

SUMMARY
Expression of the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase associated with cell proliferation and survival, is overactive in many tumors of epithelial origin. Blockade of the kinase activity of EGFR has been used for cancer therapy; however, by itself, it does not seem to reach maximum therapeutic efficacy. We report here that in human cancer cells, the function of kinase-independent EGFR is to prevent autophagic cell death by maintaining intracellular glucose level through interaction and stabilization of the sodium/glucose cotransporter 1 (SGLT1).
INTRODUCTION
As a member of the receptor tyrosine kinase family, the epidermal growth factor receptor (EGFR), upon ligand binding, dimerizes, autophosphorylates, and triggers cascades of downstream signaling, such as activation of phosphatidylinositol-3-kinase/Akt, mitogen-activated protein kinase (MAPK), Jak/Stat, and protein kinase C and modulation of ion channels, by which cell proliferation and survival are enhanced (Wells, 1999). Overexpression/activation of EGFR, which is often found in tumors of epithelial origin, is associated with metastasis, poor prognosis, and resistance to chemotherapy (Nicholson et al., 2001), which makes it an ideal target for therapy. Multiple clinical trials of using EGFR tyrosine kinase inhibitors in cancer therapy have been conducted, but blockage of tyrosine kinase activity alone does not seem to reach maximum therapeutic efficacy. The general response rates are between 10%–20% across a variety of human malignancies (Fukuoka et al., 2002; Kris et al., 2002; Cohen et al., 2003; Dancey and Freidlin, 2003).
The expression level of EGFR in cancer tissues is correlated with prognosis, but not with responsiveness, to EGFR tyrosine kinase inhibitor treatment (Arteaga, 2002), suggesting that, independent of its kinase activity, EGFR may contribute to the progression of cancer. The existence of kinase-independent prosurvival function of EGFR is supported by several studies. First of all, loss of kinase activity of EGFR does not produce similar phenotypes as to loss of EGFR protein in vivo. EGFR knockout animals die soon after birth (Miettinen et al., 1995), but animals with severely compromised kinase mutant EGFR are completely viable and display only some epithelial defects (Luetteke et al., 1994). Second, EGFR without kinase activity was shown to be able to stimulate DNA synthesis (Coker et al., 1994) and enhance cell survival (Ewald et al., 2003). Finally, inhibition of the kinase activity of EGFR by tyrosine kinase inhibitors often leads to decreased cell proliferation but not cell death (Harari and Huang, 2004), whereas knocking down the EGFR receptor protein results in cell death (Nagy et al., 2003).
In this study, we investigated the mechanism of kinase-independent prosurvival function of the EGFR and found that, independent of its kinase activity, EGFR prevents cancer cells from autophagic cell death by maintaining the basal intracellular glucose level.
SIGNIFICANCE
Overexpression/activation of EGFR, which is often found in tumors of epithelial origin, is associated with metastasis, poor prognosis, and resistance to chemotherapy. Multiple clinical trials using EGFR tyrosine kinase inhibitors in cancer therapy have been conducted; however, blockage of tyrosine kinase activity alone does not seem to reach maximum therapeutic efficacy. We report here that EGFR, independent of its kinase activity, maintains the basal intracellular glucose level, thereby preventing cells from undergoing autophagic death. This function of EGFR may endow tumor cells with an increased survival capacity even in the presence of chemotherapeutic agents and tyrosine kinase inhibitors. Thus, the inhibition of this function and of the kinase activity of EGFR may both be necessary for eradication of epithelial neoplasms.
RESULTS
Loss of Expression of EGFR, but Not Its Kinase Activity, Resulted in Autophagic Cell Death
PC-3MM2 cells werecultured in minimum essentialmedium (MEM) containing physiological glucose content of 5.5 mM (Baltzan et al., 1962). As shown in Figure 1A, EGFR tyrosine kinase inhibitor, AEE788 (Traxler et al., 2004) (5.0 μM), did not decrease the expression of EGFR but did completely inhibit its phosphorylation. In contrast, the transfection of the cells with EGFR siRNA decreased the expression of the EGFR (Figure 1B). As shown in Figure 1C, unlike control cells, treatment of PC-3MM2 cells with AEE788 (5.0 μM) for 3 days led to inhibition of cell proliferation, but not to cell death. However, incubation of PC-3MM2 cells transfected with EGFR siRNA for 3 days in MEM resulted in cell death, as indicated by the presence of sub-G1 cells. The use of the commercial EGFR kinase inhibitor, AG1478 (data not shown), and different siRNA against EGFR produced similar results (Figure S1 available online).
Figure 1. Blocking the Kinase Activity of EGFR Does Not Lead to Cell Death but Knocking Down EGFR with siRNA Does.
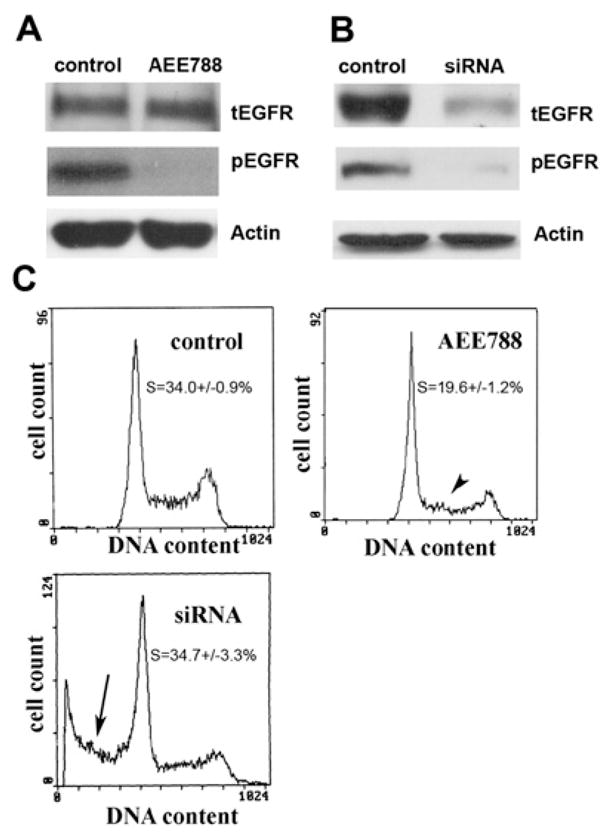
(A) PC3MM2 cells grown in MEM with 5.5 mM glucose were treated with AEE788 (5.0 μM, with AEE788 readded every 24 hr) for 72 hr. Western blot analysis revealed that pEGFR was completely blocked by AEE788 compared with the control. β-actin served as a loading control (tEGFR, total EGFR).
(B) Seventy-two hours later after cells were cultured in MEM with 5.5 mM glucose, tEGFR and pEGFR levels were both reduced by siRNA treatment compared with the control that was transfected with siRNA vector-expressing scrambled sequences. β-actin served as a loading control.
(C) Compared with the control cells (there was no difference between non-transfected control cells and control siRNA vector transfected cells), AEE788 only caused a decrease in the proportion of cells in S phase (p < 0.01, arrowhead) with no detectable sub-G1 cells (dead cells). However, the EGFR siRNA-treated cells showed a significant (p < 0.01) proportion of sub-G1 cells (arrow).
To characterize the cell death due to loss of the EGFR protein, we measured EGFR downstream signalings Akt and MAPK and apoptotic-associated caspases 9 and 3 by western blotting. Contrary to our expectation, the knocking down of EGFR by siRNA led to upregulation of phosphorylated Akt (pAkt) and phosphorylated MAPK (pMAPK) without changes in Akt and MAPK levels (Figure 2A). Only procaspases 9 and 3 were detected, but not their cleaved forms, indicating that the cell death caused by knocking down EGFR was not due to typical apoptosis.
Figure 2. Cell Death Induced by Knocking Down EGFR with siRNA Is Characterized as Autophagic Cell Death.
