

Abstract
Background
To determine if soluble pre-amyloid oligomers (PAOs) are toxic when expressed internally in the cardiomyocyte, we tested the hypothesis that cardiomyocyte-restricted expression and accumulation of a known PAO is cytotoxic and sufficient to cause heart failure.
Methods and Results
Intracellular PAOs, the entities believed to cause toxicity in many neurodegenerative diseases, have been observed in cardiomyocytes derived from mouse and human heart failure samples. Long (>50) polyglutamine (PQ) repeats form PAOs and cause neurotoxicity in Huntington’s and other neurodegenerative diseases, while shorter PQ peptides are benign. We created transgenic mice in which cardiomyocyte-autonomous expression of an 83 residue-long PQ repeat (PQ83), or a non-amyloid forming peptide of 19 PQ repeats (PQ19) as a non-pathological control, were expressed. A PQ83 line with relatively low levels of expression was generated, along with a PQ19 line that expressed approximately 9-fold the levels observed in the PQ83 line. Hearts expressing PQ83 exhibit reduced cardiac function and dilation by five months and all mice died by eight months, whereas PQ19 mice had normal cardiac function, morphology, and lifespan. PQ83 protein accumulates within aggresomes with PAO-specific staining. The PQ83 hearts showed increased autophagosomal and lysosomal content, but also showed markers of necrotic death, including inflammatory cell infiltration and increased sarcolemmal permeability.
Conclusions
The data confirm the hypothesis that expression of an exogenous PAO forming peptide is toxic to cardiomyocytes and is sufficient to cause cardiomyocyte loss and heart failure.
Introduction
Amyloidoses are well characterized in many tissues, including the heart, and are generally thought of as a heterogeneous syndrome characterized by the formation and accumulation of extracellular proteinaceous fibrils. A number of inherited forms of systemic amyloidoses such as those associated with mutations in the serum protein transthyretin1 can have diverse effects on heart function and result in dilated or restrictive cardiomyopathy or diastolic dysfunction.2,3 Some well known neurodegenerative diseases, including Alzheimer’s, Parkinson’s and Huntington’s disease share the pathological characteristic of accumulating extracellular, insoluble aggregates of misfolded amyloid proteins.4 Until recently the extracellular, insoluble protein aggregates observed in these diseases were thought to be causal for disease presentation, but multiple studies have shown that aggregate content correlates poorly with disease severity and pathological alterations often precede amyloid deposition.5–8 Recently it has been proposed that cytoplasmic, soluble oligomeric forms of amyloidogenic proteins or pre-amyloid oligomers (PAOs) are the primary toxic entities responsible for disease. PAOs are a diverse set of proteins of divergent sequences that share a common conformational structure, which may impart a common mechanism of toxicity.9,10 PAOs represent an intermediate step in amyloid fibril formation that begins when a native soluble protein becomes misfolded. The misfolded but soluble protein then self-associates to form soluble oligomers (PAO), which may subsequently go on to form protofibrils and other insoluble intermediates in the amyloid fibril pathway. PAOs from numerous amyloid-forming proteins can cause cellular toxicity in neural cells,10 consistent with the hypothesis that PAOs are the primary cytotoxic entities in several amyloidogenic diseases.11,12
It is important to draw a distinction between the presence of PAO, an intracellular, soluble, precursor amyloidogenic entity and the final extracellular amyloid plaques/fibrils themselves. Classically characterized cardiac amyloidoses describe the accumulation of the extracellular, proteinaceous amyloid fibrils between myofibers, whereas PAOs accumulate as intracellular, soluble amyloid precursors. While the majority of studies on the PAO mechanism of toxicity have been restricted largely to nerve cells and neurodegenerative diseases, our recent data suggest that a mechanism of PAO-induced toxicity may also play a role in cardiomyocyte death, cardiac disease and heart failure. In a model of desmin-related cardiomyopathy produced by cardiomyocyte-specific transgenic (TG) expression of a mutant αB-crystallin (CryABR120G), PAO-positive aggregates were observed within cardiomyocytes.13,14 The presence of intracellular PAOs in the heart closely associated with loss of cardiac function and heart failure, and silencing CryABR120G expression resulted in the rescue of cardiac function and concomitant, significant reduction in cardiomyocyte PAO levels. We have also observed high intracellular PAO levels in a number of human heart failure samples of various etiologies.14
On the basis of those data, we undertook a series of experiments to test if direct expression and cardiomyocyte accumulation of an ectopic PAO-genic protein could cause dysfunction and cardiac disease. PAOs consisting of expanded polyglutamine (PQ) repeats are toxic when expressed in neural cells10 and expanded PQ repeats are responsible for nine neurodegenerative diseases, including Huntington’s disease.15 Proteins with expanded PQ tracts confer a gain of toxic function in a length-dependent manner, with longer tracts (>50) leading to neurotoxicity, while shorter tracts are benign.16 Upon reaching a toxic length, the PQ proteins undergo a conformational change, oligomerizing into toxic PAO intermediates.15,17 To assess the role of PQ repeat peptides in the mouse heart, we generated TG mice expressing either a PAO-forming peptide of 83 PQ repeats (PQ83) or a non-PAO forming peptide of 19 PQ repeats (PQ19) under the control of the α-myosin heavy chain promoter (MyHC) to drive cardiomyocyte-specific expression. PQ83 hearts develop cardiac dysfunction and dilation with a concomitant increase in PAO-positive staining and formation of insoluble aggresomes within the cardiomyocytes, and the mice invariably died before reaching eight months of age. PAO-induced heart failure was due to cardiomyocyte loss and, while apoptotic indices were unchanged in PQ83 hearts, ultrastructural analysis revealed increased autophagic and lysosomal content indicative of increased autophagy. PQ83 hearts also showed evidence of necrotic death including inflammatory cell content and sarcolemmal permeability. The data confirm the hypothesis that PAO-forming peptides are toxic when they accumulate in cardiomyocytes and that cardiomyocyte-based PAO accumulation causes heart failure.
