fas, PNA, CD80, or CD86 was found on the intestinal B cells. Therefore, it is possible that intestinal B cells represent a unique activated B cell population that is MHCIIbright, CD69intermediate, Faslow, PNAlow, CD80−, and CD86low. To elucidate whether the phenotype of intestinal B cells represents a response to an intestinal pathogen, and to determine if these cells can be further activated by pathogens, we examined intestinal B cells after infection with Citrobacter rodentium. This infection did not induce the expression of Fas or PNA on intestinal B cells (Fig. 3 B). These data suggest that the MHCIIbright intestinal IgM+ B cell population is not elicited by a known intestinal pathogen, and these cells may represent a unique preactivated B cell population.
Figure 3.

Unique activated status of intestinal IgM+ B cells. (A) Expressions of IgM versus MHC class II, CD69, Fas, PNA, CD80, or CD86 in cells of the spleen and colon from WT mice (Day 0) and WT mice during recovery phase of DSS colitis (Day 8) are shown. The data are representative of three individual mice. (B) Mice were infected with C. rodentium and killed 6 and 10 d after infection. Expressions of IgM versus PNA or Fas on cells from the normal colon (left) and the infected colon (right) are shown. The data are representative of three individual mice/group. Representative expression patter was observed on the intestinal B cells 6 d after infection (not depicted).
Functional property of IgM+ B cells in the large intestine
Because the intestine is a major source of IgA-plasma cells (17–20, 23–25), the differentiation of activated intestinal B cells into IgA plasma cells was next investigated. Interestingly, although IgM+ B cells were markedly increased in the inflamed mucosa during the recovery phase of DSS colitis, no significant increase in the proportion of B220−IgA+ or CD138+ plasma cells was observed at this site (Fig. 4 A). ELISPOT assay also showed that the absolute number of IgA-secreting cells among total intestinal cells decreased during the recovery phase of DSS colitis (day 8; Fig. 4, B and C). In addition, expression of activation-induced deaminase an enzyme required for class switch recombination and SHM (37, 38) was slightly lower in the IgM+ B cells of the large intestine when compared with splenic IgM+ B cells (Fig. 4 D). To analyze the frequency of SHM, genomic DNA was isolated from purified IgM+ B cells from the large intestine and the spleen of WT mice and subjected to PCR/DNA sequence analysis using a primer set that amplifies rearranged Vκ4-Jκ5 light chain segments of genomic DNA (39). There was no significant restriction of Vκ4 subfamily usages between intestinal and splenic IgM+ B cells. Interestingly, the frequency of SHM within the Vκ4 segments was significantly lower in intestinal IgM+ B cells compared with splenic IgM+ B cells (Fig. 4, E and F). These findings suggest that IgM+ B cells do not appear to contribute to IgA-producing plasma cells in the large intestine, and they are characterized by the low frequency of SHM.
Figure 4.

Large intestinal IgM+ B cells are characterized by low frequency of SHM and by production of IL-12 in response to CpG. (A) Expressions of IgA and CD138 versus B220 on large intestinal cells from day 0 (n = 6) and day 8 (n = 6) of DSS colitis are shown. (B and C) ELISPOT assay shows the number of IgA-secreting cells among total large intestinal cells at day 0 and 8 of DSS colitis (B). The data are summarized in C. (D) Expressions of activation-induced deaminase in purified B cells from the colon of day 0 (open bar, n = 16) and day 8 (gray bar, n = 15) after DSS treatment and from the spleen of day 0 (black bar, n = 8) are shown. *, P < 0.1; **, P < 0.05. (E) Analysis of sequences with mutations (splenic and colonic IgM+ B cells of normal WT mice) is depicted by pie charts. Numbers outside of each pie chart are the number of mutations/clone. The size of the wedge is proportional to the percentage of clones carrying that number of mutations. Inside each inner circle is the number of clones sequenced. Data from an individual spleen (as control) and two different normal large intestines are shown. (F) Mutation frequencies are calculated by the number of mutations per 104 bp from splenic and large intestinal IgM+ B cells of WT mice. (G) Cells from the normal large intestine and spleen of WT mice were stimulated with 1 μM CpG for 17 h, including a 7-h culture period with GolgiStop. The stimulated cells were subjected to surface staining for detection of IgM and B220 and intracellular staining for detection of IL-12p70. Expression of IL-12p70 in the gated IgM+B220+ cell population is shown. (H) The mean percentage of IL-12–producing cells among IgM+ B cells from the large intestine (n = 6) and spleen (n = 6) of WT mice is shown. *, P < 0.001.
Several recent studies have suggested that, like T cells, B cells also possess the ability to produce several kinds of cytokines (40–43). We used real-time PCR to primarily screen B cells stimulated with ligands for pattern recognition receptors (including ligands for TLR 2, 4, 5, 7, and 9), and found that IL-12p35 mRNA expression was specifically induced in intestinal B cells after stimulation with CpG (unpublished data). Indeed, flow cytometric analysis clearly showed that IgM+B220+ B cells from the large intestine, but not the spleen, of WT mice produce a large amount of lL-12p70 in response to CpG (Fig. 4, G and H).
Antigen-independent polyclonal expansion of intestinal CD21−CD23− B cells
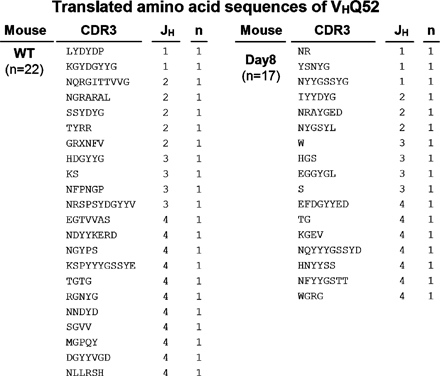
The intestine is constitutively exposed to a wide spectrum of enteric microorganisms (19). Therefore, we examined the clonality of B cells in the large intestine by analyzing the VDJ region of IgM heavy chains (VHJ558 and VHQ52) to test whether some specific enteric (microbial) antigens cause the expansion of intestinal B cells. There was no restricted diversity of the BCR repertoire of VHJ558 or VHQ52 within the B cells from the normal intestine (Fig. S3 and Fig. S4). Interestingly, a similar polyclonal pattern was also observed in B cells from the inflamed intestine of DSS colitis (Fig. S3 and Fig. S4). These findings suggest that the intestinal IgM+ B cells using VHJ558 and VHQ52, which together account for 65% of the BCR repertoire (44), expand polyclonally.