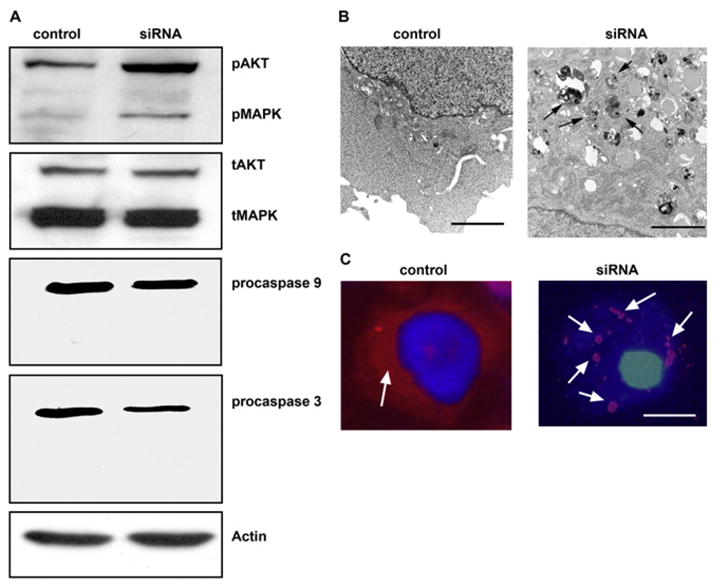
(A) Western blot analysis of the activity of Akt, MAPK, and caspases 9 and 3 in EGFR siRNA-treated cells. Both Akt and MAPK pathways were more active in siRNA-treated cells than in control cells, as indicated by the upregulation of phosphorylated Akt (pAkt) and MAPK (pMAPK); the total Akt (tAkt) and MAPK (tMAPK) were not changed. No cleaved form of caspase 9 or 3 was detected in the siRNA-treated cells; only their proforms (procaspase 9 and 3) were present, indicating that the cell death caused by knocking down EGFR was not a typical apoptosis. β-actin served as a loading control.
(B) Transmission electron microscopy of control and EGFR siRNA-treated cells. Knocking down EGFR resulted in the appearance of autophagosomes (arrows) in the cytoplasm, which were absent from the control cells. (N, nucleus; bar, 200 nm). (C) LC3 staining (arrow, red color) in control and EGFR siRNA-transfected cells. Note that in the cytoplasm of control cells LC3 is diffused, whereas LC3 is in aggregates in the EGFR-siRNA treated cells (arrows, red color; bar, 2 μm).
To further elucidate the mechanisms of this cell death, we examined the cells with transmission electron microscopy. As shown in Figure 2B, the EGFR siRNA-transfected cells contained multiple autophagosomes: that is, lysosome-infused cytoplasmic organelles in which the contents are degraded for energy production (Kroemer and Jaattela, 2005). It is worth noting here that, as a survival mechanism triggered by an intracellular energy crisis, autophagy provides cells with an energy backup mechanism, ultimately leading to death if external energetic substrates remain deprived (Lum et al., 2005). Aggregates of exogenous microtubule-associated protein 1 light chain 3 (LC3) (Tanida et al., 2004) were found in the cytoplasm of EGFR siRNA-transfected cells, but not in the control cells (Figure 2C), providing additional evidence that the cell death was due to autophagy. Treatment of EGFR siRNA-transfected cells with the autophagy inhibitor 3-methyladenine (Petiot et al., 2000) resulted in cell death with characteristics of necrosis—translocation of HMGB1 (high mobility group box 1) (Ito et al., 2007) from the nucleus to the cytoplasm and cell lysis in situ (Figure S2). The autophagic phenotype in PC-3MM2 cells treated with EGFR siRNA was also seen in other cell types, for example, MDA-MB231 human breast cancer cells and KM12C human colon cancer cells (Figures S3A and S3B).
Decreased Intracellular Glucose Level Is Responsible for Autophagic Cell Death Induced by EGFR siRNA Treatment
Because glucose is the major energy substrate for all cells and tumor tissues accumulate and consume more glucose than do normal tissues (Gatenby and Gillies, 2004), we next measured the intracellular glucose level in cells treated with AEE788 and cells transfected with EGFR siRNA. The data shown in Figure 3A reveal that 3 days of treatment with AEE788 did not alter the intracellular glucose level. In sharp contrast, in cells treated with EGFR siRNA, 3 days of culture in MEM containing 5.5 mM glucose led to a 50% decrease in the glucose level (Figure 3B). Similar data were found in EGFR siRNA-treated human breast cancer MDA-MB231 and human colon cancer KM12C cells (Figure S3C). Notably, the cell death phenotype caused by the knocking down of EGFR could be rescued by increasing the glucose content in the MEM to the same level as in Dulbecco’s modified Eagle’s medium with a high (25 mM) glucose level (Figure 3C). As shown in Figure 3D, incubating EGFR siRNA cells in MEM containing high glucose levels also reversed the autophagic phenotype as indicated by the disappearance of autophagosomes. The cell death caused by decreased EGFR expression was thus due to autophagy triggered by a decreased intracellular glucose level. High glucose MEM treatment also increased the intracellular glucose level (Figure 3E) and decreased pAKT and pMAPK (Figure 3F) of siRNA-treated cells, suggesting the increased phosphorylation of AKT and MAPK in response to EGFR knocking down (Figure 2A) may be a stress response to lower intracellular glucose level.
Figure 3. Knocking Down EGFR Leads to Decreases in Intracellular Glucose Level, and Increasing the Glucose Content in the Culture Medium Rescues the Cell from Death Caused by Knocking Down EGFR.
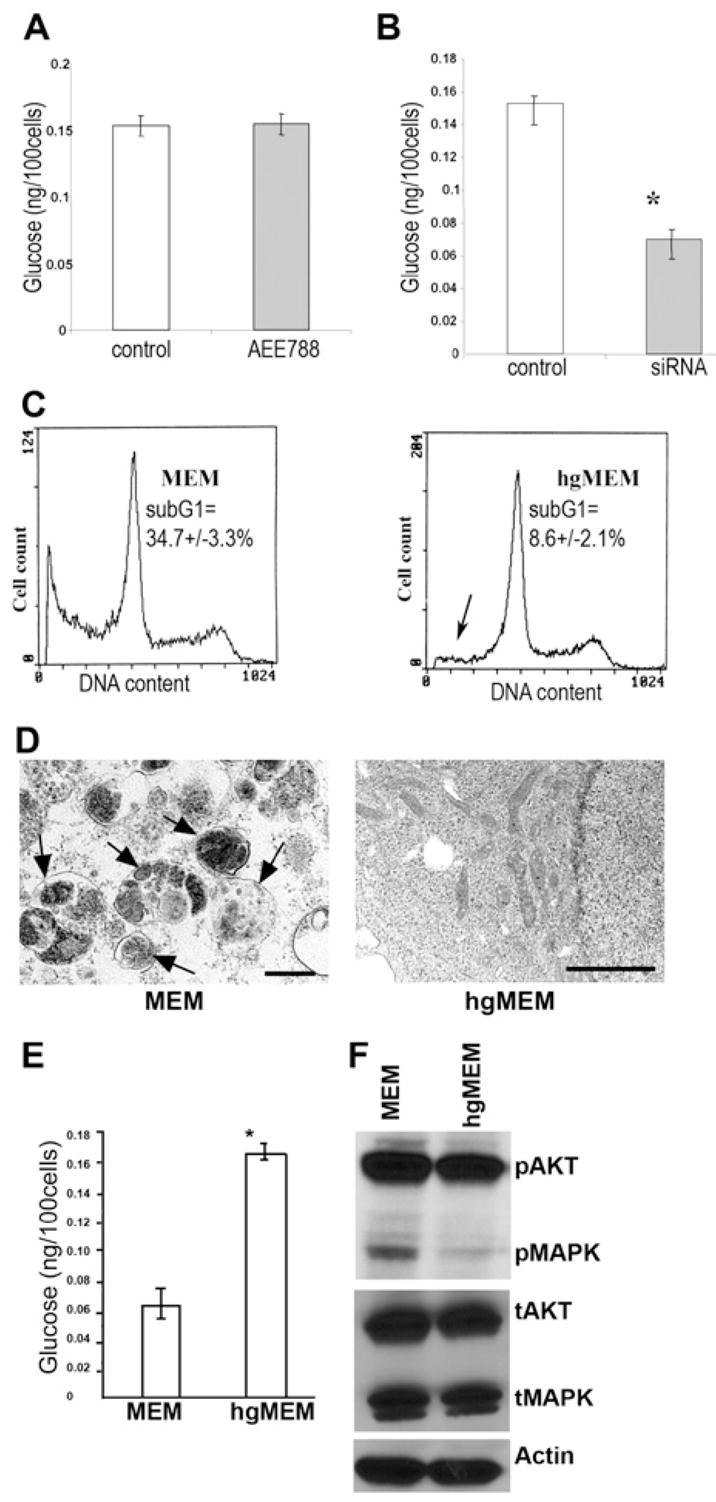
(A) Measurement of intracellular glucose content. AEE788 (5.0 μM) treatment for 72 hr did not alter the intracellular glucose content. (Error bars indicate mean ± SD.)
(B) The glucose content of EGFR siRNA-treated cells was significantly decreased (p < 0.001) compared with that of control cells after a 72 hr culture in MEM with 5.5 mM glucose. (Triplicate samples were used in each group. The asterisk indicates a significant difference with p < 0.001. Error bars indicate mean ± SD.)