Methods
Transgenic Mice
A hemagglutinin epitope tag (HA) was added at the carboxyl-terminus of each PQ construct. Those cDNA’s were placed behind the α-MyHC promoter and used to generate transgenic (TG) FVB/N mice expressing PQ19-HA or PQ83-HA. Animals were housed in an AAALAC-approved, germ-free barrier facility and all experiments were approved by the Children’s Hospital Animal Review Board.
Echocardiography
Cardiac ultrasound was performed on isoflurane-anesthetized mice from two to seven months of age using a VisualSonics Vevo 770 Imaging System (Toronto, Canada) with a 30 MHz transducer. Heart rate and body temperature were continuously monitored. Images of the left ventricle were collected along the parasternal short axis and stored on the system’s hard-drive. Two dimensional directed M-mode echocardiographic images were recorded to determine left ventricular (LV) systolic function and expressed as shortening fraction (SF). M-mode measurements of LV end-diastolic and end-systolic chamber size (LVID and LVIS, respectively) were calculated; SF=(LVIDd-LVIDs)/LVIDd.
Transcript and Protein Analyses
Total ventricular RNA was isolated using TRIREAGENT (Molecular Research Center, Cincinnati, OH) according to the manufacturer’s protocol. RNA dot blots hybridized with transcript-specific oligonucleotides were used to quantify TG expression. Levels of hypertrophic markers were measured by 5′ nuclease TaqMan assays (Applied Biosystems, Foster City, CA) following cDNA synthesis. Total ventricular protein was isolated by homogenization in RIPA buffer. SDS-PAGE and western blots were run on soluble fractions. Aggregate filter assays were performed on RIPA insoluble fractions and immunoblotted with anti-HA antibodies (Y-11, Santa Cruz Biotechnology, Santa Cruz, California) and (3F10, Roche, Indianapolis, IN), as previously described (2). RIPA insoluble fractions were further solubilized by treatment with 95% formic acid at 37°C for an hour, lyophilized, neutralized and resuspended in Laemmli buffer for SDS-PAGE and immunoblot analyses. Immunoblots were performed using antibodies against: total caspase-3, cleaved caspase-3, anti-IRE1 (Cell Signaling Technologies Boston, MA), anti-caspase-8 (Biovision), anti Grp78 (BD Biosciences, San Jose, California), anti Xbp-1 (Biolegend, San Diego, California), anti-Atg5 Cosmo Bio Co., Tokyo, Japan), anti-HA, anti-PARP, anti-Lamp1, anti ATF6, cathepsin D (C-20), anti-PERK (H-300) (Santa Cruz Biotechnology, Santa Cruz, California), anti-LC3 (Nanotools, Munich, Germany).
Immunohistochemistry
Immunohistochemical analyses were performed as described.14,18 Primary antibodies were purchased as follows: anti-HA, anti-Lamp1 and anti-cathepsin D (Santa Cruz Biotechnology, Santa Cruz, CA), anti-PAO, A11 antibody (courtesy of C. Glabe, University of CA, Irvine, CA), anti-CD45 (R&D Systems, Inc., Minneapolis, MN), anti-mac-3 (BD Pharmingen, San Jose, CA), and anti-desmin (Biomeda, Foster City, CA). To-Pro-3, phalloidin and Alexa 488 or 568-conjugated antibodies were from Invitrogen (Carlsbad, CA). Stained sections were examined by confocal microscopy (PCM 2000, Nikon, Melville, NY) and images captured with Simple PCI version 4 (Compix Inc., Sewickley, PA). CD45 and Mac-3-positive cells were quantified using MetaMorph 7 software from >10 images/genotype.
Apoptosis Assays
To quantify apoptotic nuclei via immunofluorescence, an in situ cell death detection kit (Roche, Indianapolis, IN) was used for direct TUNEL (TdT-mediated dUTP-nick end labeling) staining. Paraffin whole-heart sections (n=3/genotype) were de-paraffined, and boiled in 0.1 mol/L sodium citrate buffer (pH 6.0) for 20 minutes. Following antigen retrieval, sections underwent the labeling reaction with fluorescein-dUTP. Sections were counterstained with desmin and TOPRO to detect myocytes and nuclei and visualized by confocal microscopy. To quantify the DNA laddering due to apoptotic endonucleases, genomic DNA isolated from three hearts per genotype. One microgram of DNA was ligated with adaptor primers overnight followed by PCR for 30 or 40 cycles. Amplified products were separated on a 2% TAE gel, along with positive and negative controls. The PCR-based DNA laddering kit was purchased from Maxim Biotech (San Francisco, CA).
Mitochondrial Swelling
Whole hearts were collected from 4.5 month old mice (n = 4/genotype) to analyze mitochondrial swelling. Mitochondrial swelling assays were performed as described.19
Evans Blue Staining
Evans Blue dye was diluted in sterile saline to 10 mg/ml and sterile-filtered. Isoflurane-anesthetized mice were given intraperitoneal injections of 5 μl dye/gm of body weight. Six hours after injection mice were anesthetized again and hearts were perfused with cardioplegic buffer in 4% paraformaldehyde. Following perfusion, hearts were drop fixed in the same buffer for 1 hour and fixed overnight in 4% paraformaldehyde in PBS at 4°C. Fixed hearts were bisected and incubated in 15% sucrose for two hours and then 30% sucrose overnight at 4°C for cytoprotection. Hearts were mounted in OCT and cut into 5 μm cryosections and visualized by fluorescence microscopy.