To further test the antigen specificity of intestinal B cell development, we generated IghelMD4 Tg x RAG1 KO mice; these mice contain only a monoclonal B cell population that expresses a restricted BCR specific for hen egg lysozyme. IgM+ B cells were still detected in the intestines of these mice in the absence of any inflammatory stimulus, and a marked expansion of these B cells occurred in the inflamed mucosa during the recovery phase from DSS-induced colitis (Fig. 5 A). As seen in WT mice, the expanded IgM+ B cells formed clusters within the inflamed mucosa (Fig. 5 B). In addition, the majority of IgM+ B cells were of the CD21−CD23− phenotype (Fig. 5 C). These findings suggest that antigen/BCR interactions may not be essential for intestinal CD21−CD23− B cell development. It however cannot be ruled out that some self-antigen that cross-reacts with hen egg lysozyme exists. To further explore the role of antigens and the BCR, we investigated KO mice deficient in Bruton's tyrosine kinase (Btk) or Lyn, both of which play a crucial role in the BCR signaling cascade (45–47). Interestingly, CD21−CD23−IgM+ B cells were detectable at normal levels in the intestines of these mice in the absence of DSS (Fig. 5 D), and they were significantly increased within the inflamed intestine during the recovery phase of DSS colitis (Fig. 5 E).
Figure 5.

Development of intestinal CD21−CD23− B cells through BCR-independent response without T cell help. (A) Absolute number of IgM+ cells in the large intestine from lghelMD4 tg x RAG1 KO mice at day 0 (normal, open bar; n = 10) and day 8 (recovery phase of DSS colitis, shaded bar; n = 10) are shown. (B) Colonic tissues from WT mice at day 8 and from lghelMD4 tg x RAG1 KO mice at days 0 and 8 were subjected to immunohistochemical analysis for the detection of IgM+ cells. The findings are representative of >12 individual experiments. (C) Expressions of IgM versus CD21 or CD23 on the gated B220+ cells of normal large intestine (Day 0) from lghelMD4 tg x RAG1 KO mice are shown. (D) Expressions of IgM versus CD21 or CD23 on the gated B220+ cells from normal large intestine of Btk KO mice (n = 5) and Lyn KO mice (n = 5) are shown. (E) Absolute numbers of large intestinal IgM+ cells from Btk KO and Lyn KO mice at day 0 (normal, open bar, n = 5) and day 8 of DSS colitis (recovery, solid bar, n = 5–7) are shown. (F) Inflamed intestinal mucosa (Day 8 of DSS colitis, top) and colonic patch within normal mucosa (bottom) of WT mice were subjected to immunohistochemical analysis for the detection of IgM (left), CD3 (middle), and FDC (right). (G) Expressions of IgM versus CD21 or CD23 on the gated B220+ cells of the normal large intestine from TCRβδ DKO mice are shown. (H) Absolute numbers of IgM+ cells in the large intestine of WR and TCRβδ DKO mice at day 0 (normal, open bar, n = 5–7) and day 8 (recovery of DSS colitis, solid bar; n = 5–7) are shown. (I) Large intestinal tissues of TCRβδ DKO mice at day 8 of DSS colitis were subjected to immunohistochemical analysis for the detection of IgM+ cells. Statistical significances are indicated in by ** (P < 0.001). Bars, 100 μm.
T cell help is not required for intestinal CD21−CD23− B cell development or expansion
Interestingly, expansion of intestinal B cells after an inflammatory stimulus resulted in the formation of B cell clusters with a unique pattern of T cell localization (Fig. 5 F); T cells were present around B cell clusters rather than in well-defined T cell areas, as normally seen in resident organized lymphoid tissues such as colonic patches (Fig. 5 F) (48). In addition, follicular dendritic cells (FDCs) were not detectable within the B cell clusters, whereas scattered FDCs were normally present within the follicles of organized lymphoid tissues (Fig. 5 F). Therefore, we next examined the role of T cells during intestinal B cell development by using TCRβδ double KO (DKO) mice that have no TCRαβ or TCRγδ T cells. Just as was seen in WT mice, CD21−CD23− B cells represented the majority of IgM+ B cells in the intestine of TCRβδ DKO mice (Fig. 5 G). Interestingly, intestinal B cells were significantly increased in TCRβδ DKO mice as compared with WT mice both in the absence of inflammation and under inflammatory conditions (Fig. 5 H). Large IgM+ B cell clusters were also recognized in the inflamed intestine of TCRβδ DKO mice (Fig. 5 I). These findings suggest that T cells are not required for intestinal CD21−CD23− B cell development in a state of health and during inflammation, but may contribute to the suppression of the development or recruitment of intestinal IgM+ B cells.
Intestinal CD21−CD23− B cells develop in the absence of both the spleen and organized lymphoid tissues
The LT-α1β2 signaling cascade is required for the development of GALT (20–22). Because some intestinal IgA+ plasma cells have been shown to be derived from GALT, particularly from PP (17, 20, 23), we examined the role of GALT in CD21−CD23− B cell development by using LTα KO mice that lack GALT (20–22). Interestingly, CD21−CD23− B cells were still detectable in LTα KO mice (Fig. 6 A), and a marked expansion of these B cells was induced in the inflamed intestine after exposure to DSS (Fig. 6, B and C). IgM+ B cell clusters were still detectable in LTα KO mice (Fig. 6 D). These findings indicate that intestinal CD21−CD23− B cells develop even in the absence of GALT, which is consistent with previous reports showing that naive B cells can directly migrate into the small intestinal mucosa through a pathway different from GALT-primed B cells (24, 25).
Figure 6.

Intestinal CD21−CD23− B cell development is independent of spleen and organization lymphoid tissues. Intestinal inflammation was induced in WT (Day 0, n = 8; Day 8, n = 8) and LTα KO (Day 0, n = 5; Day 8, n = 5) and splenectomized LTα (SP[-] LTα; Day 0, n = 5; Day 8, n = 5) mice. (A) Expressions of IgM versus CD21 or CD23 on large intestinal cells from LTα KO mice are shown. (B) Proportions (IgM/CD3ε) in large intestinal cells at day 8 are shown. (C) Absolute numbers of large intestinal IgM+ cells at day 0 (normal, open bar) and day 8 (recovery, shaded bar) are shown. (D and E) The inflamed intestine from LTα KO (D) and splenectomized LTα KO (E) mice were subjected to immunohistochemical analysis for the detection of IgM+ cells. (F) Expressions of IgM versus CD21 or CD23 on the gated B220+ cells from the normal colon (Day 0) of splenectomized LTα KO mice are shown. Bars, 100 μm.