(C) Culturing EGFR siRNA-transfected cells in high glucose (25 mM) MEM (hgMEM) significantly reduced (p < 0.01) the cell death proportion, as indicated by the decrease in sub-G1 cells (arrow). (Triplicate samples were used in this experiment.)
(D)The hgMEM-treated cells also lead to disappearance of autophagosomes on transmission electron microscopy (bar, 2 μm).
(E) The intracellular glucose level was also restored by hgMEM treatment. (Error bars indicate mean ± SD.)
(F) Western blotting shows that hgMEM treatment was also able to inhibit the enhanced activation of AKT and MAPK induced by knocking down EGFR.
Consequential Loss of SGLT1 following EGFR Knocking Down Leads to a Decrease in Intracellular Glucose Level
Glucose is transported into cells by two families of transporter—a facilitative-type glucose transporter family (GLUT) and an active-type glucose transporter family (sodium/glucose cotransporter [SGLT]) that in human cells consists of two major members (SGLT1 and SGLT2). In response to stress or stimuli, such as insulin and hormones, GLUTs translocate from the intracellular compartment to the cell membrane, thus transporting glucose along a glucose gradient (inward or outward) (Wood and Tryhurn, 2003). GLUT1 is the most wildly distributed, serving many cell types for glucose uptake (Zierler, 1999). In contrast, SGLT transports glucose into cells regardless of glucose concentration in the medium and cells depend on SGLT to accumulate and maintain higher intracellular glucose levels (Wright et al., 1994). SGLT1 is the major active glucose transporter in the body, and SGLT1 (but not SGLT2 measured by RT-PCR; data not shown) was expressed in PC-3MM2 cells.
To investigate which glucose transporter systems may contribute to the cell death phenotype caused by the EGFR knockdown, we measured the expression of GLUT1 and SGLT1. In the EGFR siRNA-treated cells, the expression of SGLT1 was reduced to undetectable levels while the expression of GLUT1 was not suppressed by the treatment (Figure 4A). In addition, knocking down SGLT1 by SGLT1 siRNA was sufficient to produce autophagic cell death in low glucose (5.5 mM) MEM (Figures 4B–4D), which can be rescued by high glucose (25 mM) MEM (Figure 4C). Together, these results raise an interesting possibility that EGFR knocking down-induced SGLT1 downregulation contributed to the autophagic cell death.
Figure 4. Knocking Down EGFR Leads to Downregulation of SGLT1, and Knocking Down SGLT1 Results in a Similar Cell Death Phenotype as Knocking Down EGFR.
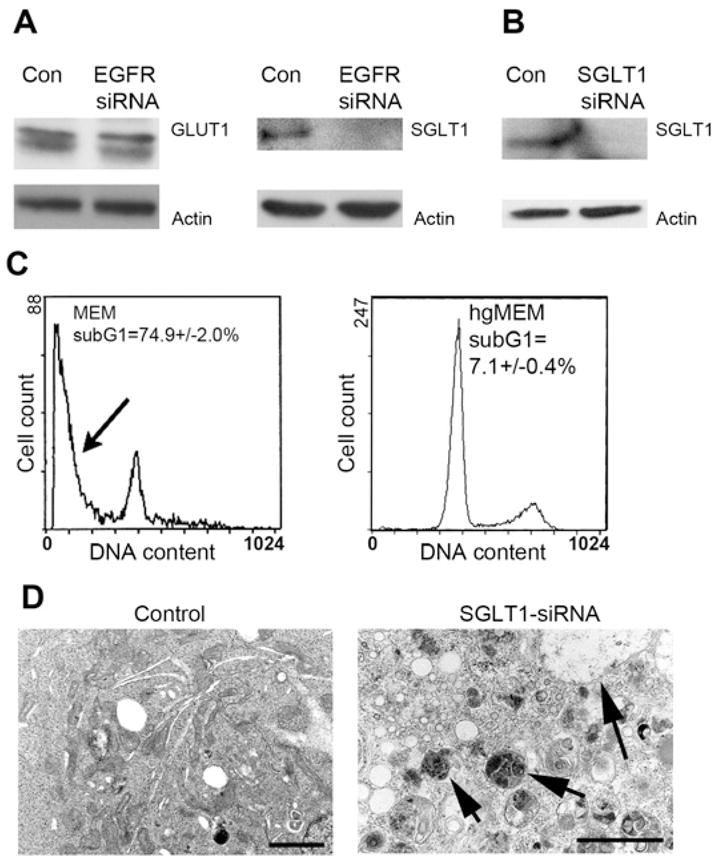
(A) No changes on GLUT1 expression were detected by knocking down EGFR, but SGLT1 was downregulated to an undetectable level. β-actin served as a loading control.
(B) Cells treated with SGLT siRNA led to downregulation of SGLT1 after 4 days of culture in MEM.
(C) This treatment also led to a significant proportion of cell deaths (p < 0.01) as evidenced by the presence of sub-G1 cells (arrow), and hgMEM was able to prevent cells from death. (The flow cytometry data of control vector-transfected cells were similar to those of the control cells used in the EGFR siRNA experiment in Figure 2.)
(D) As with the EGFR siRNA-treated cells, cells that underwent SGLT1 siRNA treatment also showed a large amount of autophagosomes (arrows) on transmission electron microscopy (bar, 2 μm).
EGFR Interacts with SGLT1 Independent of the Kinase Activity of EGFR
Next, we measured the protein and mRNA expression of SGLT1 in PC-3MM2 cells over time after knocking down EGFR by using siRNA. As shown in Figure 5A, the protein level of EGFR decreased at 24 hr and even more at 48 hr after transient transfection with EGFR siRNA. Similar results were obtained for the SGLT1 protein and intracellular glucose level. The mRNA level of EGFR decreased in response to EGFR siRNA treatment, whereas the mRNA level of SGLT1 did not (both measured by reverse transcriptase-polymerase chain reaction [RT-PCR]) (Figure 5B). These results suggest that the downregulation of SGLT1 in cells treated with EGFR siRNA occurred at the protein level. To test whether the decrease of SGLT1 was due to its degradation, we added the proteasome inhibitor MG132 to the medium with the EGFR siRNA-treated cells. As shown in Figure 5C, the addition of MG132 rescued the level of SGLT1, indicating that the decrease in SGLT1 in response to knocking down EGFR is due to degradation.
Figure 5. EGFR Interacts with and Stabilizes SGLT1 Independent of EGFR Kinase Activity.
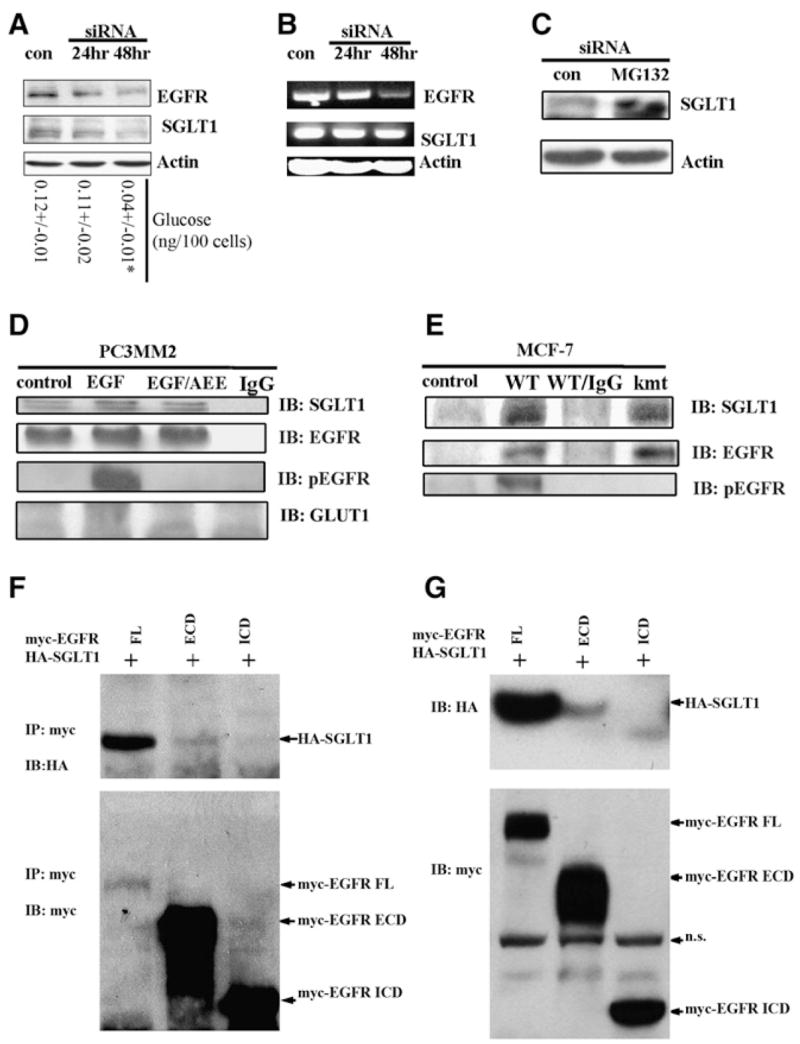
(A) Western blot revealed a time-dependent downregulation of SGLT1 by knocking down EGFR with siRNA (con, control). At each time point, levels of glucose were measured. *p < 0.05.