Statistical Analyses
Data are expressed as mean±SEM and analyzed using one-way analysis of variance, where a significant difference was P<0.05, following a Tukey’s post-hoc adjustment. Longitudinal echo data were analyzed with 2-way analysis of variance with repeated measures. The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
RESULTS
To test the hypothesis that PAO expression in cardiomyocytes leads to cell dysfunction and heart failure, we generated TG mice with cardiomyocyte-specific expression of either PQ19 (as a negative control) or PQ83 under control of the α-MyHC promoter (Figure 1A). Both constructs contained a hemagglutinin (HA) tag at the carboxyl terminus for identification. Several TG PQ19 founders were obtained and the highest expressing PQ19 line (~9-fold relative to the PQ83 line used) was analyzed in parallel with the PQ83 mice. Six TG PQ83 founders were produced, but only a single TG founder line survived to breeding age and yielded TG progeny before dying of dilated cardiomyopathy. The five other mice positive for the presence of the transgene all died of heart failure prior to reaching breeding age. PQ83 expression by RNA dot blot in the surviving line was barely measurable under normal conditions but could be detected if five-fold the normal amount of RNA was applied to the membrane for hybridization analysis (Figure 1B). Neither PQ19 nor PQ83 protein could be detected in the soluble cytoplasmic fraction using western blots probed with an anti-HA antibody (Figure 1C). However, insoluble PQ83 peptide could be quantified on a filter-trap assay (Figure 1D) and formic acid solubilization of the aggregates enabled us to detect HA-positive protein in the PQ83 hearts (Figure 1E).
Figure 1.

Transgenic expression of PQ83 and PQ19 in the mouse heart. A, Construct design. The mouse α-MyHC promoter was used to drive PQ peptides of 19 or 83 repeats, fused with a hemagglutinin (HA) tag and flanked by a human growth hormone polyadenylation signal (hGH polyA). B, RNA dot blots of PQ19 and PQ83 transgene expression. Ventricular RNA was probed for hGH. GAPDH levels were used for normalization (n = 6/genotype). *, significant difference vs NTG (P<0.05) by one-way ANOVA. Note that to detect the low levels of transcript expressed in the PQ83 line, 10 μg of RNA as opposed to 2 μg for the PQ19 sample was used. C, Immunoblotting with an anti-HA antibody shows that the bulk of PQ83 protein is insoluble. The PQ19 polypeptides were not retained on the gel under the electrophoresis and transfer conditions used. D, Aggregate filter-trapped protein probed with anti-HA antibody shows that PQ83 protein is retained in the insoluble aggregates. E, Formic acid treatment of insoluble protein fractions revealed HA-specific bands for PQ83 samples, confirming protein expression despite the very low levels of transcript that were detected.
Histological analysis of PQ83 mice showed progressive cardiac dilation from 5–7 months with no morphological changes in PQ19 hearts relative to nontransgenic (NTG) animals (Figure 2A). Echocardiography revealed a significant loss in cardiac contractile function and chamber dilation in the PQ83 hearts by seven months (Figure 2B). Cardiomyocyte loss, hypertrophy and infiltrating inflammatory cells were all apparent as early as 3 months in PQ83 hearts, none of which were observed in either the PQ19 or NTG hearts (Figure 2C). Trichrome staining showed modest increases in fibrosis and interstitial space in the dilated PQ83 hearts (Figure 2D). Those hearts showed activation of hypertrophic markers at the molecular level (Figure 2E) and overt hypertrophy as measured by a significant increase in the heart weight/body weight ratios (Figure 2F).
Figure 2.
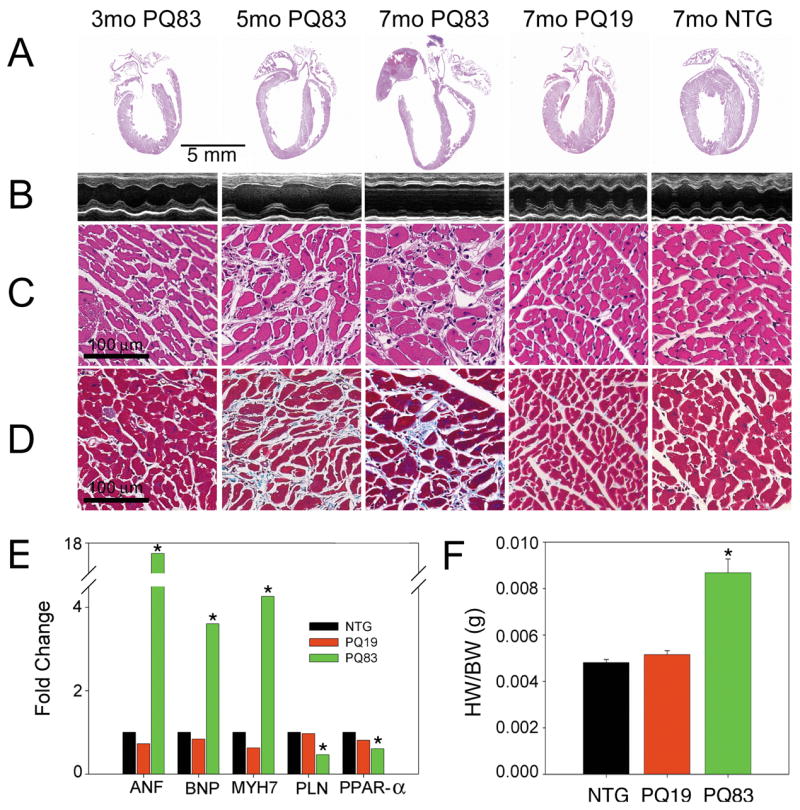
Histology of PQ19 and PQ83 hearts. A, Representative whole heart sections from 3, 5 or 7 month old mice of mixed gender show increasing dilation in PQ83 expressing hearts with age relative to 7 month PQ19 and NTG hearts (x4). B, M-mode echocardiographic tracings show the left ventricular chamber and confirm increasing PQ83 chamber size. C, Hematoxylin-eosin staining (×20) shows myocyte degeneration, hypertrophy and infiltrating cells in PQ83 vs PQ19 and NTG sections. D, Sections stained with Gomori’s trichrome reveal mild fibrosis and increased interstitial spacing PQ83 hearts (×20). E, Molecular markers of hypertrophy: atrial natriuretic factor; (ANF), brain natriuretic protein; (BNP), β-MyHC; (MYH7) were elevated, while the expected decreases in phospholamban (PLN) and the peroxisome proliferator-activated receptor α (PPAR-α) were observed in PQ83 samples relative to PQ19 and NTG controls (n = 6/genotype). F, Heart (wet) weight to body weight ratios (HW/BW) confirm the hypertrophy of PQ83 hearts. *, significant difference vs NTG (P<0.05) by one-way ANOVA.