Peripheral B cells complete their maturation through transitional stages in the spleen (1–6, 8, 34). Like recirculating follicular B cells, intestinal AA4.1−CD21−CD23− B cells also appear to mature from AA4.1+ immature B cells. Therefore, to test the role of the splenic environment during intestinal B cell development, we used splenectomized LTα KO mice in which both the spleen and GALT were absent. Like WT mice, the majority of intestinal B cells in splenectomized LTα KO mice were of the CD21−CD23− phenotype (Fig. 6 F), and a marked expansion of these B cells with cluster formation was observed during the recovery from DSS colitis (Fig. 6, C and E). These results indicate that neither the spleen nor GALT is required for the development of intestinal CD21−CD23− B cells, and that in the absence of spleen, the intestinal B cells may be recruited from recirculating naive B cells that mature in the BM (7) and/or from BM-derived immature AA4.1 B cells.
Insufficient evidence to suggest a linkage between intestinal CD21−CD23− B cells and B1 lineage cells
IgA+ plasma cells in the small intestine have previously been demonstrated to originate from peritoneal B1 cells (18, 23). Therefore, we sought to determine if CD21−CD23− B cells in the large intestine might be derived from B1 B cells. To test this possibility, RAG-1 KO mice were reconstituted with peritoneal CD11b+ cells from WT mice, which include B1 cells and macrophages, and the mice were subsequently treated with DSS. Although CD11b+ IgM+ B1 cells could be recognized in the peritoneal cavity of the recipient RAG KO mice, no IgM+ cells were detectable in the inflamed intestine (Fig. 7 A). We next examined IL-7 receptor α (IL-7R) KO mice in which the development of B2, but not B1, cells is impaired (49, 50) and Rac2 (guanosine triphosphatase) KO mice that show the impaired development of B1 cells (51). Only a few IgM+ cells were detectable in the normal and inflamed intestine of IL-7R KO mice (Fig. 7, B and C). In addition, no B cell clusters were observed in the inflamed mucosa of IL-7R KO mice exposed to DSS (Fig. 7 D). In contrast, there were similar numbers of IgM+ B cells in the normal intestine between Rac2 KO and WT mice, and significant B cell expansion was observed in the inflamed mucosa of both mouse groups after exposure to DSS (Fig. 7, B and C). These findings, together with the phenotype of intestinal B cells (CD11b−, CD5−, and CD43−) shown in Fig. 1 E, and the data from Btk KO mice, all suggest that the majority of IgM+ B cells in the large intestine may originate from conventional B2 lineage cells.
Figure 7.

Intestinal CD21−CD23− IgM+ B cells are not associated with B1 lineage cell. (A) RAG1 KO mice that had been reconstituted with peritoneal B1 cells from WT mice were treated with 4% DSS for 4 d and killed at day 8. The DSS-treated recipient mice show sufficient reconstitution of CD11blow IgM+ B 1 cells in the peritoneal cavity (PC, left), whereas no IgM+ B cells are detectable in the inflamed mucosa (LP, right) of these mice. The data are representative of four individual recipient mice. (B–D) Intestinal inflammation was induced in WT (Day 0, n = 6; Day 8, n = 6), Rac2 KO (Day 0, n = 5; Day 8, n = 5), and IL-7Rα KO (Day 0, n = 5; Day 8, n = 5) mice by oral administration of 4% DSS for 4 d. Proportion (IgM/CD3ε) at day 8 (B) and absolute number of IgM+ cells in the large intestine at day 0 (normal, open bar) and day 8 (recovery, shaded bar; C) are shown. The large intestine from IL-7Rα KO mice at day 8 was subjected to immunohistochemical analysis for detection of IgM+ cells (D). Presence of statistical significance (P < 0.001) compared with day 8 WT mice is indicated by **. Bars, 100 μm.
Dependence of intestinal CD21−CD23− B cells on BAFF-R signaling cascade
The rapid expansion of intestinal B cells by DSS-induced intestinal inflammation provided an opportunity to closely examine the molecular events involved in intestinal B cell development and/or recruitment. We therefore performed a real-time PCR-based screening approach using purified intestinal IgM+ B cells from day 0 (normal), 4 (acute colitis), and 8 (recovery phase of colitis with B cell expansion) of DSS colitis to analyze the expression of 276 molecules that have been demonstrated to play crucial roles in the genesis, differentiation, maturation, apoptosis/survival, homing and activation of B cells (1–6, 46, 47, 52, 53). We found that receptor of B cell activation factor from the TNF family (BAFF-R) is specifically up-regulated in intestinal B cells on day 8 as compared with day 4 and 0 (Fig. 8 A). In contrast, intestinal IgM+ B cells did not express BCMA (B cell maturation antigen) during the course of inflammation (Fig. 8 A). BAFF-R engagement promotes processing of NF-κB2 protein p100 to p52, which is dependent on NF-κB–inducing kinase (NIK) (54, 55). Recent studies indicate that activation of the BAFF-R signaling cascade up-regulates Pim2 (a prosurvival kinase) and Bim (a BH3-only member of the Bcl-2 family) (56), and that BAFF-R signaling is negatively regulated by an adaptor protein, Act1 (57). Indeed, a significant up-regulation of NIK, Pim2, and Bim was observed in purified intestinal IgM+ B cells during the recovery, but not the acute, phase of intestinal inflammation (Fig. 8 A). In contrast, expression of Act-1 was significantly down-regulated in B cells during the course (both day 4 and 8) of this inflammation (Fig. 8 A). In addition, enhanced processing of NF-κB2 protein p100 to p52 in intestinal B cells was specifically observed in the recovery phase of inflammation (Fig. 8 B).
Figure 8.

Requirement of BAFF-R–mediated signaling for intestinal CD21−CD23− B cell development. (A) Expressions of BAFF signaling-related molecules in purified B cells from the large intestine of day 0 (open bars), 4 (gray bars), and 8 (black bars) after DSS treatment and from the spleen of day 0 (hatched bars) are shown. The results represent the means ± the SD from six individual mice in each group. *, P < 0.00 1 (comparison between colonic B cells at day 0 versus day 8). (B) Western blot analysis shows the expression levels of NF-κB2 p100 and p52 in purified B cells (>97% are IgM+) of the large intestine from 4–30 pooled WT mice at day 0 (D0, normal) and 8 (D8, recovery of DSS colitis), and of the spleen from day 0. The data are representative of two individual experiments. (C) Expressions of IgM versus CD21 or CD23 on the gated B220+ cells from normal large intestine of control and BAFF-R mutant mice are shown. (D–G) Intestinal inflammation was induced in BAFF-R mutant (A/WySnJ; Day 0, n = 5; Day 8, n = 16) and control (A/J) mice (Day 0, n = 5; Day 8, n = 10) by oral administration of 4% DSS for 4 d. (D and E) Proportion (CD3/IgM; D) and absolute numbers of IgM+ B cells (E) in the colon of control and BAFF-R mutant mice at day 8 of DSS colitis are shown. Presence of statistically significant (P < 0.001) compared with control mice at day 8 is indicated by **. (F and G) Large intestine from control mice (F) and BAFF-R mutant mice (G) at day 8 were subjected to immunohistochemical analysis for the detection of IgM+ cells. (H) Apoptosis of intestinal IgM+ B cells in control mice and BAFF-R mutant mice at day 8 of DSS colitis as judged by Annexin V staining is shown. The mean percentages of Annexin V+ cells among IgM+ cells are 36.6 ± 1.09% in control mice (n = 7) and 75.6 ± 1.64% in BAFF-R mutant mice (n = 7; P < 0.001). (I) Expression of AA4.1 versus B220 on the cells from normal intestine of control (left) and BAFF-R mutant (right) are shown. Bars, 100 μm.