(B) RT-PCR analysis of EGFR mRNA and SGLT1 mRNA in response to EGFR siRNA treatment (con, control). The EGFR mRNA became downregulated at 48 hr after EGFR siRNA treatment, whereas the SGLT1 mRNA level remained unchanged.
(C) Proteasome inhibitor MG132 blocked the downregulation of SGLT1 by EGFR siRNA; siRNA and MG132 treatments were for 24 hr. Actin was used as an internal control.
(D) EGFR physically interacted with SGLT1, independent of EGFR kinase activity, as revealed by coimmunoprecipitation with the anti-EGFR antibody C225. PC3MM2 cells were cultured in serum-free medium for 12 hr before being treated with 40 ng/ml EGF or with EGF and AEE788 (EGF/AEE). The blotting of immunoprecipitates was against SGLT1, total EGFR, pEGFR, and GLUT1. Normal mouse IgG was used as a negative control).
(E) kmtEGFR also interacted with SGLT1 as did WT-EGFR in MCF-7 cells co-transfected with WT-EGFR/SGLT1 or kmtEGFR/SGLT1.
(F) HA-SGLT1 was coimmunoprecipitated with myc-EGFR and myc-ECD, but not myc-ICD.
(G) Western blotting detection of HA-SGLT1 coexpressed with myc-tagged full-length (FL), the extracellular domain (ECD containing the transmembrane domain), and intracellular domain (ICD) of EGFR in HEK293 cells. Note that HA-SGLT1 was only efficiently coexpressed with full-length EGFR, to much less extent with ECD, but not with ICD of EGFR (IP, immunoprecipitation; IB, immunoblotting; n.s., nonspecific band).
Because both EGFR and SGLT1 are membrane proteins, one possibility is that they physically interact with each other. To test this, we performed an immunoprecipitation assay. Using the anti-EGFR specific antibody C225 (Goldstein et al., 1995), we were able to coprecipitate SGLT1 with EGFR, independent of EGFR phosphorylation (Figure 5D). To further test the kinase independence of the EGFR-SGLT1 interaction, we coexpressed wild-type EGFR (WT-EGFR) or kinase domain-mutated EGFR (kmtEGFR) (Chan and Gill, 1996) with SGLT1 in human MCF-7 cells, which express very low level of EGFR protein (Davidson et al., 1987). As shown in Figure 5E, immunoprecipitation of EGFR with the C225 antibody coprecipitated SGLT1 with either WT-EGFR or kmtEGFR. These results support the conclusion that the interaction of EGFR with SGLT1 was independent of EGFR kinase activity.
To illustrate which domains of EGFR, intracellular or extracellular-transmembrane domain, interacts with SGLT1, we used two truncated forms of EGFR (Hsu and Hung, 2007): one has only the intracellular domain (ICD) and the other has both the transmembrane and extracellular domains (ECD). These two truncated forms of EGFR contain myc tags at their C termini. We also created C-terminal HA-tagged SGLT1. We coexpressed HA-tagged SGLT1 with myc-tagged full-length ICD or ECD of EGFR individually in HEK293 cells. As shown in Figure 5F, the full-length EGFR was coprecipitated with SGLT1. To a lesser extent, SGLT1 was also coprecipitated with ECD, but not with ICD. Consistently, HA-SGLT1 was efficiently coexpressed with full-length EGFR, to a much less extent with ECD, but not expressed with ICD (Figure 5G). Together, the results suggest that ECD of EGFR is required for interaction with SGLT1 and the full-length EGFR is required to efficiently stabilize SGLT1.
Because both WT-EGFR and kmtEGFR interacted with SGLT1, we reasoned that both should be able to rescue the autophagic death phenotype in cells transfected with EGFR siRNA by stabilizing SGLT1. We therefore designed siRNA to target the 5′UTR of the EGFR mRNA. The use of expression vectors lacking the 5′UTR sequence of EGFR allowed the reexpression of WT-EGFR or kmtEGFR in the PC-3MM2 cells. As shown in Figure 6A, the 5′UTR siRNA dramatically downregulated the EGFR level in treated versus control vector-transfected PC-3MM2 cells. Moreover, the transient expression of either WT-EGFR or kmtEGFR preserved SGLT1 (Figure 6A) and rescued the cells from death (Figure 6B).
Figure 6. Rescue Cells Treated with EGFR siRNA from Death by Re-Expression of WT-EGFR or kmtEGFR.
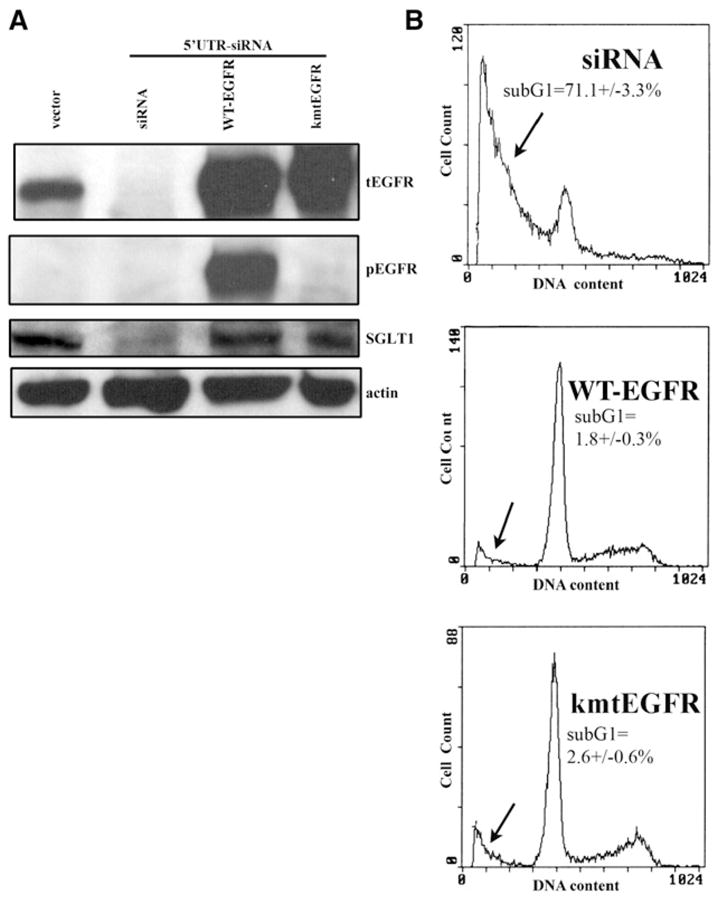
(A) PC-3MM2 cells were treated with siRNA targeting the 5′UTR region of EGFR mRNA, which downregulated the EGFR level after a 4-day siRNA treatment. Both WT-EGFR and kmtEGFR were re-expressed in the siRNA-treated cells and were able to upregulate SGLT1 expression. The absence of kinase activity of kmtEGFR was shown by western blotting with an anti-pEGFR antibody. β-actin served as a loading control.
(B) The re-expression of WT-EGFR and kmtEGFR also rescued the EGFR siRNA-treated cells from death. The sub-G1 proportion is indicated by the arrows. (Duplicate samples were used in each group of samples.)