Cardiac function was measured longitudinally using echocardiography (Tables 1 and 2). PQ83 hearts undergo a progressive loss of cardiac function between 4.5 and 7 months while PQ19 and NTG hearts maintained normal function (Figure 3A). All PQ83 mice died between 5.5 and 8 months while the PQ19 mice appeared functionally and anatomically normal in all respects and had normal life spans of approximately 2 years with no heart failure apparent at 22 months (data not shown). (Figure 3B). Thus, PQ83 expressing mice die due to diminished cardiac function and dilated cardiomyopathy.
Table 1.
M-mode Measurements for Parasternal Short Axis Protocol
| Age (months) | IVS/d (mm) | LVID/d (mm) | LV PW/d (mm) | IVS/s (mm) | LVID/s (mm) | LV PW/s (mm) | % EF | % FS | |
|---|---|---|---|---|---|---|---|---|---|
| NTG | 3.5 | 0.764±0.017 | 4.20±0.08 | 0.635±0.019 | 1.165±0.040 | 2.72±0.10 | 1.000±0.035 | 64.5±1.5 | 35.0±1.1 |
| PQ19 | 3.5 | 0.743±0.026 | 4.12±0.12 | 0.633±0.025 | 1.113±0.051‡ | 2.62±0.09 | 0.965±0.030 | 66.1±1.1 | 36.1±0.8 |
| PQ83 | 3.5 | 0.714±0.018 | 4.24±0.09 | 0.603±0.019 | 1.081±0.040 | 2.76±0.09 | 0.910±0.045 | 64.4±1.4 | 34.9±1.0 |
| NTG | 5 | 0.776±0.035 | 4.10±0.08 | 0.684±0.029 | 1.234±0.043 | 2.58±0.07 | 1.016±0.019 | 67.3±1.1 | 37.0±0.8 |
| PQ19 | 5 | 0.767±0.058 | 4.25±0.11 | 0.707±0.018 | 1.182±0.091 | 2.67±0.03 | 1.037±0.029 | 67.1±1.3 | 37.0±1.1 |
| PQ83 | 5 | 0.836±0.039‡ | 4.30±0.08 | 0.701±0.035 | 1.238±0.047 | 2.97±0.12* | 0.994±0.036 | 58.6±2.8* | 31.3±1.8 |
| NTG | 6.5 | 0.801±0.037 | 4.11±0.08 | 0.650±0.016 | 1.189±0.054 | 2.58±0.07 | 1.004±0.016 | 67.6±1.3 | 37.3±1.0 |
| PQ19 | 6.5 | 0.766±0.032 | 4.09±0.12 | 0.616±0.028 | 1.206±0.017 | 2.44±0.19 | 0.986±0.047 | 71.3±3.9 | 40.6±3.0 |
| PQ83 | 6.5 | 0.706±0.033§ | 4.89±0.15*†‡§ | 0.530±0.019*†§ | 1.045±0.046§ | 3.98±0.17*†‡§ | 0.666±0.032*†‡§ | 37.3±5.2*†‡§ | 18.5±2.9*†‡§ |
M-mode measurements of the left ventricle from a parasternal long-axis view. Data are presented as the mean value±standard error. IVS/d = interventricular septal thickness during diastole; LVID/d = left ventricular internal dimension during diastole; LV PW/d = posterior wall thickness during diastole; IVS/s = interventricular septal thickness during systole; LVID/s = left ventricular dimension during systole; LV PW/s = posterior wall thickness during systole; %EF = ejection fraction; %FS = fractional shortening.
, significant difference vs NTG (P<0.05).
, significant difference vs PQ19 (P<0.05),
, significant difference vs 3.5 months PQ83 (P<0.05),
, significant difference vs 5 months PQ83 (P<0.05) by two-way ANOVA with repeated measures.
Table 2.
B-mode measurements for Endocardial Protocol
| Age (months) | Endocardial Area D (mm2) | Endocardial Area S (mm2) | Endocardial FAC (%) | Endocardial Area Change (mm2) | |
|---|---|---|---|---|---|
| NTG | 3.5 | 12.4±0.3 | 7.89±0.42 | 49.2±1.7 | 7.80±0.34 |
| PQ19 | 3.5 | 12.3±0.4 | 7.71±0.73 | 46.4±1.4 | 6.87±0.43 |
| PQ83 | 3.5 | 12.4±0.4 | 7.71±0.49 | 45.8±1.6 | 6.95±0.33 |
| NTG | 5 | 14.8±0.5 | 7.53±0.45 | 49.3±1.9 | 7.28±0.28 |
| PQ19 | 5 | 14.4±0.7 | 7.13±0.53 | 50.6±2.5 | 7.28±0.45 |
| PQ83 | 5 | 16.0±0.9 | 9.50±0.79‡ | 41.3±1.8*† | 8.68±2.22 |
| NTG | 6.5 | 13.8±0.8 | 6.90±0.64 | 50.6±2.0 | 6.94±0.26 |
| PQ19 | 6.5 | 11.6±1.0 | 5.43±0.60 | 52.1±4.1 | 6.39±0.64 |
| PQ83 | 6.5 | 19.8±0.9*†‡§ | 14.60±0.82*†‡§ | 26.2±2.7*†‡§ | 5.21±0.56*‡§ |
B-mode measurements were made to increase resolution of cardiac structures. D=during diastole; S = during systole; FAC = fractional area change.
, significant difference vs NTG (P<0.05).
, significant difference vs PQ19 (P<0.05),
, significant difference vs 3.5 months PQ83 (P<0.05),
, significant difference vs 5 months PQ83 (P<0.05) by two-way ANOVA with repeated measures.
Figure 3.