To further examine the role of the BAFF-R signaling cascade in intestinal CD21−CD23− B cell development, we analyzed BAFF-R mutant (A/WySnJ) and A/J (control) mice (54–56, 58, 59). Interestingly, intestinal B cells exhibiting CD21−CD23− phenotype were significantly reduced in BAFF-R mutant mice as compared with control mice (Fig. 8 C). In addition, DSS-induced colitis failed to induce a significant expansion of IgM+CD21−CD23− B cells in BAFF-R mutant mice (Fig. 8, D and E). Indeed, B cell clusters within the inflamed mucosa were detectable in control (Fig. 8 F) but not BAFF-R mutant mice (Fig. 8 G). In addition, significantly enhanced apoptosis of intestinal B cells was observed in BAFF-R mutant as compared with control mice (Fig. 8 H), which is consistent with an important role of BAFF-R signaling in B cell survival (54–59). In addition, immature AA4.1+ B cells in the large intestine were significantly reduced in BAFF-R mutant mice as compared with control mice (Fig. 8 I). These findings suggest that the BAFF-R–associated signaling pathway plays a crucial role in intestinal B cell development and homeostasis. They are consistent with a role for BAFF-R in all B2 populations after the T1 stage and the absence of a role for BAFF-R in B1 B cells (58, 60).
DISCUSSION
In this study, we have described a previously unidentified, intestine-specific, IgM+ B cell subset that is characterized by a unique AA4.1−CD21−CD23−MHCIIbright surface phenotype, low frequency of SHM, and the ability to secrete IL-12 in response to a TLR ligand. This B cell subset exists in the normal mucosa of the large intestine and expands in the inflamed mucosa of mice with acute or chronic colitis. The LTα1β2 signaling cascade is required for the development of organized lymphoid tissues (such as PP) that provide a site of origin of some of IgA+ plasma cells in small intestine (17, 18, 20, 23, 24). Most other small intestinal plasma cells originate from peritoneal B1 cells (17, 18, 20, 23, 24, 26–28). We herein describe an additional pathway for intestinal IgM+ B cell development, in which AA4.1−CD21−CD23− B cells in the large intestine are supplied from B2 lineage cells that do not require organized lymphoid tissues for their development. Indeed, recent studies have revealed that some IgM+ B cells in the small intestine are derived from the BM through a pathway distinct from that used by gut-primed B cells (24, 25). Although the phenotype of BM-derived small intestinal B cells has not been characterized in previous studies, it is possible that the AA4.1−CD21−CD23−MHCIIbright B cells in the large intestine characterized in this study may have a counterpart in the small intestine.
Recirculating B cells mature through transitional stages in the spleen, as well as the BM (2, 3, 5, 8). Our BrdU labeling and cell transfer studies suggest that intestinal AA4.1−IgM+ B cells can be derived from the AA4.1+ immature B cell pool. In addition, intestinal AA4.1−CD23− B cells were virtually absent in BAFF-R mutant mice in which development of AA4.1+ CD23+ T2 transitional B cells is defective and T1 transitional/newly formed B cells accumulate (61–63). Therefore, it is likely that recirculating T2 B cells that mature from T1 cells in the spleen seed the intestinal IgM+CD21− B cell compartment in the steady state, down-regulating CD23 when they enter the intestinal milieu. Soon after they seed the intestine, they may continue to express AA4.1, which is a marker of recent maturation. In addition, CD21−CD23− B cells may accumulate in the intestine after the recruitment to the inflamed intestine of cells from the recirculating IgDhigh naive B cell pool that contains both T2 cells and more mature recirculating follicular B cells. Therefore, it may be inferred that inflammatory chemokines and cytokines participate in the recruitment of recirculating B cells to the intestine during inflammation.
Peripheral B cell maturation in the spleen and BM depends in part on BCR-linked positive selection events (2, 5, 9). Interestingly, although inflammatory conditions generally induce a restriction of BCR clonality (12), our data showed that intestinal AA4.1−IgM+ B cells polyclonally expand even in the presence of intestinal inflammation (Fig. S3 and Fig. 4). In addition, B cells with a restricted BCR expanded readily in normal and inflamed intestine (Fig. 3). Furthermore, T cell help was not required for the intestinal AA4.1−IgM+ B cell development. Importantly, a large number of intestinal AA4.1−IgM+ B cells were still detectable even in the absence of Btk or Lyn, which are major signaling molecules involved in BCR-mediated signaling. In addition, SHM that is often induced in mature B cells after encounter with specific antigens (38) was detectable at very low frequency in the intestinal IgM+ B cells. These data suggest that intestinal AA4.1−IgM+ B cells that are constitutively exposed to enteric microorganisms may develop or expand through a BCR-independent pathway.
Our data demonstrated that intestinal IgM+ B cells express remarkably high levels of MHC class II. In contrast, several conventional markers expressed on activated splenic B cells, were not expressed by intestinal B cells. The unique expression profile of activation markers was not altered under several intestinal inflammatory conditions, including spontaneous chronic colitis, a chemically induced acute colitis, and Citrobacter rodentium infection. Therefore, it is possible that the intestinal IgM+ B cell population may represent a unique preactivated B cell population presumably caused by the constitutive exposure to enteric microorganisms in the intestine. B cells have generally been believed not to produce IL-12p70, as this cytokine was detectable only in EBV-transformed B cells but not normal systemic B cells (64). However, recent accumulating studies have identified the potential ability of B cells to produce IL-12p70 under certain inflammatory and stimulatory (e.g., combination of CpG and CD40) conditions (41–43). Interestingly, intestinal IgM+ B cells showed the ability to easily produce IL-12p70 in response to a microbial product CpG. Because IL-12p70 is a well known cytokine required for safeguarding against microorganisms (65), it is possible that the intestinal IgM+ B cells may contribute to preserve appropriate host–microbe interaction.