Survival Advantage of EGFR/SGLT1 Expressing Cells in Medium with Low Level of Glucose
Considering the status of EGFR overexpression in malignant tumors and the stability dependency of SGLT1 on EGFR expression, we argue that the more EGFR/SGLT1 tumor cells harbor, the less they depend on the level of extracellular glucose for survival. To test it, we compared the sensitivity of three cell lines to glucose starvation: A431, PC3-MM2, and MCF-7 representing high, medium, and low/no EGFR expression, respectively (Figure 7A). Both EGFR-expressing cells A431 and PC3-MM2 expressed SGLT1, but MCF-7 did not. Each type of cell was cultured in three kinds of medium containing high (25 mM), physiological (5 mM), and subphysiological (3 mM) glucose for 3 days, and cell death was measured by flow cytometry. As shown in Figure 7B, the EGFR-expressing cells A431 and PC3-MM2 are resistant to glucose starvation-induced cell death, while EGFR low cells, MCF-7, could not survive even in 5 mM glucose-containing medium (Figure 7B). In addition, overexpression of either EGFR or SGLT1 resulted in enhanced survival of MCF-7 cells in low glucose MEM (Figure 7C). Due to very low expression level of SGLT1 in MCF-7 cells (undetectable by WB in Figure 7A but detectable mRNA by RT-PCR; data not shown), the SGLT1 expression in the EGFR-transfected MCF-7 cells is not as high as SGLT1-transfected MCF-7 cells (Figure 7D, lane 2 versus lane 3). It is worthwhile to mention that transfection of EGFR in MCF-7 cells showed better prosurvival effect than transfection of SGLT1 alone (Figure 7C versus Figure 7D). Thus, in addition to the EGFR-stabilized SGLT1, other mechanisms induced by traditional EGFR-mediated pathway may also contribute to the prosurvival phenotype shown in Figure 7C. Together, the results support survival advantage of EGFR/SGLT1 expression for cells cultured in the low glucose medium.
Figure 7. Survival Advantage of EGFR/SGLT1-Expressing Cells in a Low Level of Glucose Medium.
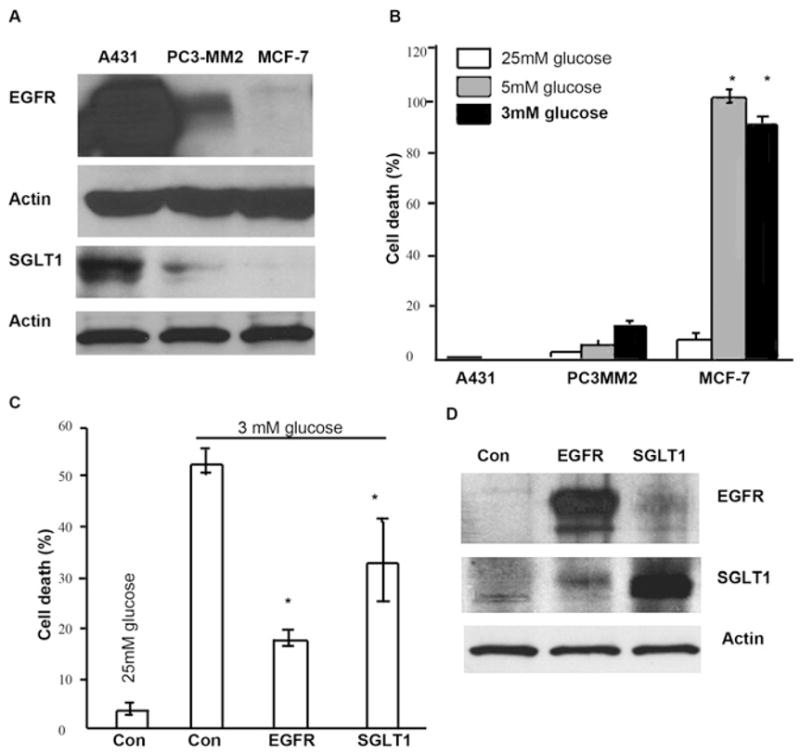
(A) Western Blotting analysis demonstrated that SGLT1 was expressed in EGFR-positive cells, A431 and PC3-MM2, but not in MCF-7.
(B) When A431, PC3-MM2, and MCF-7 cells were cultured in media containing different levels of glucose, 25 mM, 5 mM, and 3 mM, for 3 days, only MCF-7 cells showed significant degree of cell death. (Error bars indicate mean ± SD.) (C and D) Either EGFR or SGLT1 was transiently expressed in MCF-7 cells cultured in 25 mM containing MEM. Forty-eight hours later after the transfection, medium was changed to MEM containing 3 mM glucose. After a 2 day incubation in low glucose MEM, cells were either fixed for cell death analysis by flow cytometry (C) or harvested for western blotting analysis of EGFR and SGLT1 (D). (Triplicate samples were used in each group of samples. Asterisks represent statistical significance as compared with cells grown in high glucose [25 mM] medium, p < 0.05. Error bars indicate mean ± SD.)
DISCUSSION
Activation of EGFR has been reported to transiently increase glucose transport (Inman and Colowick, 1985). We reproduced this transient increase in glucose uptake following the activation of EGFR in PC-3MM2 cells by exposure to EGF in serum-free medium. This activation was abrogated by the presence of the EGFR tyrosine kinase inhibitor AEE788 (Traxler et al., 2004) (Figure S4). Inhibition of EGFR phosphorylation, however, only blocked the peak glucose uptake and did not decrease the level of intracellular glucose to below that found in cells whose EGFR was not activated (Figure S4). These data suggest that peak glucose intake into cells requires EGFR kinase activity, but maintenance of a basal level of intracellular glucose does not. Indeed, expression of nonphosphorylated EGFR is often observed in normal human tissues (Hudelist et al., 2006) as well as in multiple tumor samples (Piazzi et al., 2006), where the role of EGFR may likely be maintaining basal glucose uptake required for survival.
Although kinase-independent functions of EGFR have been reported previously (Coker et al., 1994; Ewald et al., 2003), efforts to understand the role of EGFR have been largely directed to its kinase-related activity. The yet unimpressive clinical outcomes of EGFR tyrosine kinase inhibitors for treatment of multiple types of cancer suggest that kinase independent functions of EGFR may be a significant contributor for cancer progression.
The prosurvival and proproliferation roles of EGFR might be mediated by at least two separated pathways. Activation of the EGFR by its ligands results in increased cell proliferation, which is often supported by data of deceleration of cell proliferation by inhibitions of tyrosine kinase activity of EGFR (Mendelsohn, 2001; Rodeck et al., 1997; Peng et al., 1996). However, very rarely (Wu et al., 1995), inhibition of the tyrosine kinase activity of EGFR leads to cell death. The lack of cytotoxicity of inhibitors of EGFR tyrosine kinase may partially explain the clinical outcome of using tyrosine kinase inhibitors in cancer treatment (Dancey and Freidlin, 2003).
Our present study shows that EGFR is a stabilizer of an active glucose transporter, SGLT1, empowering cancer cells with the ability to uptake the basic energy substrate, glucose, regardless the level of extracellular glucose, for their survival. Maintaining a sufficient level of intracellular ATP is required to prevent cells from dying. There is at least one commonality among different kinds of cell deaths, apoptosis, necrosis, and autophagic cell deaths, which is an energy crisis triggered at different levels along their death pathways. During apoptosis, ATP level sharply decreases when mitochondria lose their transmembrane potential. In hypoxia-induced necrosis, the most common cause of necrosis in vivo, depletion of ATP precedes mitochondrial permeability alteration. Autophagy, a process of self-degradation to complement environmental energy/nutrient paucity, is also characterized with ATP insufficiency occurring prior to cell death (Lemasters et al., 2002; Skulachev, 2006). The presence of SGLT1 allows cancer cells to uptake enough glucose for ATP generation via glycolysis (Nishimura et al., 1998). In general, when mitochondria are dysfunctional, high levels of glucose or ATP can prevent/delay cell deaths, such as apoptosis (Malhotra and Brosius, 1999; Palaga et al., 2004; Honda et al., 2003) and necrosis (Kinzer and Lehmann, 1991).
In normal tissues where active glucose uptake is critical for the body, such as the epithelium of intestine, tubules of the kidney (Lee et al., 1994), and vascular endothelium of the brain (Elfeber et al., 2004), EGFR and SGLT1 are coexpressed. In fact, activation of EGFR in the epithelium of intestine leads to active transport of glucose (Hardin et al., 1996). Using EGFR/SGLT1 double-negative (at mRNA level) HEK293 cells, we found that exogenous SGLT1 could only be expressed when EGFR was co-transfected (Figure S5), supporting the notion that EGFR may stabilize SGLT1. The higher content of glucose in tumor cells, as compared to normal cells (Gatenby and Gillies, 2004) requires the active glucose transport system SGLT. Considering the fact that EGFR is overexpressed in tumors of epithelial origin and our current finding that EGFR stabilizes SGLT1, we postulate that SGLT1 might also be overexpressed in EGFR-positive tumors. Indeed, it was reported that SGLT1 is overexpressed in preneoplastic and neoplastic lesions of the head and neck (Helmke et al., 2004) (i.e., in cells that express high levels of EGFR [Ford and Grandis, 2003]). Whether SGLT1 is also overexpressed in other types of epithelial neoplasms remains to be determined.