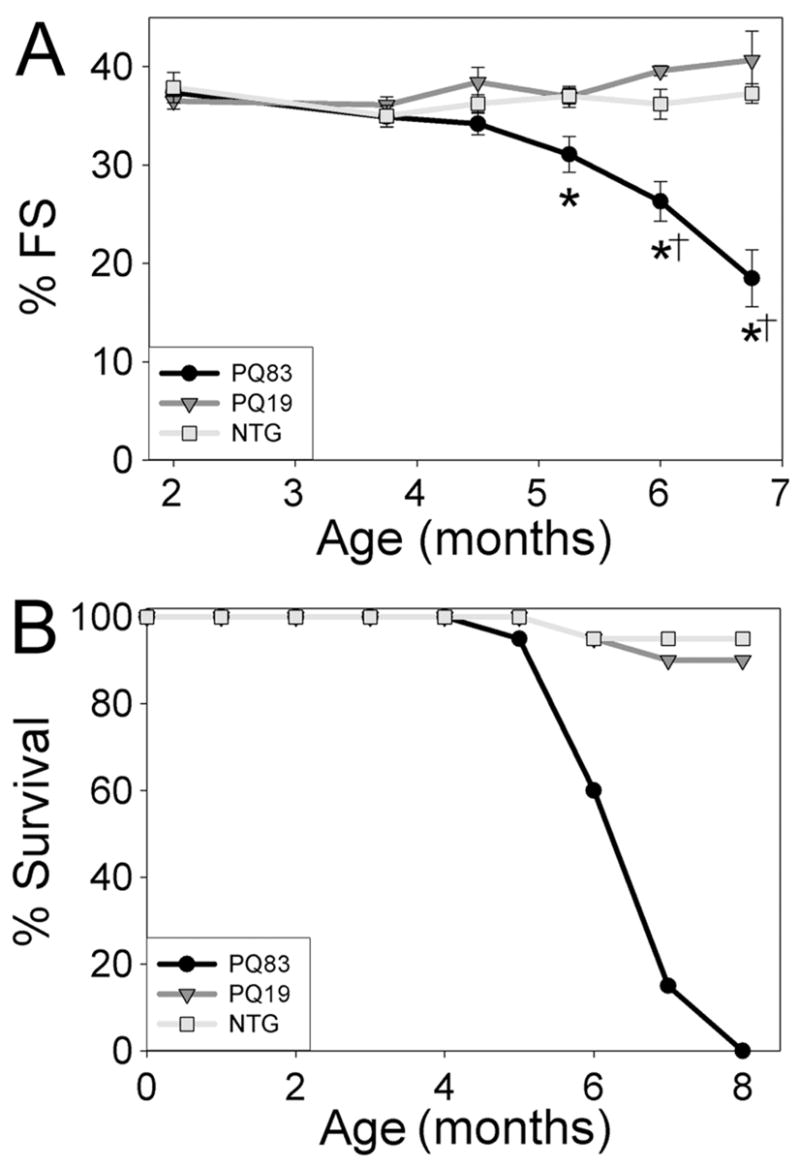
Cardiac function and survival curve. Echocardiograms were performed on PQ19, PQ83, and NTG mice of both genders from 2–7 months of age (n = 8–12/genotype). A, Percent fractional shortening across genotypes. *, significant difference vs NTG (P<0.05), †, significant difference vs PQ19 (P<0.05) by two-way ANOVA with repeated measures. B, Kaplan-Meier survival curve. No gender differences were observed (n = 12–20/genotype).
We used immunohistochemistry to detect the presence of PAO within the cardiomyocytes. While neither the NTG nor PQ19 cardiomyocytes contained significant amounts of PAO, PAO-positive material accumulated in the PQ83 cardiomyocytes (Figure 4A) in the characteristic aggregates observed in other cardiomyopathic models with high levels of PAO.14 Immunohistochemistry confirmed that PQ83 expressing hearts generate HA-positive intracellular aggregates as early as one month of age (Figure 4B).
Figure 4.

Immunohistochemistry. A, Whole heart sections from 1, 3, 5 and 7 month old mice were treated with anti-desmin (red) and TOPRO (blue) to detect cardiomyocytes and nuclei respectively. PAOs were detected using the A-11 antibody14 (green, arrows) where it localized in degenerating myocytes in and around intrasarcoplasmic aggregates (×20). B, staining with an anti-HA antibody (green, arrows) confirms early PQ83 aggregation occurs within the cardiomyocyte.
In our previously characterized CryABR120G cardiomyopathic model, which is also characterized by high PAO levels, we noted activation of apoptosis in the cardiomyocytes.19 To determine if this was a common characteristic for PAO-induced cardiomyocyte toxicity, we assayed for indices of apoptosis in the PQ83 hearts. We could not detect any increases in caspase-3, caspase-8 or PARP activation in the PQ83 samples relative to PQ19 and NTG controls (Figure 5A) and PCR-based DNA laddering assays confirmed the absence of apoptosis in the PQ83 samples (Figure 5B). Cardiomyocyte-specific TUNEL staining showed no apoptotic activation in the PQ83 cardiomyocytes (Figure 5C). We conclude that apoptotic-based cell death is not necessarily a commonly shared pathway for PAO-based cardiomyocyte death but rather may be dependent upon the specific primary pathogenic entity responsible for triggering cardiomyocyte PAO accumulation.
Figure 5.

Cell death in PQ83 cardiomyocytes is not due to apoptosis, mitochondrial swelling or ER stress. A, Immunoblots against total and cleaved caspase-3, caspase-8, total and cleaved PARP. GAPDH was used as a loading control. The positive control (+) consisted of HeLa cells treated with staurosporine (n = 4/genotype). *, significant difference vs NTG (P<0.05) by one-way ANOVA. B, PCR-based DNA laddering shows no evidence of apoptotic laddering in PQ83 hearts after 30 amplification cycles. Extended amplification (40 cycles) was also negative (data not shown). C, TUNEL staining shows no evidence of apoptotic activation in the PQ83 cardiomyocytes. The rare TUNEL-positive cells are denoted (arrows). D, Mitochondrial swelling assays show no impairment in the PQ83 samples; hearts were assayed at 4.5 months of age (n = 4/genotype). E, Immunoblots detecting markers of ER stress failed to show increases in protein levels in the PQ83 samples (n = 4/genotype). F, Bar graphs of ER stress protein densitometry normalized to GAPDH levels. *, significant difference vs NTG (P<0.05), †, significant difference vs PQ19 (P<0.05) within an age group by two-way ANOVAs.