In summary, we have identified an intestine-specific AA4.1−CD21−CD23−MHCIIbright B cell subset that is derived from AA4.1+ immature B cells and from recirculating naive B cells. This maturation of these cells represents a distinct pathway of B cell development that occurs in a tissue site, rather than in the BM or the spleen.
MATERIALS AND METHODS
Mice.
TCRβδ DKO, RAG-1 KO, IL-7Rα KO, Rac2 KO, Lyn KO, Btk KO, LTα KO, IghelMD4 Tg, GFP Tg mice of C57BL/6 background, BAFF-R mutant (A/WySnJ), and control (A/J) mice were obtained from The Jackson Laboratory and maintained in specific pathogen–free facilities at Massachusetts General Hospital. IghelMD4 Tg mice were crossed with RAG1 KO mice as previously described (40) to generate a mouse strain that possesses monoclonal B cells. Splenectomy and sham operations were performed on anesthetized LTα KO mice at 3 d after birth, as previously described (35). Mice were used according to the protocol approved by the Massachusetts General Hospital Animal Care and Use Committee.
DSS-induced intestinal inflammation and C. rodentium infection.
Female (10–12 wk of age) mice were administered 4% DSS (MW: 36,000–50,000, ICN Biomedicals) in drinking water for 4 d, and the DSS administration was then terminated to induce recovery from acute inflammation by changing the DSS water to normal drinking water as previously described (33). These mice were killed on day 0, 4, 8, 14, and 30 after induction of DSS-induced acute intestinal inflammation. Mice were orally inoculated with C. rodentium (strain DBS100; American Type Culture Collection), as previously described (66). Bacteria were grown overnight in Luria broth and resuspended in PBS before infecting mice (0.5 ml/mouse; ∼5 × 108 CFU of C. rodentium). Mice were killed on day 5 and 10 after infection.
Cell preparation and B cell isolation.
Large intestine and parts of small intestine containing PP were incubated with 1.5 mg/ml dispase to remove epithelial cells. The remaining tissues were then incubated with 0.8 mg/ml collagenase for 20 min. After passing them through a glass wool column, the mucosal cells were further enriched by Percoll gradient (40/72%) centrifugation. MLNs and spleen were dispersed using 27-gauge needles, and red blood cells were removed using ACK solution. For purifying B cells, cells were incubated with PE-conjugated anti-Ig (Invitrogen) at 4°C for 30 min, and then incubated with the anti-PE microbeads at 4°C for 30 min. After washing, the labeled cells were isolated using the MACS system (Miltenyi Biotec).
Cell transfer.
Immature AA4.1+ B220+ B cells or recirculating naive IgDhigh B cells were purified from the spleens of GFP transgenic mice (5–20 mice). After enrichment of B cells using the MACS system, B cell populations were purified using flow cytometry and sorting. 5 × 106 purified B cells were intravenously transferred into WT mice with or without DSS treatment. Peritoneal CD11b+ cells were purified from pooled WT mice (day 5) using a magnetic cell separation (MACS) system (Miltenyi Biotec). 5 × 106 purified cells were transferred intraperitoneally into recipient RAG1 KO mice that subsequently received DSS orally.
Phenotypic analysis.
Cells were isolated from the large intestine (excluding the cecum), MLN, spleen, PP, and BM as previously described (33). The mAbs used were FITC-, PE-, PerCP-, APC-, or biotin (followed by incubation with PerCP-streptavidin)-conjugated anti-CD1d (1B1), -CD3ε (145-2C11), -CD5 (53–7.3), -CD9 (KMC8), -CD11b (M1/70), -CD21 (7G6), -CD23 (B3B4), -CD24 (M1/69), -CD43 (S7), -CD69 (H1,2F3), -B220 (RA3-6B2), -CD62L (hen egg lysozyme-14), -CD138 (282–2), -IgD (11-26C2a), -Fas (Jo2; BD Biosciences), -IgA (SouthernBiotech), -AA4.1, -CD80 (16-10A1), -CD86 (GL1), MHC-II (M5/114, 15.2; eBioscience), -Igμ (Caltag), -IL-12p70 (BioLegend), and FITC-PNA (Vector Laboratories). Annexin V staining was performed according to the manufacturer's instructions (BD Biosciences). BrdU pulse labeling and nuclear staining for the detection of BrdU+ cells were performed as previously described (35). Immunohistochemical staining was performed using the avidin–biotin complex method, as previously described (67). The mAbs used were purified anti-CD3ε, -IgM, -IgD, and -FDC.
Intracellular cytokine staining.
Spleen and large intestinal cells were stimulated either with or without 1 μM CpG (InvivoGen) for 17 h. GolgiStop (BD Bioscience) was added during the last 7 h. The cells were first stained with FITC-conjugated anti-B220 (RA3-6B2) and APC-conjugated anti-Igμ antibody, and then they were fixed and permeabilized with Cytofix/Cytoperm solution. The cells were then stained intracellularly with PE-conjugated anti–IL-12p70. After washing, the cells were immediately subjected to flow cytometric analysis.
Ig secretion and proliferation of B cells.
Cells were subjected to ELISPOT assay for the detection of IgA-secreting cells as previously described (32). For proliferation assay, B cells were enriched by negative sorting for depletion of CD3-, Gr1-, DX5-, and CD11c-expressing cells, as previously described (40). The enriched B cells (5 × 104/well) were incubated with 50 μg/ml F(ab')2 goat anti-IgM (Jackson ImmunoResearch Laboratories) or LPS (5 μg/ml, Invivogen) for 48 h, as previously described (34). After pulsing with 1 μCi of [3H] thymidine for 18 h, uptake was assessed by scintillation counting.
Analysis of SHM.
SHM analysis was performed as previously described (39). Genomic DNA was prepared from purified IgM+ B cells using a DNeasy Tissue kit (QIAGEN) and amplified using a Vκ4 light chain primer set (Vκ4-68F: 5′-GATTTTCAGCTTCCTGCTAATGAGTGCC-3′ and Jκ5int3-R; 5′-TGATAATGAGCCCTCTCCAT-3′) and Platinum Taq DNA polymerase High Fidelity (Invitrogen). Amplified products were cloned into pCR2.1-TOPO vector (Invitrogen) and subjected to DNA sequence analysis.
Quantitative real-time PCR.