In summary, we report that EGFR, independent of its kinase activity, maintains the basal intracellular glucose level, thereby preventing cells from undergoing autophagic death. This function of EGFR may endow tumor cells with an increased survival capacity even in the presence of chemotherapeutic agents and tyrosine kinase inhibitors (Ikari et al., 2005). Thus, the inhibition of this function and of the kinase activity of EGFR may both be necessary for eradication of epithelial neoplasms.
EXPERIMENTAL PROCEDURES
Cell Line and Major Reagents
The human metastatic prostate cancer cell line PC-3MM2 was selected from the parental PC3 line in our laboratory (Pattaway et al., 1996). The breast cancer cell lines MCF-7 (originally from the American Type Culture Collection, Manassas, VA) and MDA-MB-231, the skin cancer cell line A431, and the colon cancer cell line KM12C were obtained from The University of Texas M.D. Anderson Cancer Center (Houston, TX). The GeneJuice transfection reagent was from Novagen (San Diego, CA). The U6 promoter-driven siRNA vector with green fluorescent protein (GFP) expression (pRNAT-U6.1/Neo for EGFR, SGLT1, and their scrambled controls) was constructed by Genscript Corp. (Piscataway, NJ). The target sequences for EGFR siRNA were CGCAAAGT GTGTAACGGAATA within exon 13 of the EGFR gene and CTGACTCCGTC CAGTATTGAT within the 5′UTR region of EGFR mRNA. Validated commercial control (cat. no. 4635) and two different EGFR siRNAs (Cat. 51334) (used in experiments shown in Figure S1 were from Ambion [Austin, TX]). The target sequence for SGLT1 siRNA was TCTTCCGCATCCAGGTCAAT. The negative control siRNA sequence was GAACAATGTTGACCAGGTGA. The SGLT1-expressing vector SGLT1- pCMV6-XL4 was from OriGene Technologies (Rockville, MD). LC3 cDNA was a gift of Dr. Seiji Kondo (M.D. Anderson Cancer Center). Antibodies against total EGFR (cat. no. 2232), pEGFR (cat. no. 2434L), total Akt (cat. no. 9277), phosphorylated AKT (cat. no. 9271L), total MAPK (cat. no. 9102), and phosphorylated MAPK (9101L) were from Cell Signaling Technology (Danvers, MA). Antibodies against SGLT1 (cat. no. ab14685) and GLUT1 (cat. no. Ab652) were from Abcam, Inc. (Cambridge, MA). Antibody against HMGB1 (cat. no. H9537) was from Sigma-Aldrich (St. Louis, MO). The monoclonal antibody against C225 was a gift from Dr. Liana Adam (M.D. Anderson Cancer Center). Mouse anti-myc-tag antibody (cat. no. 05–724) and anti-HA-tag antibody (cat. no. 05–904) were from Millipore (Billerica, MA). Rabbit anti-caspase 9 (cat. no. sc-7885); goat anti-caspase 3 (cat. no. sc-1225); rabbit anti-actin (cat. no. sc-7210); and secondary antibodies against rabbit, mouse, or goat labeled with horseradish peroxidase and protein A/G conjugated agarose beads (cat. no. sc-2003) were from Santa Cruz Biotechnology (Santa Cruz, CA). EGFR tyrosine kinase inhibitor AEE788 was from Novartis Pharma (Basel, Switzerland). Commercial EGFR kinase inhibitor AG1478 was from EMD Bioscience (San Diego, CA). Autophagy inhibitor 3-methyladenine was from Sigma-Aldrich (St. Louis, MO). Proteasome inhibitor MG132 was from Sigma-Aldrich.
Construction of N-Terminal HA-Tagged SGLT1
To construct the HA-tagged SGLT1, the human SGLT1 full-length cDNA was amplified by PCR using SGLT1-pCMV6-XL4 as a template with the following primers: 5′-CTAGTTAAGCTTG GATGGACAGTAGCACCTGGAGC-3′ and 5′-A GCGGCCGCCAGCAGGCAAAATATGCATGGC-3′. The DNA fragments were digested with HindIII and NotI and cloned into the corresponding sites of the modified pcDNA6A vector (Invitrogen, CA), in which myc epitope has been replaced with hemaglutinin (HA). The cDNA inserts were confirmed by DNA sequencing.
Treatment with Tyrosine Kinase Inhibitor AEE788 and AG1478
AEE788 and AG1478 were dissolved in dimethyl sulfoxide (vehicle) and then added to the medium in the indicated concentrations, respectively. Control PC-3MM2 cells were treated with medium containing the same volume of the vehicle. For glucose uptake experiments, triplicate cultures of PC-3MM2 cells grown in serum-free medium for 12 hr were treated with the vehicle, EGF (40 ng/ml), or EGF plus AEE788 (0.1 μM), and cells were harvested at 5, 10, 20, 30, 50, and 60 min after treatment and then assayed for glucose uptake (data are shown in Figure S4).
Cell Transfections
To knock down EGFR or SGLT1 expression by using siRNA, PC-3MM2 cells were cultured in MEM supplemented with 10% fetal bovine serum, sodium pyruvate, nonessential amino acids, L-glutamine, and a two-fold vitamin solution in 5% CO2%–95% air at 37°C. Vectors expressing siRNA (targeting exon 13 of the EGFR gene) for EGFR, SGLT1, and their corresponding controls were transfected into the PC-3MM2, MDA-MB-436, and KM12C cells using Gene-Juice. With GFP used as a tracer, the cells were sorted 24 hr later with a cell-sorting machine (BD FACS Vantage SE system; BD Biosciences, Palo Alto, CA) that used green fluorescence as a selector. The effects of siRNA on the expression of the target gene were evaluated by western blotting 24 hr after the sorted cells were reseeded and cultured. Treatment of PC-3MM2 cells with commercial validated control and EGFR siRNAs was done by transient transfection of cells with 100 nM of each siRNA. EGFR expression and analysis of cell death were determined 96 hr after transfection. For each set of experiments, 1.0 × 106 cells with GFP expression were used in each triplicate sample. For 3-methyladenine treatment, a final concentration of 1 μM 3- methyladenine was added to the medium of EGFR siRNA transfected cells 6 hr after sorting. The immunocytochemical staining of HMGB1 was conducted 24 hr later after the 3-methyladenine treatment. The morphological changes of 3-methyladenine-treated cells were monitored by a converted light microscope.
To re-express the WT-EGFR or kmtEGFR, we first knocked down EGFR in PC-3MM2 cells with siRNA (5′UTR-siRNA), targeting the 5′UTR region of EGFR mRNA, which allowed us to use an EGFR-expressing vector that does not contain the 5′UTR region of EGFR. Triplicate cultures of PC-3MM2 cells were then transfected with 5′UTR-siRNA, and 24 hr later, the cells were sorted by using a GFP as a selection marker. The sorted cells were then transfected with either an empty vector (pcDNA3.1[−]) or a vector containing WT-EGFR or kmtEGFR. For LC3 overexpression in control and EGFR siRNA-transfected cells, twelve hours after the siRNA treatment, we transiently transfected 1 μg cDNA of LC3 into 1.0 × 106 cells. For immunocytochemical staining of LC3, the cells were fixed with 70% ethanol after a 72 hr culture in MEM.
To test the interaction between WT-EGFR or kmtEGFR and SGLT1, we used MCF-7 low EGFR-expressing cells. The cells (duplicate) were cultured for 24 hr in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum prior to cotransfection with empty vectors (pcDNA3.1 and pCMV6-SPORT6), WT-EGFR/SGLT1, kmtEGFR/SGLT1, or only SGLT1. The cells were harvested 24 hr after transfection and subjected to immunoprecipitation with a C225 antibody. The precipitates were analyzed for EGFR, phosphorylated EGFR, and SGLT1 by western blotting.
To test which domain of EGFR interact with SGLT1, 1 μg cDNA of myc-tagged EGFR with either intracellular domain truncation or extracellular domain truncation was transiently transfected into PC3MM2 cells culture in 6-well plate. Control cells were transfected an equal amount of vector DNA. Forty-eight hr after transfection, cells were harvested for immunoprecipitation with a mouse anti-myc antibody. A positive control was also included, which is protein extracts of PC3MM2 cells immunoprecipitated with a mouse anti-EGFR C225. The precipitates were analyzed for the presence of SGLT1 by western blotting.