The CryABR120G model of cardiomyopathy previously was shown to have impaired mitochondrial swelling.19 Neither the PQ19 or PQ83 heart mitochondria demonstrated any swelling defect (Figure 5D). Elevated markers of endoplasmic reticulum (ER) stress have also been noted in some aggregate-forming diseases,20,21 but multiple markers of the ER stress pathway were not activated as heart failure developed in the PQ83 hearts (Figure 5E and 5F).
With apoptosis ruled out as a primary cause for cardiomyocyte death, we examined other potential death pathways. Ultrastructural analyses revealed that PQ83 hearts had an increased number of small mitochondria. The cardiomyocytes showed anatomical hallmarks of autophagy, with autophagic vesicles, multilamellar bodies, and lysosomes (Figure 6A). The PQ83 aggresomes frequently could be observed being engulfed by autophagosomes, further suggesting an autophagic means of degradation. Lysosomal markers cathepsin D and LAMP1 were significantly increased in failing PQ83 hearts (Figure 6B and 6C). The active forms of cathepsin D, a lysosomal proteolytic enzyme, have molecular weights of ~44 and ~33 kDa22 and the ~44kDa species was increased 2.8-fold in the seven month old PQ83 hearts relative to NTG or PQ19 (Figure 6B and 6C). LC3-II, a protein that integrates into autophagosomal membranes, was also significantly elevated 2.4 fold in the seven month old PQ83 hearts relative to NTG samples (P<0.05). However, other autophagic markers such as Atg5 were unchanged. Immunohistochemical analyses showed high levels of cathepsin D staining in degenerating PQ83 cardiomyocytes, consistent with lysosomal permeabilization, and elevation of LAMP1 staining was apparent (Figure 6D). On the basis of all of the above data and particularly, the ultrastructural identification of autophagosomes, we conclude that PQ83-mediated PAO accumulation leads to the formation of PQ-aggregates, which are engulfed via macroautophagy with corresponding increases in lysosomal content, leading to autophagic degradation.
Figure 6.
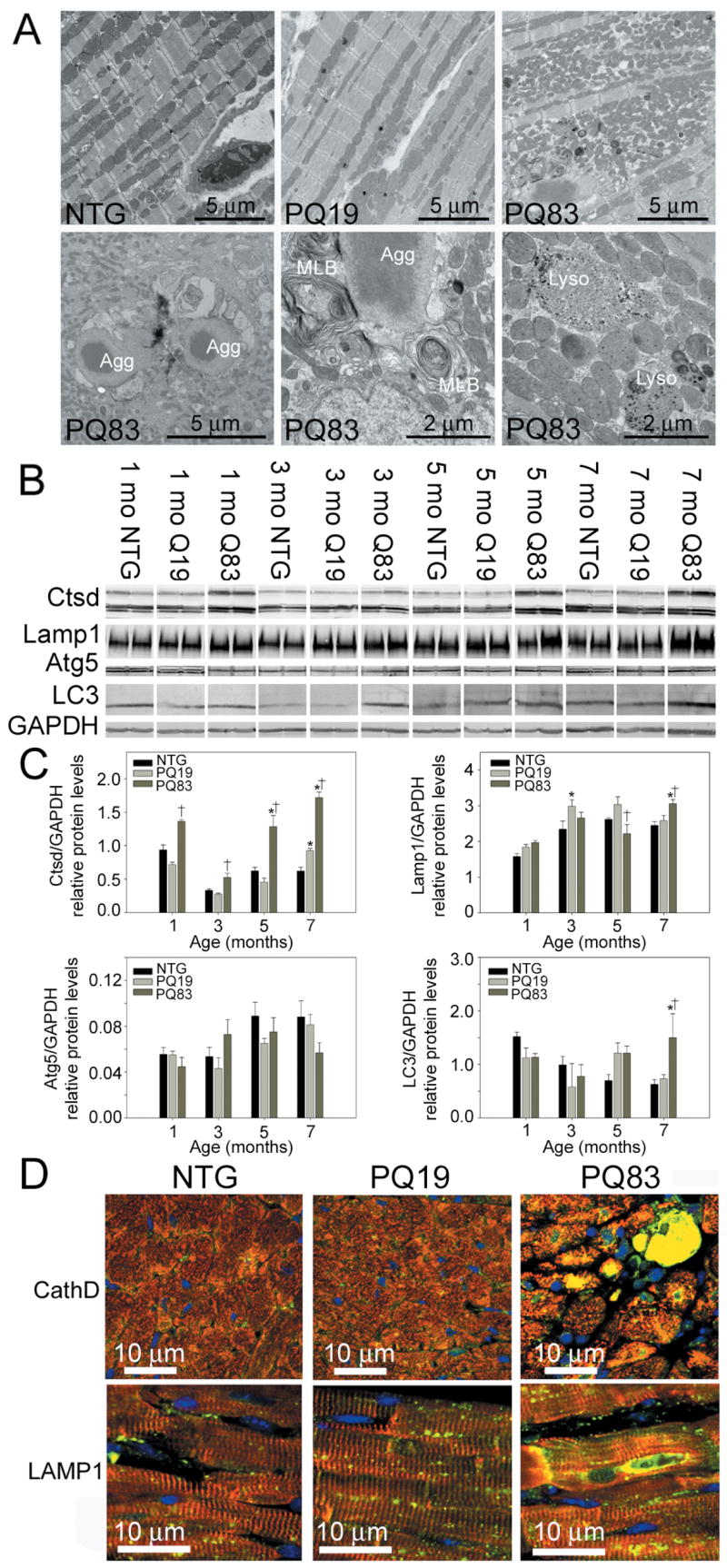
Autophagy and lysosomal activation. A, Upper panels depict cardiomyocyte ultrastructure derived from 7 month old hearts (10,000X). Lower panels show higher magnification of autophagic consumption of aggregates (lower left, 10,000X), increased number of multilamellar bodies (MLBs) (lower middle, 30,000X), and lysosomes (lower right, 30,000X). B, Immunoblots against lysosomal and autophagic markers cathepsin D (Ctsd), LAMP1 and LC3 were increased significantly, while Atg5 was not elevated in PQ83 cardiomyocytes (n = 4/genotype). C, Bar graphs of lysosomal and autophagy protein densitometry normalized to GAPDH levels. *, significant difference vs NTG (P<0.05), †, significant difference vs PQ19 (P<0.05) within an age group by two-way ANOVAs. D, Immunohistochemistry using antibodies against cathepsin D and LAMP1 (green). Desmin (red) was used to identify cardiomyocytes and TOPRO (blue) used to identify nuclei (60X).