After RNA purification from the purified B cells using QIAshredder and RNeasy Mini kit (QIAGEN), the first strand of the cDNA was synthesized by SuperScript (Invitrogen) according to the manufacturer's instruction. Real-time PCR was performed as previously described (33). cDNA was initially amplified by using SYBR green PCR core reagent kit (Stratagene) with a primer set specific for β-actin. The real-time PCR reaction was performed by MX3000p QPCR machine (Stratagene), and the cDNA concentration was normalized as the CT value of β-actin. The normalized cDNA samples were subsequently subjected to real-time PCR with Rox dye as a reference dye for the measurement of expression levels of specific molecules.
BCR repertoire.
For DNA sequence of CDR3 within IgM heavy chain, cDNA from purified B cells were amplified using VHJ558 or VHQ52 and Cμ primer set, as previously described (68). The amplified products were cloned using TOPO-TA cloning system. Positive clones that were randomly picked up were subjected to DNA sequence.
Detection of NF-κB2.
After B cells were positively purified from spleen and colon of pooled mice (4–30 mice) under MACS system, purified IgM+ cell (2 × 106) B cells were immediately lysed with RIPA buffer. 3.5 μg of protein was subjected to immunoblot using rabbit polyclonal anti-p52 (k-27; Santa Cruz Biotechnology, Inc.) and anti–rabbit HRP (Thermo Fisher Scientific), as previously described (55).
Online supplemental material.
Fig. S1 shows the expression of B1 cell markers on colonic IgM+ B cells. Fig. S2 shows the BrdU-incorporated AA4.1+ and AA4.1−IgM+ cells under inflammatory condition. Fig. S3 shows BCR repertoire (VHJ558) of colonic B cells in steady state and under inflammatory condition. Fig. S4 shows the BCR repertoire (VHQ52) of colonic B cells in steady state and under inflammatory condition. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071572/DC1.
Supplementary Material
Acknowledgments
We thank Drs. Deanna Nguyen, Katsunori Shirane, and Kiyotaka Nagahama for helpful discussion, and Ms. Ichiko Kata for technical assistance.
This study was supported mainly by National Institutes of Health grant DK64351 (A. Mizoguchi) and partially by the Eli and Edythe L. Broad Foundation (A. Mizoguchi), DK47677 (A.K. Bhan), and DK64289 (E. Mizoguchi) and Center for the Study of Inflammatory Bowel Disease at Massachussetts General Hospital.
The authors have no conflicting financial interests.
Abbreviations used: BAFF-R, receptor of B cell activation factor from the TNF family; Btk, Bruton's tyrosine kinase; DSS, dextran sodium sulfate; FDC, follicular dendritic cell; GALT, gut-associated lymphoid tissue; LT, lymphotoxin; MLN, mesenteric lymph node; PP, Peyer's patch; SHM, somatic hypermutation.
References
- 1.Hardy, R.R., and K. Hayakawa. 2001. B cell development pathways. Annu. Rev. Immunol. 19:595–621. [DOI] [PubMed] [Google Scholar]
- 2.Su, T.T., B. Guo, B. Wei, J. Braun, and D. Rawlings. 2004. Signaling in transitional type 2 B cells is critical for peripheral B-cell development. Immunol. Rev. 197:161–178. [DOI] [PubMed] [Google Scholar]
- 3.Allman, D., B. Srivastava, and R.C. Lindsley. 2004. Alternative routes to maturity: branch points and pathways for generating follicular and marginal zone B cells. Immunol. Rev. 197:147–160. [DOI] [PubMed] [Google Scholar]
- 4.Cancro, M.P. 2004. Peripheral B-cell maturation: the intersection of selection and homeostasis. Immunol. Rev. 197:89–101. [DOI] [PubMed] [Google Scholar]
- 5.Pillai, S., A. Cariappa, and S. Moran. 2005. Marginal zone B cells. Annu. Rev. Immunol. 23:161–196. [DOI] [PubMed] [Google Scholar]
- 6.Matthias, P., and A. Rolink. 2005. Transcriptional networks in developing and mature B cells. Nat. Rev. Immunol. 5:497–508. [DOI] [PubMed] [Google Scholar]
- 7.Cariappa, A., C. Chase, H. Liu, P. Russell, and S. Pillai. 2007. Naive recirculating B cells mature simultaneously in the spleen and bone marrow. Blood. 109:2339–2345. [DOI] [PubMed] [Google Scholar]
- 8.Loder, F., B. Mutschler, R. Ray, C. Paige, P. Sideras, R. Torres, M. Lamers, and R. Carsetti. 1999. B cell development in the spleen takes place in discrete steps and is determined by the quality of B cell receptor-derived signals. J. Exp. Med. 190:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wen, L., J. Brill-Dashoff, S. Shinton, M. Asano, R. Hardy, and K. Hayakawa. 2005. Evidence of marginal-zone B cell-positive selection in spleen. Immunity. 23:297–308. [DOI] [PubMed] [Google Scholar]
- 10.Merrell, K.T., R.J. Benschop, S.B. Gauld, K. Aviszus, D. Decote-Ricardo, L.J. Wysocki, and J.C. Cambier. 2006. Identification of anergic B cells within a wild-type repertoire. Immunity. 25:953–962. [DOI] [PubMed] [Google Scholar]
- 11.Ruddle, N.H. 1999. Lymphoid neo-organogenesis: lymphotoxin's role in inflammation and development. Immunol. Res. 19:119–125. [DOI] [PubMed] [Google Scholar]
- 12.Schroder, A.E., A. Greiner, C. Seyfert, and C. Berek. 1996. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 93:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cupedo, T., W. Jansen, G. Kraal, and R. Mebius. 2004. Induction of secondary and tertiary lymphoid structures in the skin. Immunity. 21:655–667. [DOI] [PubMed] [Google Scholar]
- 14.Ueda, Y., K. Yang, S. Foster, M. Kondo, and G. Kelsoe. 2004. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J. Exp. Med. 199:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagaoka, H., G. Gonzalez-Aseguinolaza, M. Tsuji, and M. Nussenzweig. 2000. Immunization and infection change the number of recombination activating gene (RAG)-expressing B cells in the periphery by altering immature lymphocyte production. J. Exp. Med. 191:2113–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cariappa, A., I. Mazo, C. Chase, H. Shi, H. Liu, Q. Li, H. Rose, H. Leung, B. Cherayil, P. Russell, U. von Andrian, and S. Pillai. 2005. Perisinusoidal B cells in the bone marrow participate in T-independent responses to blood-borne microbes. Immunity. 23:397–407. [DOI] [PubMed] [Google Scholar]