Measurement of Intracellular Glucose and ATP
Prior to harvesting, adherent cultures of control and EGFR siRNA-treated cells in MEM containing 1 mg/ml glucose were washed twice with cold phosphate-buffered saline (PBS) and then lysed with ion-free H2O for 5 min on ice. The glucose content was measured with D-glucose measurement kit (GAHK-20, Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s protocol. Intracellular ATP level was measured using Bioluminescent Somatic Cell Assay Kit (cat. no. FLASC, Sigma-Aldrich) according to the protocol provided by the manufacturer. The level of ATP is reflected by the amount of generated bioluminescence measured by a Luminescence Meter (Monolight 2010, San Diego, CA).
Measurement of Cell Survival in Medium with Low and High Glucose Medium
PC-3MM2, A431, and MCF-7 cells were cultured in MEM containing low glucose (3 or 5 mM) or in MEM supplemented with an additional 3.5 mg/ml D-glucose (high glucose MEM, containing a total of 25 mM glucose). Triplicate of sorted siRNA-expressing cells cultured for 3 or 4 days (indicated in the figure legends) in either MEM or high-glucose MEM were used to test survival in response to changes in the environment. The population of sub-G1 cells (as an indicator of cell death) was determined by flow cytometry. Briefly, trypsinized cells were washed once with MEM containing serum and then washed three times with cold PBS and fixed for 3 hr in cold ethanol (70% v/v). The cells were then centrifuged at 2,000 × g, resuspended in PBS containing 0.05% propidium iodide and 10 μg/ml RNase A, and incubated for 30 min at 37°C before analysis with a fluorescence-activated cell sorter (FACS Elite; BD Biosciences).
Western Blot Analysis
For western blot analysis, PC-3MM2 cells were incubated for 10 min at 0°C in a lysis buffer (50 mM HEPES, pH 7.4; 150 mM NaCl; 0.1% Triton X-100; 1.5 mM MgCl2; 1 mM EDTA; and 1 mM phenylmethlysulfonyl fluoride). Equal amounts of proteins pooled from triplicate samples separated by 7% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) were trans-blotted to nitrocellulose, blocked with 5% nonfat dry milk for 2 hr at room temperature, and then incubated overnight with primary antibodies (all at a 1:2000 dilution, except for SGLT1 with 1:200 dilution). The primary antibody-bound membranes were washed for 10 min with a washing buffer (PBS solution containing 0.1% NP40) before incubation with corresponding secondary antibodies conjugated with horseradish peroxidase (all at a 1:3000 dilution). After a 30 min washing, immunoreactive signals were visualized by enhanced chemiluminescence.
Immunoprecipitation
The physical interaction between EGFR and SLGT1 was detected by immunoprecipitation. Briefly, cells were lysed by scraping them with a rubber policeman into a lysis buffer (50 mM HEPES, pH 7.4; 150 mM NaCl; 0.1% Triton X-100; 1.5 mM MgCl2; 1 mM EDTA; and 1 mM phenylmethlysulfonyl fluoride) and then incubated for 10 min at 0°C, followed by a 5 s sonication (Sonic Dismembrator; Fisher Scientific, Pittsburgh, PA). The lysates were then cleared by centrifugation for 10 min at 14,000 × g. Protein extracts containing 500 μg protein were subsequently incubated for 12 hr at 4°C with the anti-EGFR monoclonal antibody C225 (10 ng), mouse anti-myc (10 ng), mouse anti-HA (10 ng), or with nonspecific normal mouse immunoglobulin G (IgG) (10 ng) (as a negative control). At that time, 50 μl protein A/G beads were added to precipitate the EGFR complex. The precipitates were washed twice with a lysis buffer and then denatured by heating in sample buffer. Immunoprecipitates were resolved on a 7.5% SDS-PAGE gel followed by transfer to a nitrocellulose membrane. The presence of EGFR, phosphorylated EGFR, SGLT1, myc-EGFR, myc-ECD, myc-ICD, and HA-SG LT1 was detected by western blotting.
RT-PCR to Analyze Changes of EGFR and SGLT1 mRNA in Response to Treatment with EGFR
PC-3MM2 cells treated with EGFR siRNA were cultured for 24 and 48 hr in the presence or absence of the proteasome inhibitor MG132 (5 μM). At each time, total RNA prepared using Trizol reagent (Ambion, Inc., Austin, TX) was collected from triplicate samples. RT-PCR was performed with a one-step RT-PCR kit (BD Biosciences) to detect the mRNA levels of SGLT1, SGLT2, and EGFR. The mRNA level of β-actin was used as an internal control. The sequences of primers for EGFR were 5′-TTTCGATACCCAGGACCAAGCCA CAGCAGG-3′ and 5′-AATATTCTTGCTGGATGCGTTTCTGTA-3′, for SGLT1 were 5′-TGGCAGGCCGAAGTATGGTGT-3′ and 5′-ATGAATATGGCCCCC GAGAAGA-3′, for SGLT2 were 5′ACACGGACACGGTACAGACCTT-3′ and 5′-GAACAGCACAATGGCGAAGTAGA-3, and for β-actin were 5′-ATCTGGCACCACACCTTCTACAATG-3′ and 5′-CGTCATACTCCTGCTTGCTG-3′. The RT-PCR reaction program was set to 50°C for 1 hr and 94°C for 5 min, followed by 30 cycles of 94°C for 40 s, 56°C for 40 s, and 72°C for 50 s with an extension at 72°C for 10 min. The PCR products were analyzed with a 1% agarose gel stained with ethidium bromide and visualized under ultraviolet light.
Transmission Electron Microscopy
Transmission electron microscopy was conducted by Kenneth Dunner, Jr., in the High Resolution Electron Microscopy Facility at M.D. Anderson (www.mdanderson.org/HREMF).
Statistical Analysis
The Student’s t test was used to assess the difference of glucose levels of cells treated with AEE788 and EGFR siRNA and the difference of cell counts at each “phase” of cell cycle of treated and control cells. p < 0.01 was defined as the statistical significance.
Supplementary Material
The Supplemental Data include five supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/13/5/385/DC1/.
Acknowledgments
This work was supported in part by SPORE in Prostate Cancer CA-90270 (to I.J.F.), SPORE in Breast Cancer CA-116199 (to M-C. Hung) and Cancer Center Support Core grant CA-16672, National Cancer Institute, National Institutes of Health. Z.W. was partially supported by an Odyssey Fellowship Award from The University of Texas M.D. Anderson Cancer Center. We thank David Zhiwei He and Karen Ramirez for excellent technical support.