A unique characteristic of necrotic death is the infiltration of inflammatory cells. The infiltrating cells apparent in the histological analyses of the PQ83 hearts were confirmed to be leukocytes and macrophages, using antibodies against CD45 and Mac-3, respectively. Increased staining for both types of inflammatory markers was evident in PQ83 hearts when compared with the PQ19 and NTG controls (Figure 7A and 7B). PQ83 hearts had significantly more CD45-positive cells/view (241±18 vs 122±18 and 121±6, respectively) than PQ19 and NTG hearts (P<0.001). Mac-3-positive cells were also significantly increased (P<0.05) in PQ83 hearts relative to NTG (55±5 vs 35±4), but did not differ from PQ19 (48±5 cells/view). In addition to increased infiltration of inflammatory cells (Figure 7), another early feature of necrotic cell death is the loss of plasma membrane integrity. To assess cardiac sarcolemmal integrity, adult mice were injected with Evans blue dye six hours before death. The dye is taken up by cardiomyocytes with leaky sarcolemmal membranes and excluded by cells with intact membranes. No cardiomyocytes from PQ19 or NTG hearts took up the dye, while numerous cells in the PQ83 hearts stained positively (Figure 7C).
Figure 7.

Necrosis and inflammation in PQ83 hearts. A, Inflammatory cells were detected using a leukocyte marker, CD45 (green). Anti-phalloidin (red) and TOPRO staining identified cardiomyocytes and nuclei, respectively. B, Mac-3 antibody (green) identified macrophages with anti-phalloidin (red) and TOPRO used to identify cardiomyocytes and nuclei, respectively (×20). C, Evans Blue dye (EB) uptake detected as red fluorescence indicates impaired sarcolemmal permeability in the PQ83 hearts.
Discussion
Our previously characterized CryABR120G model developed heart failure as a result of cardiomyocyte-specific expression of the mutant CryAB.13,14,18 The PQ83 model represents a second model of cardiomyocyte-specific PAO expression leading to heart failure and confirms that intracellular PAO accumulations can contribute to cardiomyocyte death and heart failure. Toxicity of the PQ repeat is a function of both the sequence and number of repeats10,15,16 and the data are consistent with cardiomyocyte toxicity being a function of the PAO-genic properties of the construct rather than depending simply on the size of the protein being expressed. While our experiments do not formally rule out the possibility that the observed toxic effects are due to the relative size of the PQ83 peptide rather than its property to form PAO, we have expressed numerous larger proteins in these cells via transgenesis and at much higher levels, without seeing PAO accumulation or cardiomyocyte toxicity.23 The observation of intracellular PAO staining in cardiomyocytes from failing human hearts of different etiologies supports the idea that intracellular PAOs are toxic and pertinent to cardiac disease in humans and it is therefore critical to better understand the distribution and pathogenicity of cardiomyocyte-based PAOs during the development of cardiovascular disease.
The recent focus of studies on amyloid-based pathogenic processes and mechanisms has shifted to intracellular events preceding visible plaque accretion rather than extracellular accumulation of amyloid plaques. Although the inherent toxicity of the amyloid-containing aggregates has been assumed in a wide range of human conditions such as light chain amyloidosis, the spongiform encephalopathies, Alzheimer’s disease, Parkinson’s disease and others,24 it is now generally thought that amyloid plaques are a consequence of a long pathogenic process, representing “tombstones” rather than being directly cytotoxic.9,25 Our recent studies show a strong correlation between PAO levels in cardiomyocytes and cardiac dysfunction in both mice and humans,13,14 and confirm that PAO-induced toxicity and pathology are not restricted to neurodegenerative conditions, but can also cause cardiomyocyte-based pathogenesis and lead to heart failure. Both previous and current TG mouse models confirm the hypothesis that PAO expression and accumulation leads to cardiomyocyte loss resulting in heart failure.
Cardiomyocyte-specific expression of CryABR120G led to high intracellular PAO levels and subsequent activation of apoptosis.19 Similarly the PQ83 data support the idea that PAO-forming proteins can induce toxicity in multiple cell-types but illustrates that not all PAOs activate apoptosis as a mode of cell death. Many proteins are potentially amyloidogenic,9,26 and therefore are potentially pathogenic if the protein accumulates as oligomeric amyloidogenic intermediates and is not cleared by the normal degradation pathways. Proteasome-based degradation and recycling via autophagy and lysosomal digestion represent two distinct pathways for clearance of misfolded proteins. The narrow barrel structure of the proteasome precludes the entry of large protein oligomers or aggregates for degradation, and thus these substrates must necessarily be processed via autophagic/lysosomal degradation.27 Autophagy is the catabolic process whereby cellular contents are sequestered by double-membrane vacuoles, termed autophagosomes, and delivered to the lysosomes for degradation. Autophagy can function as a survival mechanism in starving cells but under certain circumstances, also serves as a form of programmed cell death, and dysregulated or upregulated autophagy can contribute materially to pathogenic processes in a variety of cell types under stress, including the heart.28 We observed increased autophagic and lysosomal content in PQ83 hearts. However, whether it is beneficial or maladaptive in this system is presently unclear; further studies will be needed to determine whether autophagy is contributing to PAO-induced cardiomyocyte toxicity and death.