- 17.Brandtzaeg, P., and F.E. Johansen. 2005. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol. Rev. 206:32–63. [DOI] [PubMed] [Google Scholar]
- 18.Fagarasan, S., and T. Honjo. 2003. Intestinal IgA synthesis: regulation of front-line body defences. Nat. Rev. Immunol. 3:63–72. [DOI] [PubMed] [Google Scholar]
- 19.Macdonald, T.T., and G. Monteleone. 2005. Immunity, inflammation, and allergy in the gut. Science. 307:1920–1925. [DOI] [PubMed] [Google Scholar]
- 20.Mowat, A.M. 2003. Anatomical basis of tolerance and immunity to intestinal antigens. Nat. Rev. Immunol. 3:331–341. [DOI] [PubMed] [Google Scholar]
- 21.Neutra, M.R., N. Mantis, and J. Kraehenbuhl. 2001. Collaboration of epithelial cells with organized mucosal lymphoid tissues. Nat. Immunol. 2:1004–1009. [DOI] [PubMed] [Google Scholar]
- 22.Hamada, H., T. Hiroi, Y. Nishiyama, H. Takahashi, Y. Masunaga, S. Hachimura, S. Kaminogawa, H. Takahashi-Iwanaga, T. Iwanaga, H. Kiyono, et al. 2002. Identification of multiple isolated lymphoid follicles on the antimesenteric wall of the mouse small intestine. J. Immunol. 168:57–64. [DOI] [PubMed] [Google Scholar]
- 23.Macpherson, A.J., D. Gatto, E. Sainsbury, G. Harriman, H. Hengartner, and R. Zinkernagel. 2000. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 288:2222–2226. [DOI] [PubMed] [Google Scholar]
- 24.Kang, H.S., R. Chin, Y. Wang, P. Yu, J. Wang, K. Newell, and Y. Fu. 2002. Signaling via LTbetaR on the lamina propria stromal cells of the gut is required for IgA production. Nat. Immunol. 3:576–582. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki, K., B. Meek, Y. Doi, T. Honjo, and S. Fagarasan. 2005. Two distinctive pathways for recruitment of naive and primed IgM+ B cells to the gut lamina propria. Proc. Natl. Acad. Sci. USA. 102:2482–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kroese, F.G., E.C. Butcher, A.M. Stall, and L.A. Herzenberg. 1989. A major peritoneal reservoir of precursors for intestinal IgA plasma cells. Immunol. Invest. 18:47–58. [DOI] [PubMed] [Google Scholar]
- 27.Kroese, F.G., E.C. Butcher, A.M. Stall, P.A. Lalor, S. Adams, and L.A. Herzenberg. 1989. Many of the IgA producing plasma cells in murine gut are derived from self-replenishing precursors in the peritoneal cavity. Int. Immunol. 1:75–84. [DOI] [PubMed] [Google Scholar]
- 28.de Waard, R., P.M. Dammers, J.W. Tung, A.B. Kantor, J.A. Wilshire, N.A. Bos, L.A. Herzenberg, and F.G. Kroese. 1998. Presence of germline and full-length IgA RNA transcripts among peritoneal B-1 cells. Dev. Immunol. 6:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berg, D.J., J. Zhang, J.V. Weinstock, H.F. Ismail, K.A. Earle, H. Alila, R. Pamukcu, S. Moore, and R.G. Lynch. 2002. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 123:1527–1542. [DOI] [PubMed] [Google Scholar]
- 30.Brenner, O., D. Levanon, V. Negreanu, O. Golubkov, O. Fainaru, E. Woolf, and Y. Groner. 2004. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proc. Natl. Acad. Sci. USA. 101:16016–16021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlsen, H.S., E. Baekkevold, F. Johansen, G. Haraldsen, and P. Brandtzaeg. 2002. B cell attracting chemokine 1 (CXCL13) and its receptor CXCR5 are expressed in normal and aberrant gut associated lymphoid tissue. Gut. 51:364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizoguchi, A., E. Mizoguchi, C. Chiba, G. Spiekermann, S. Tonegawa, C. Nagler-Anderson, and A. Bhan. 1996. Cytokine imbalance and autoantibody production in T cell receptor-α mutant mice with inflammatory bowel disease. J. Exp. Med. 183:847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hokama, A., E. Mizoguchi, K. Sugimoto, Y. Shimomura, Y. Tanaka, M. Yoshida, S. Rietdijk, Y.P. de Jong, S. Snapper, C. Terhorst, et al. 2004. Induced reactivity of intestinal CD4(+) T cells with an epithelial cell lectin, galectin-4, contributes to exacerbation of intestinal inflammation. Immunity. 20:681–693. [DOI] [PubMed] [Google Scholar]
- 34.Allman, D., R.C. Lindsley, W. DeMuth, K. Rudd, S.A. Shinton, and R.R. Hardy. 2001. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J. Immunol. 167:6834–6840. [DOI] [PubMed] [Google Scholar]
- 35.Srivastava, B., W. Quinn, K. Hazard, J. Erikson, and D. Allman. 2005. Characterization of marginal zone B cell precursors. J. Exp. Med. 202:1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardy, R.R., K. Hayakawa, D.R. Parks, and L.A. Herzenberg. 1983. Demonstration of B-cell maturation in X-linked immunodeficient mice by simultaneous three-colour immunofluorescence. Nature. 306:270–272. [DOI] [PubMed] [Google Scholar]
- 37.Fagarasan, S., K. Kinoshita, M. Muramatsu, K. Ikuta, and T. Honjo. 2001. In situ class switching and differentiation to IgA-producing cells in the gut lamina propria. Nature. 413:639–643. [DOI] [PubMed] [Google Scholar]
- 38.Barreto, V.M., A.R. Ramiro, and M.C. Nussenzweig. 2005. Activation-induced deaminase: controversies and open questions. Trends Immunol. 26:90–96. [DOI] [PubMed] [Google Scholar]
- 39.Han, J.H., S. Akira, K. Calame, B. Beutler, E. Selsing, and T. Imanishi-Kari. 2007. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity. 27:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizoguchi, A., E. Mizoguchi, H. Takedatsu, R. Blumberg, and A. Bhan. 2002. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 16:219–230. [DOI] [PubMed] [Google Scholar]
- 41.Harris, D.P., L. Haynes, P.C. Sayles, D.K. Duso, S.M. Eaton, N.M. Lepak, L.L. Johnson, S.L. Swain, and F.E. Lund. 2000. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat. Immunol. 1:475–482. [DOI] [PubMed] [Google Scholar]
- 42.Liu, N., N. Ohnishi, L. Ni, S. Akira, and K.B. Bacon. 2003. CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nat. Immunol. 4:687–693. [DOI] [PubMed] [Google Scholar]