References
- Arteaga CL. Epidermal growth factor receptor dependence in human tumors: More than just expression? Oncologist. 2002;7 (Suppl 4):31–39. doi: 10.1634/theoncologist.7-suppl_4-31. [DOI] [PubMed] [Google Scholar]
- Baltzan MA, Andres R, Cader G, Zierler KL. Heterogeneity of forearm metabolism with special reference to free fatty acids. J Clin Invest. 1962;41:116–125. doi: 10.1172/JCI104453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Gill GN. Mutational analysis of the nucleotide binding site of the epidermal growth factor receptor and v-Src protein-tyrosine kinases. J Biol Chem. 1996;271:22619–22623. doi: 10.1074/jbc.271.37.22619. [DOI] [PubMed] [Google Scholar]
- Cohen EE, Rosen F, Stadler WM, Recant W, Stenson K, Huo D, Vokes EE. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21:1980–1987. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- Coker KJ, Staros JV, Guyer CA. A kinase-negative epidermal growth factor receptor that retains the capacity to stimulate DNA synthesis. Proc Natl Acad Sci USA. 1994;91:6967–6971. doi: 10.1073/pnas.91.15.6967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancey JE, Freidlin B. Targeting epidermal growth factor receptor–are we missing the mark? Lancet. 2003;362:62–64. doi: 10.1016/S0140-6736(03)13810-X. [DOI] [PubMed] [Google Scholar]
- Davidson NE, Gelmann EP, Lippman ME, Dickson RB. Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol Endocrinol. 1987;1:216–223. doi: 10.1210/mend-1-3-216. [DOI] [PubMed] [Google Scholar]
- Elfeber K, Köhler A, Lutzenburg M, Osswald C, Galla HJ, Witte O, Koepsell H. Localization of the Na+-D-glucose cotransporter SGLT1 in the blood-brain barrier. Histochem Cell Biol. 2004;121:201–207. doi: 10.1007/s00418-004-0633-9. [DOI] [PubMed] [Google Scholar]
- Ewald JA, Wilkinson JC, Guyer CA, Staros JV. Ligand- and kinase activityindependent cell survival mediated by the epidermal growth factor receptor expressed in 32D cells. Exp Cell Res. 2003;282:121–131. doi: 10.1016/s0014-4827(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Ford AC, Grandis JR. Targeting epidermal growth factor receptor in head and neck cancer. Head Neck. 2003;25:67–73. doi: 10.1002/hed.10224. [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Yano S, Giaccone G. Final results from a phase II trial of ZD1839 for patients with advanced non-small lung cancer. Proc Am Soc Clin Oncol. 2002;21:298a. [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1:1311–1318. [PubMed] [Google Scholar]
- Harari PM, Huang SM. Combining EGFR inhibitors with radiation or chemotherapy: Will preclinical studies predict clinical results? Int. J Radiat Oncol Biol Phys. 2004;58:976–983. doi: 10.1016/j.ijrobp.2003.09.097. [DOI] [PubMed] [Google Scholar]
- Hardin JA, Wong JK, Cheeseman CI, Gall DG. The effect of luminal epidermal growth factor on enterocyte glucose and proline transport. Am J Physiol Gastrointest Liver Physiol. 1996;271:G509–G515. doi: 10.1152/ajpgi.1996.271.3.G509. [DOI] [PubMed] [Google Scholar]
- Helmke BM, Reisser C, Idzko M, Dyckhoff G, Herold-Mende C. Expression of SGLT-1 in preneoplastic and neoplastic lesions of the head and neck. Oral Oncol. 2004;40:28–35. doi: 10.1016/s1368-8375(03)00129-5. [DOI] [PubMed] [Google Scholar]
- Honda K, Kato K, Dairaku N, Iijima K, Koike T, Imatani A, Sekine H, Ohara S, Matsui H, Shimosegawa T. High levels of intracellular ATP prevent nitric oxideinduced apoptosis in rat gastric mucosal cells. Int J Exp Pathol. 2003;84:281–288. doi: 10.1111/j.0959-9673.2003.00364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007;282:10432–10440. doi: 10.1074/jbc.M610014200. [DOI] [PubMed] [Google Scholar]
- Hudelist G, Köstler WJ, Czerwenka K, Kubista E, Attems J, Müller R, Gschwantler-Kaulich D, Manavi M, Huber I, Hoschützky H, et al. Her-2/neu and EGFR tyrosine kinase activation predict the efficacy of trastuzumab-based therapy in patients with metastatic breast cancer. Int J Cancer. 2006;118:1126–1134. doi: 10.1002/ijc.21492. [DOI] [PubMed] [Google Scholar]
- Ikari A, Nagatani Y, Tsukimoto M, Harada H, Miwa M, Takagi K. Sodiumdependent glucose transporter reduces peroxynitrite and cell injury caused by cisplatin in renal tubular epithelial cells. Biochim Biophys Acta. 2005;1717:109–117. doi: 10.1016/j.bbamem.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Inman WH, Colowick SP. Stimulation of glucose uptake by transforming growth factor beta: evidence for the requirement of epidermal growth factor-receptor activation. Proc Natl Acad Sci USA. 1985;82:1346–1349. doi: 10.1073/pnas.82.5.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N, DeMarco RA, Mailiard RB, Han J, Rabinowich H, Kalinski P, Stolz DB, Zeh HJ, III, Lotze MT. Cytolytic cells induce HMGB1 release from melanoma cell lines. J Leukoc Biol. 2007;81:75–83. doi: 10.1189/jlb.0306169. [DOI] [PubMed] [Google Scholar]
- Kinzer D, Lehmann V. Extracellular ATP and adenosine modulate tumor necrosis factor-induced lysis of L929 cells in the presence of actinomycin D. J Immunol. 1991;146:2708–2711. [PubMed] [Google Scholar]
- Kris MG, Natale RB, Herbst RS. A Phase II trial of ZD1839 in advanced non-small cell lung cancer patients who had failed platinum- and docetaxelbased regiemens. Proc Am Soc Clin Oncol. 2002;21:292a. [Google Scholar]
- Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5:886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- Lee WS, Kanai Y, Wells R, Hediger M. The high affinity Na+/glucose cotransporter. J Biol Chem. 1994;269:12032–12039. [PubMed] [Google Scholar]
- Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, Jenkins NA, Lee DC. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes Dev. 1994;8:399–413. doi: 10.1101/gad.8.4.399. [DOI] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: Cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, He L, Kim JS, Elmore SP, Cascio WE, Brenner DA. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4:769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- Malhotra R, Brosius FC., III Glucose uptake and glycolysis reduce hypoxiainduced apoptosis in cultured neonatal rat cardiac myocytes. J Biol Chem. 1999;274:12567–12575. doi: 10.1074/jbc.274.18.12567. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J. The epidermal growth factor receptor as a target for cancer therapy. Endocr Relat Cancer. 2001;8:3–9. doi: 10.1677/erc.0.0080003. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- Nagy P, Arndt-Jovin DJ, Jovin TM. Small interfering RNAs suppress the expression of endogenous and GFP-fused epidermal growth factor receptor (erbB1) and induce apoptosis in erbB1-overexpressing cells. Exp Cell Res. 2003;285:39–49. doi: 10.1016/s0014-4827(02)00050-2. [DOI] [PubMed] [Google Scholar]
- Nicholson RJ, Gee JMW, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37 (Suppl 4):S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- Nishimura Y, Romer LH, Lemasters JJ. Mitochondrial dysfunction and cytoskeletal disruption during chemical hypoxia to cultured rat hepatic sinusoidal endothelial cells: The pH paradox and cytoprotection by glucose, acidotic pH, and glycine. Hepatology. 1998;27:1039–1049. doi: 10.1002/hep.510270420. [DOI] [PubMed] [Google Scholar]
- Palaga T, Kataoka T, Nagai K. Extracellular ATP inhibits apoptosis and maintains cell viability by inducing autocrine production of interleukin-4 in a myeloid progenitor cell line. Int Immunopharmacol. 2004;4:953–961. doi: 10.1016/j.intimp.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Pattaway CA, Pathak S, Greene G, Ramirez E, Wilson MR, Killion JJ, Fidler IJ. Selection of highly metastatic variants of different human prostatic carcinomas using orthotopic implantation in nude mice. Clin Cancer Res. 1996;2:1627–1636. [PubMed] [Google Scholar]
- Peng D, Fan Z, Lu Y, DeBlasio T, Scher H, Mendelsohn J. Anti-epidermal growth factor receptor monoclonal antibody 225 up-regulates p27KIP1 and induces G1 arrest in prostatic cancer cell line DU145. Cancer Res. 1996;56:3666–3669. [PubMed] [Google Scholar]
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- Piazzi G, Paterini P, Ceccarelli C, Pantaleo MA, Biasco G. Molecular determination of epidermal growth factor receptor in normal and neoplastic colorectal mucosa. Br J Cancer. 2006;95:1525–1528. doi: 10.1038/sj.bjc.6603441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeck U, Jost M, Kari C, Shih DT, Lavker RM, Ewert DL, Jensen PJ. EGF-R dependent regulation of keratinocyte survival. J Cell Sci. 1997;110:113–121. doi: 10.1242/jcs.110.2.113. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis. 2006;11:473–485. doi: 10.1007/s10495-006-5881-9. [DOI] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler P, Allegrini PB, Brandt R, Brueggen J, Cozens R, Fabbro D, Grosios K, Lane HA, McSheehy P, Mestan J, et al. AEE788: A dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004;64:4931–4941. doi: 10.1158/0008-5472.CAN-03-3681. [DOI] [PubMed] [Google Scholar]
- Wells A. EGF receptor. Int J Biochem Cell Biol. 1999;31:637–643. doi: 10.1016/s1357-2725(99)00015-1. [DOI] [PubMed] [Google Scholar]
- Wood IS, Tryhurn P. Glucose transporters (GLUT and SGLT): Expanded families of sugar transport proteins. Br J Nutr. 2003;89:3–9. doi: 10.1079/BJN2002763. [DOI] [PubMed] [Google Scholar]
- Wright EM, Loo DD, Panayotova-Heiermann M, Lostao MP, Hirayama BH, Mackenzie B, Boorer K, Zampighi G. ‘Active’ sugar transport in eukaryotes. J Exp Biol. 1994;196:197–212. doi: 10.1242/jeb.196.1.197. [DOI] [PubMed] [Google Scholar]
- Wu X, Fan Z, Masui H, Rosen N, Mendelsohn J. Apoptosis induced by an antiepidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J Clin Invest. 1995;95:1897–1905. doi: 10.1172/JCI117871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierler K. Whole body glucose metabolism. Am J Physiol. 1999;276:E409–E426. doi: 10.1152/ajpendo.1999.276.3.E409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Supplemental Data include five supplemental figures and can be found with this article online at http://www.cancercell.org/cgi/content/full/13/5/385/DC1/.