Although up-regulation or dysregulation of autophagy in the cardiomyocytes may be potentially pathogenic, particularly when the heart is stressed, autophagy appears to be essential for basal cardiac function. When the autophagy-related gene atg5 was ablated in mouse cardiomyocytes, the cells exhibited decreased autophagy but rapidly hypertrophied, and the animals developed severe heart failure, illustrating the essential nature of this process in conserving cardiomyocyte function and homeostasis.29 While the exact mechanism of PAO toxicity remains unclear, several PAOs have been shown to create pores in cellular membranes, thereby causing a loss in membrane permeability.9,24 Our data are consistent with this primary pathogenic mechanism and PQ83 expressing hearts show increased sarcolemmal permeability.
Polyglutamine PAO expression in the cardiomyocytes is clearly toxic and leads to increased sarcolemmal permeability, inflammation and cell death via autophagy and necrosis. While autophagy and necrosis have largely been studied as independent pathways of death, recent data have implicated autophagy in necrotic cell death.30–32 Consistent with our observations in the PQ83 model, a study in C. elegans demonstrated a marked increase in autophagosome formation early during necrotic processes and demonstrated that both genetic and pharmacological inhibition of autophagy effectively suppressed necrotic cell death.30 Further work will be needed to dissect the relationships between autophagy and lysosomal proteolytic mechanisms as contributors to necrotic death within the general context of cardiomyocyte loss and heart failure. The data in our current study underscore the importance of understanding the mechanism(s) by which intracellular PAOs cause cardiomyocyte death. Considering the high levels of PAO that we have detected in human hearts that failed from a variety of etiologies,13 further study of these toxic entities in cardiomyocyte-based models and human heart failure is warranted.
Acknowledgments
This work was supported by NIH research grants P01HL69799, P50HL074728, P50HL077101, P01HL059408, R01HL087862, the International Collaboration Research Project with the Japanese Health Science Ministry (J.R.), and T32 HL07752, F32 HL087478 and an American Heart Association Post-Doctoral Fellowship (J.S.P.).
References
- 1.Comenzo RL. Primary systemic amyloidosis. Curr Treat Options Oncol. 2000;1:83–89. doi: 10.1007/s11864-000-0018-9. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy RE, 3rd, Kasper EK. A review of the amyloidoses that infiltrate the heart. Clin Cardiol. 1998;21:547–552. doi: 10.1002/clc.4960210804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narang R, Chopra P, Wasir HS. Cardiac amyloidosis presenting as ischemic heart disease. A case report and review of literature. Cardiology. 1993;82:294–300. doi: 10.1159/000175878. [DOI] [PubMed] [Google Scholar]
- 4.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 5.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 7.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- 10.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 11.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 12.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 13.Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–10136. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanbe A, Osinska H, Villa C, Gulick J, Klevitsky R, Glabe CG, Kayed R, Robbins J. Reversal of amyloid-induced heart disease in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2005;102:13592–13597. doi: 10.1073/pnas.0503324102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunawardena S, Goldstein LS. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch Neurol. 2005;62:46–51. doi: 10.1001/archneur.62.1.46. [DOI] [PubMed] [Google Scholar]
- 16.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 17.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 18.Maloyan A, Gulick J, Glabe CG, Kayed R, Robbins J. Exercise reverses preamyloid oligomer and prolongs survival in alphaB-crystallin-based desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2007;104:5995–6000. doi: 10.1073/pnas.0609202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation. 2005;112:3451–3461. doi: 10.1161/CIRCULATIONAHA.105.572552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seki T, Takahashi H, Adachi N, Abe N, Shimahara T, Saito N, Sakai N. Aggregate formation of mutant protein kinase C gamma found in spinocerebellar ataxia type 14 impairs ubiquitin-proteasome system and induces endoplasmic reticulum stress. Eur J Neurosci. 2007;26:3126–3140. doi: 10.1111/j.1460-9568.2007.05933.x. [DOI] [PubMed] [Google Scholar]
- 21.de Almeida SF, Picarote G, Fleming JV, Carmo-Fonseca M, Azevedo JE, de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem. 2007;282:27905–27912. doi: 10.1074/jbc.M702672200. [DOI] [PubMed] [Google Scholar]
- 22.Huang JS, Huang SS, Tang J. Cathepsin D isozymes from porcine spleens. Large scale purification and polypeptide chain arrangements. J Biol Chem. 1979;254:11405–11417. [PubMed] [Google Scholar]
- 23.Krenz M, Sanbe A, Bouyer-Dalloz F, Gulick J, Klevitsky R, Hewett TE, Osinska HE, Lorenz JN, Brosseau C, Federico A, Alpert NR, Warshaw DM, Perryman MB, Helmke SM, Robbins J. Analysis of myosin heavy chain functionality in the heart. J Biol Chem. 2003;278:17466–17474. doi: 10.1074/jbc.M210804200. [DOI] [PubMed] [Google Scholar]
- 24.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 25.Carrell RW. Cell toxicity and conformational disease. Trends Cell Biol. 2005;15:574–580. doi: 10.1016/j.tcb.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 27.Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol. 2006;76:89–101. doi: 10.1016/S0070-2153(06)76003-3. [DOI] [PubMed] [Google Scholar]
- 28.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 30.Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 2007 doi: 10.1038/sj.cdd.4402231. [DOI] [PubMed] [Google Scholar]
- 31.Yue Z. Regulation of neuronal autophagy in axon: implication of autophagy in axonal function and dysfunction/degeneration. Autophagy. 2007;3:139–141. doi: 10.4161/auto.3602. [DOI] [PubMed] [Google Scholar]
- 32.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]