- 43.Wagner, M., H. Poeck, B. Jahrsdoerfer, S. Rothenfusser, D. Prell, B. Bohle, E. Tuma, T. Giese, J.W. Ellwart, S. Endres, and G. Hartmann. 2004. IL-12p70-dependent Th1 induction by human B cells requires combined activation with CD40 ligand and CpG DNA. J. Immunol. 172:954–963. [DOI] [PubMed] [Google Scholar]
- 44.Kantor, A.B., C.E. Merrill, L.A. Herzenberg, and J.L. Hillson. 1997. An unbiased analysis of V(H)-D-J(H) sequences from B-1a, B-1b, and conventional B cells. J. Immunol. 158:1175–1186. [PubMed] [Google Scholar]
- 45.Aoki, Y., K.J. Isselbacher, and S. Pillai. 1994. Bruton tyrosine kinase is tyrosine phosphorylated and activated in pre-B lymphocytes and receptor-ligated B cells. Proc. Natl. Acad. Sci. USA. 91:10606–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauld, S.B., P.J.M. Dal, and J. Cambier. 2002. B cell antigen receptor signaling: roles in cell development and disease. Science. 296:1641–1642. [DOI] [PubMed] [Google Scholar]
- 47.Niiro, H., and E. Clark. 2002. Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2:945–956. [DOI] [PubMed] [Google Scholar]
- 48.de Boer, N., F. Kroese, J. Sharp, and G. Perry. 1992. Immunohistological characterization of proximal colonic lymphoid tissue in the rat. Anat. Rec. 233:569–576. [DOI] [PubMed] [Google Scholar]
- 49.Carvalho, T.L., T. Mota-Santos, A. Cumano, J. Demengeot, and P. Vieira. 2001. Arrested B lymphopoiesis and persistence of activated B cells in adult interleukin 7−/− mice. J. Exp. Med. 194:1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sitnicka, E., C. Brakebusch, I. Martensson, M. Svensson, W. Agace, M. Sigvardsson, N. Buza-Vidas, D. Bryder, C. Cilio, H. Ahlenius, et al. 2003. Complementary signaling through flt3 and interleukin-7 receptor alpha is indispensable for fetal and adult B cell genesis. J. Exp. Med. 198:1495–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Croker, B.A., D. Tarlinton, L. Cluse, A. Tuxen, A. Light, F. Yang, D. Williams, and A. Roberts. 2002. The Rac2 guanosine triphosphatase regulates B lymphocyte antigen receptor responses and chemotaxis and is required for establishment of B-1a and marginal zone B lymphocytes. J. Immunol. 168:3376–3386. [DOI] [PubMed] [Google Scholar]
- 52.Busslinger, M. 2004. Transcriptional control of early B cell development. Annu. Rev. Immunol. 22:55–79. [DOI] [PubMed] [Google Scholar]
- 53.Vos, Q., A. Lees, Z. Wu, C. Snapper, and J. Mond. 2000. B-cell activation by T-cell-independent type 2 antigens as an integral part of the humoral immune response to pathogenic microorganisms. Immunol. Rev. 176:154–170. [DOI] [PubMed] [Google Scholar]
- 54.Patke, A., I. Mecklenbrauker, and A. Tarakhovsky. 2004. Survival signaling in resting B cells. Curr. Opin. Immunol. 16:251–255. [DOI] [PubMed] [Google Scholar]
- 55.Claudio, E., K. Brown, S. Park, H. Wang, and U. Siebenlist. 2002. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 3:958–965. [DOI] [PubMed] [Google Scholar]
- 56.Lesley, R., Y. Xu, S. Kalled, D. Hess, S. Schwab, H. Shu, and J. Cyster. 2004. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 20:441–453. [DOI] [PubMed] [Google Scholar]
- 57.Qian, Y., J. Qin, G. Cui, M. Naramura, E. Snow, C. Ware, R. Fairchild, S. Omori, R. Rickert, M. Scott, et al. 2004. Act1, a negative regulator in CD40- and BAFF-mediated B cell survival. Immunity. 21:575–587. [DOI] [PubMed] [Google Scholar]
- 58.Mackay, F., P. Schneider, P. Rennert, and J. Browning. 2003. BAFF AND APRIL: a tutorial on B cell survival. Annu. Rev. Immunol. 21:231–264. [DOI] [PubMed] [Google Scholar]
- 59.Thompson, J.S., S. Bixler, F. Qian, K. Vora, M. Scott, T. Cachero, C. Hession, P. Schneider, I. Sizing, C. Mullen, et al. 2001. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 293:2108–2111. [DOI] [PubMed] [Google Scholar]
- 60.Rolink, A.G., and F. Melchers. 2002. BAFFled B cells survive and thrive: roles of BAFF in B-cell development. Curr. Opin. Immunol. 14:266–275. [DOI] [PubMed] [Google Scholar]
- 61.Hsu, B.L., S. Harless, R. Lindsley, D. Hilbert, and M. Cancro. 2002. Cutting edge: BLyS enables survival of transitional and mature B cells through distinct mediators. J. Immunol. 168:5993–5996. [DOI] [PubMed] [Google Scholar]
- 62.Sasaki, Y., S. Casola, J. Kutok, K. Rajewsky, and M. Schmidt-Supprian. 2004. TNF family member B cell-activating factor (BAFF) receptor-dependent and -independent roles for BAFF in B cell physiology. J. Immunol. 173:2245–2252. [DOI] [PubMed] [Google Scholar]
- 63.Batten, M., J. Groom, T.G. Cachero, F. Qian, P. Schneider, J. Tschopp, J.L. Browning, and F. Mackay. 2000. BAFF mediates survival of peripheral immature B lymphocytes. J. Exp. Med. 192:1453–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kobayashi, M., L. Fitz, M. Ryan, R.M. Hewick, S.C. Clark, S. Chan, R. Loudon, F. Sherman, B. Perussia, and G. Trinchieri. 1989. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 170:827–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trinchieri, G. 1995. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 13:251–276. [DOI] [PubMed] [Google Scholar]
- 66.Chen, C.C., S. Louie, B.A. McCormick, W.A. Walker, and H.N. Shi. 2006. Helminth-primed dendritic cells alter the host response to enteric bacterial infection. J. Immunol. 176:472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mizoguchi, E., R. Xavier, H. Reinecker, H. Uchino, A. Bhan, D. Podolsky, and A. Mizoguchi. 2003. Colonic epithelial functional phenotype varies with type and phase of experimental colitis. Gastroenterology. 125:148–161. [DOI] [PubMed] [Google Scholar]
- 68.LeMaoult, J., S. Delassus, R. Dyall, J. Nikolic-Zugic, P. Kourilsky, and M.E. Weksler. 1997. Clonal expansions of B lymphocytes in old mice. J. Immunol. 159:3866–3874. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}